

Targeting Natural Plant Metabolites for Hunting SARS-CoV-2 Omicron BA.1 Variant Inhibitors: Extraction, Molecular Docking, Molecular Dynamics, and Physicochemical Properties Study

 , ,
, ,  ,
,  , ,
, ,  ,
,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Extraction and Isolation
2.3. Protein Preparation
2.4. Inhibitor Preparation
2.5. Molecular Docking
2.6. Molecular Dynamics Simulations
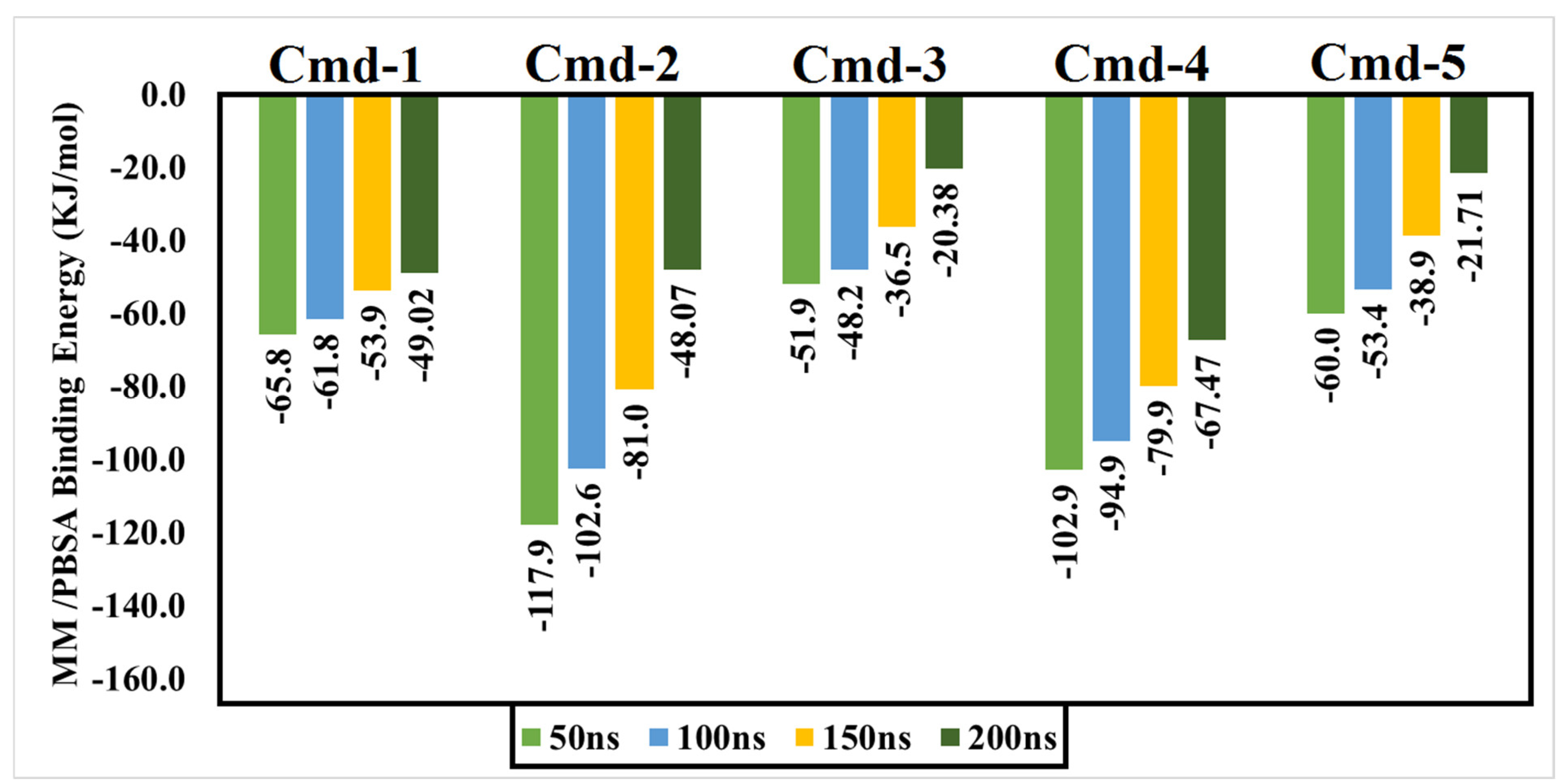
2.7. Binding Energy Calculations
2.8. Assessment of Drug-Relevant Properties
3. Results and Discussion
3.1. Identification of Phytoconstituents
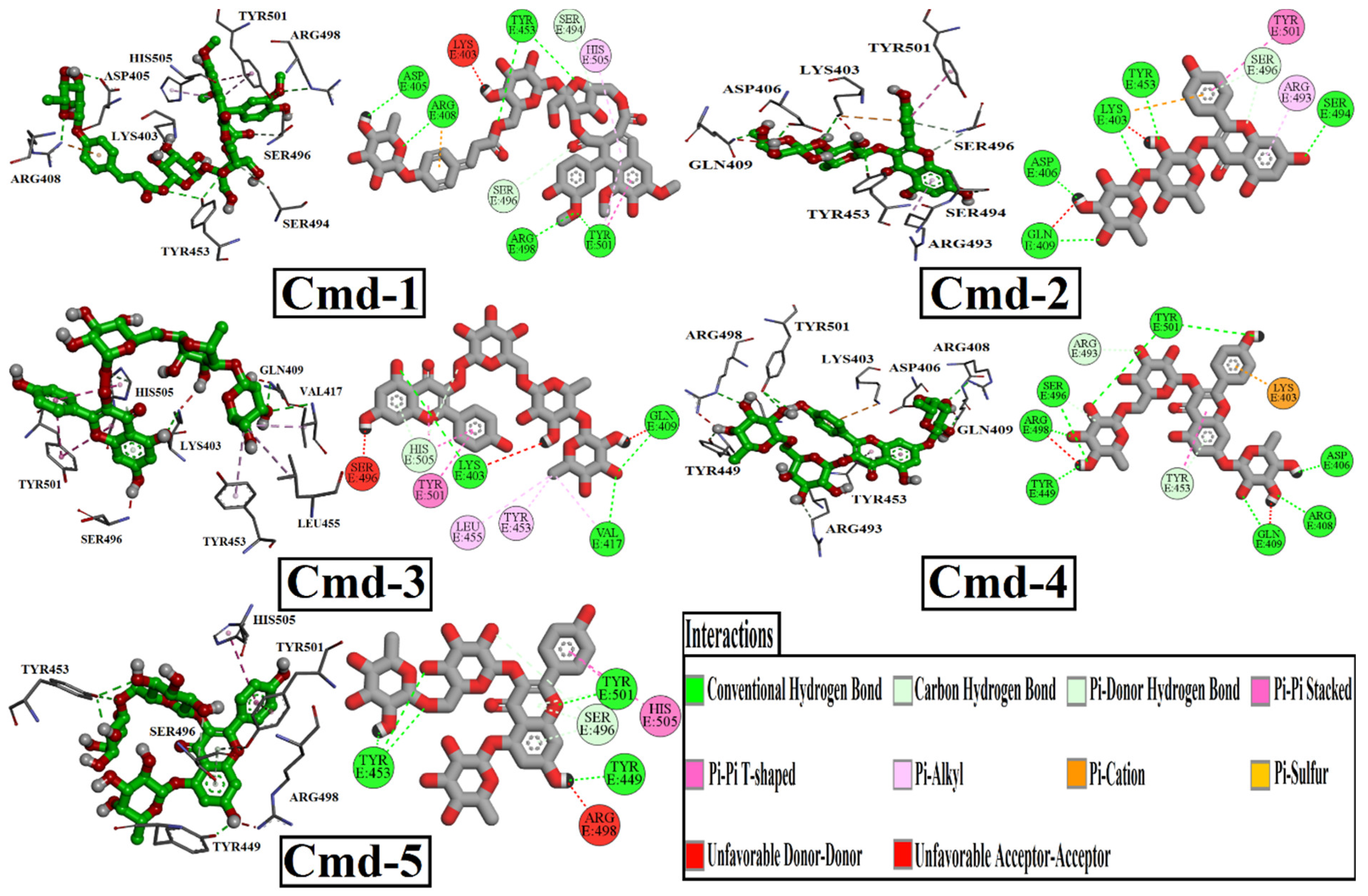
3.2. Molecular Docking
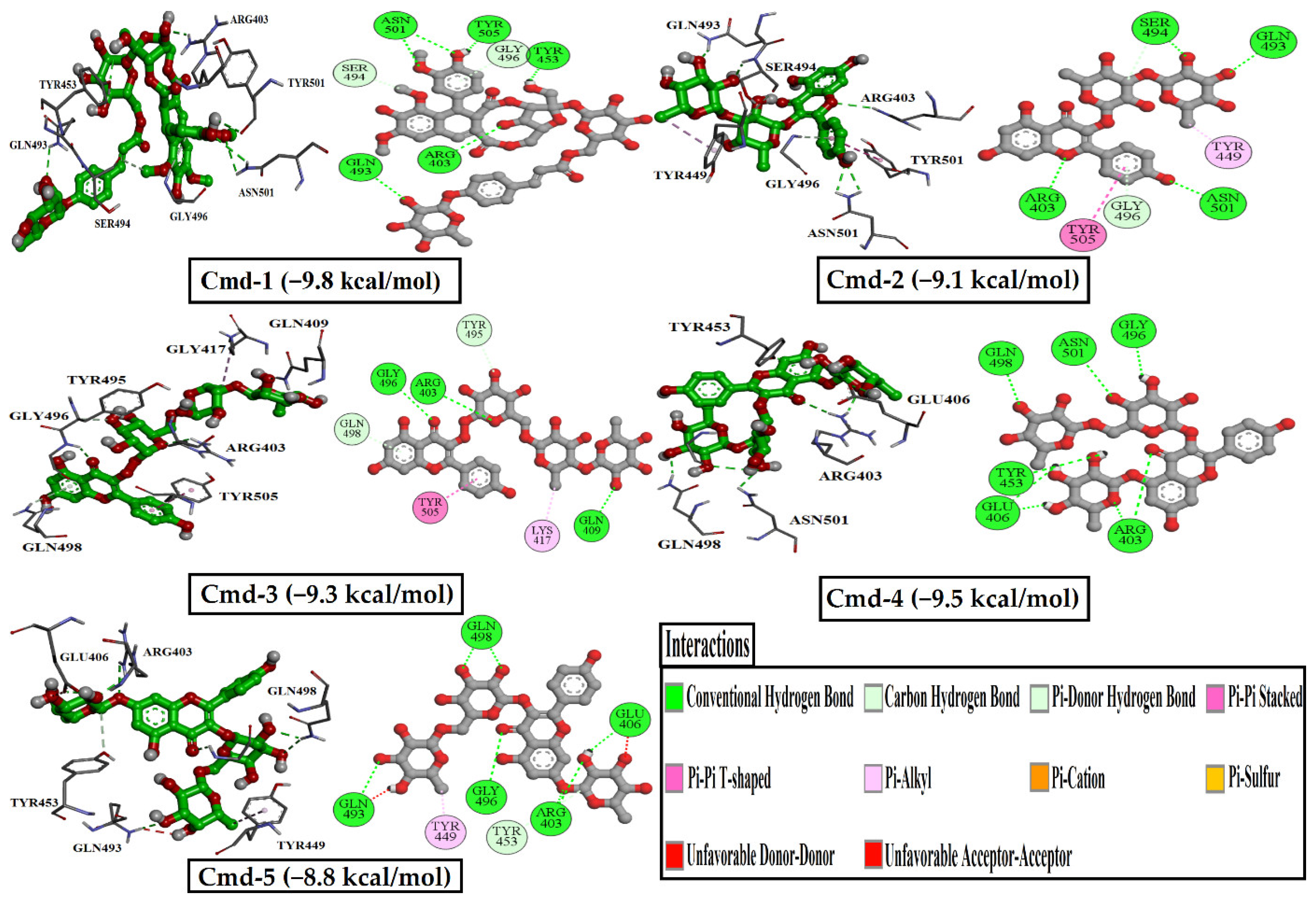
3.3. Molecular Dynamics (MD) Simulations
3.4. Post-MD Analyses
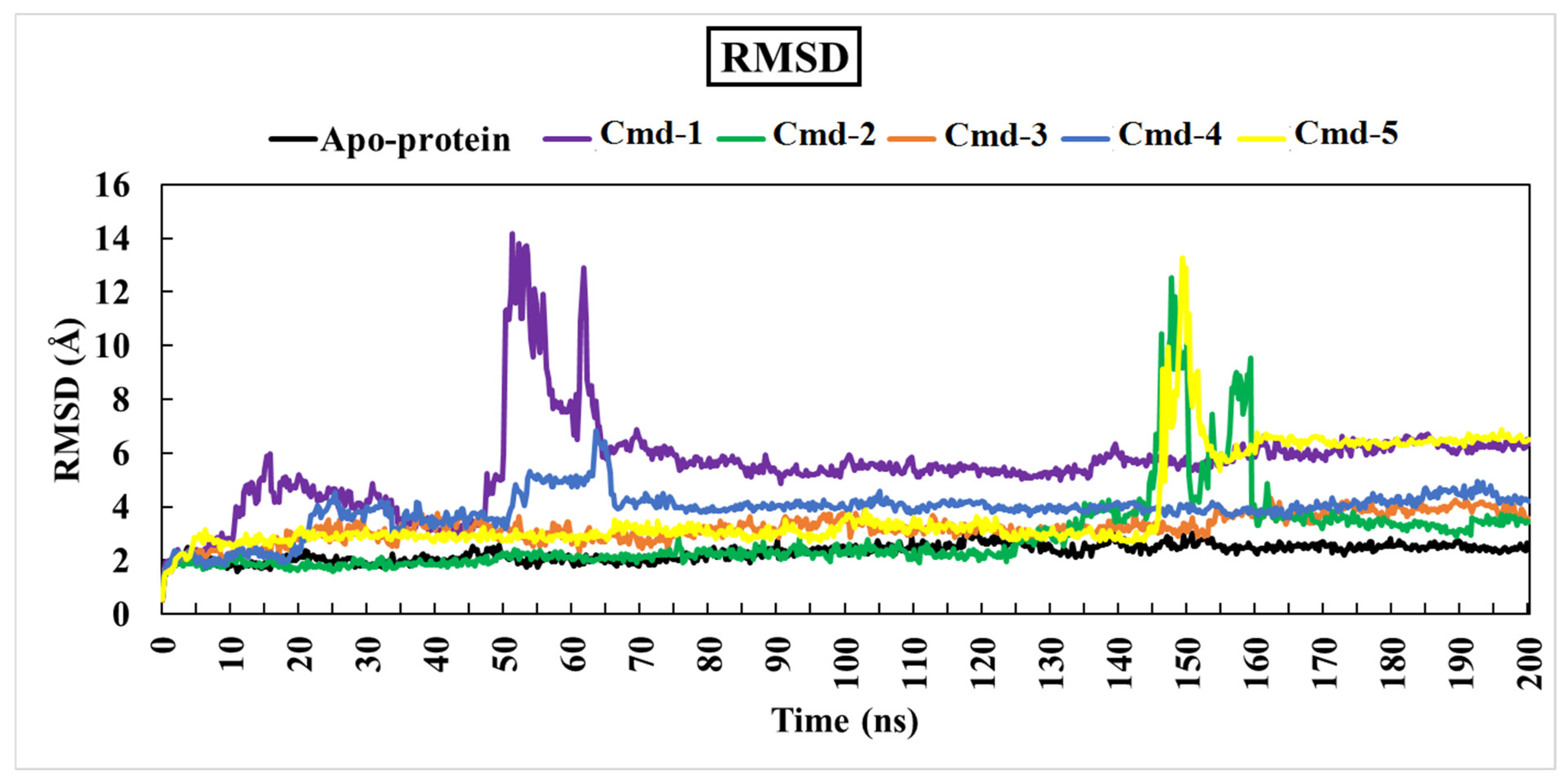
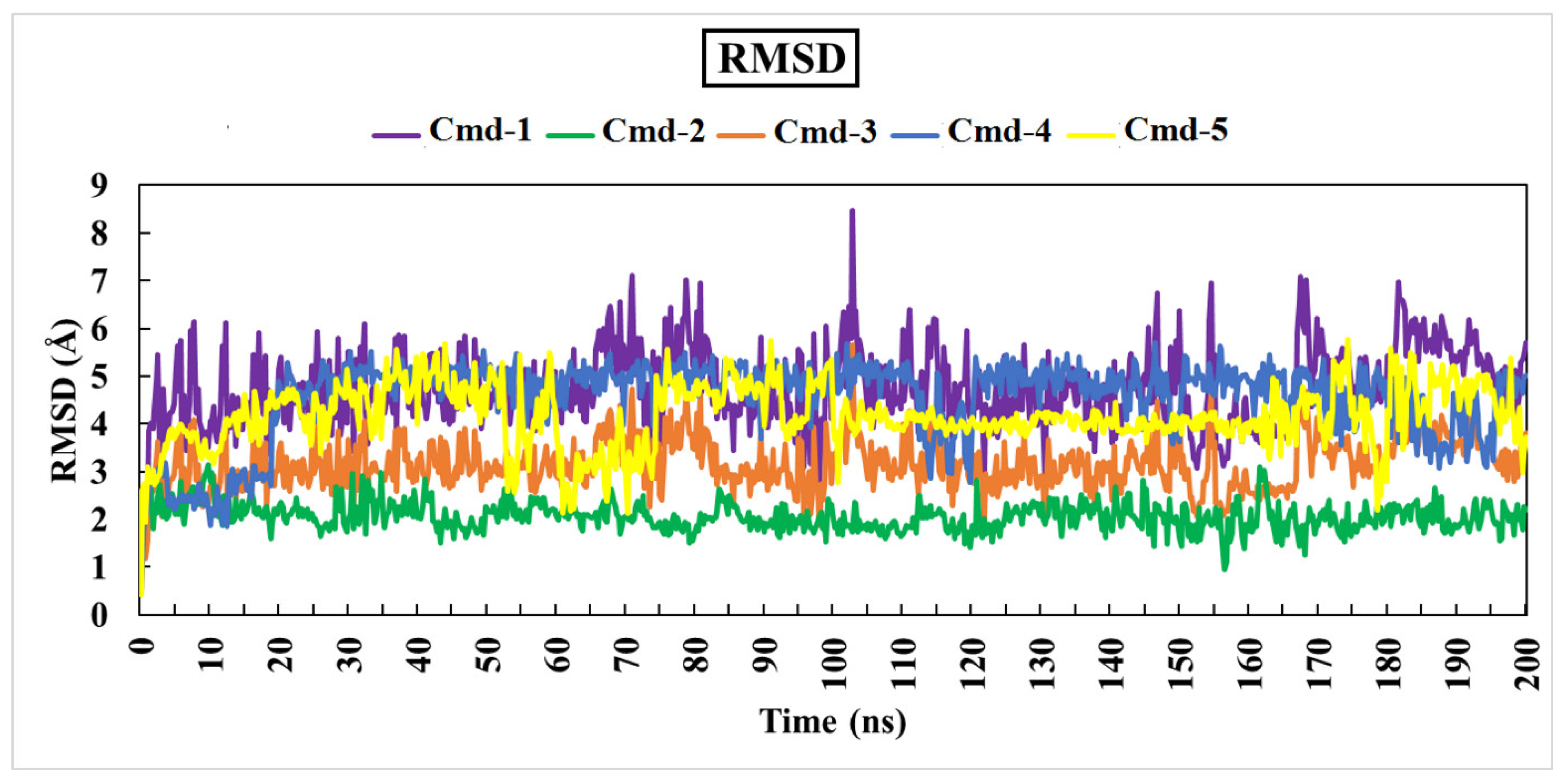
3.4.1. Root-Mean-Square Deviation (RMSD)
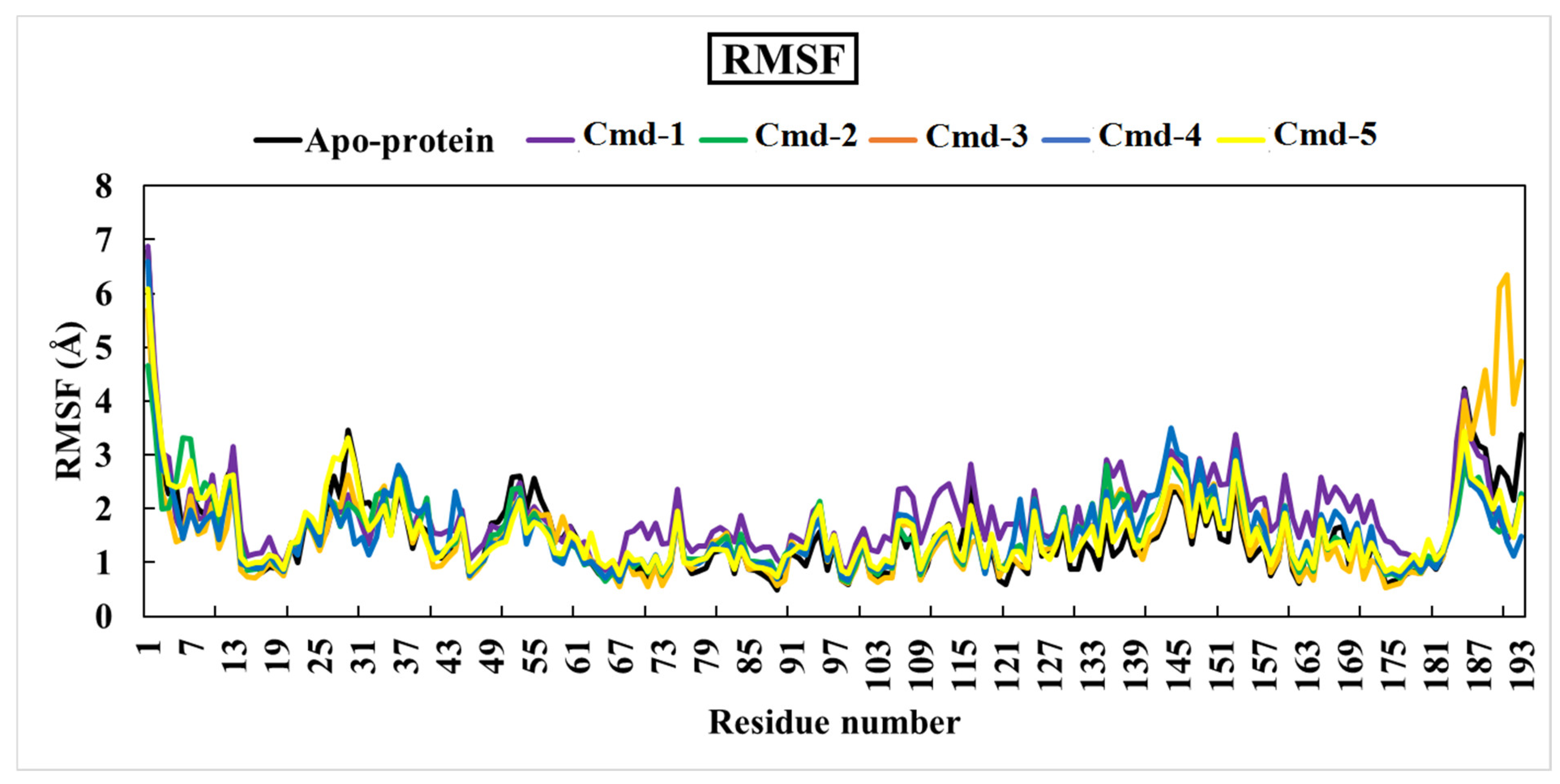
3.4.2. Root-Mean-Square Fluctuation (RMSF)
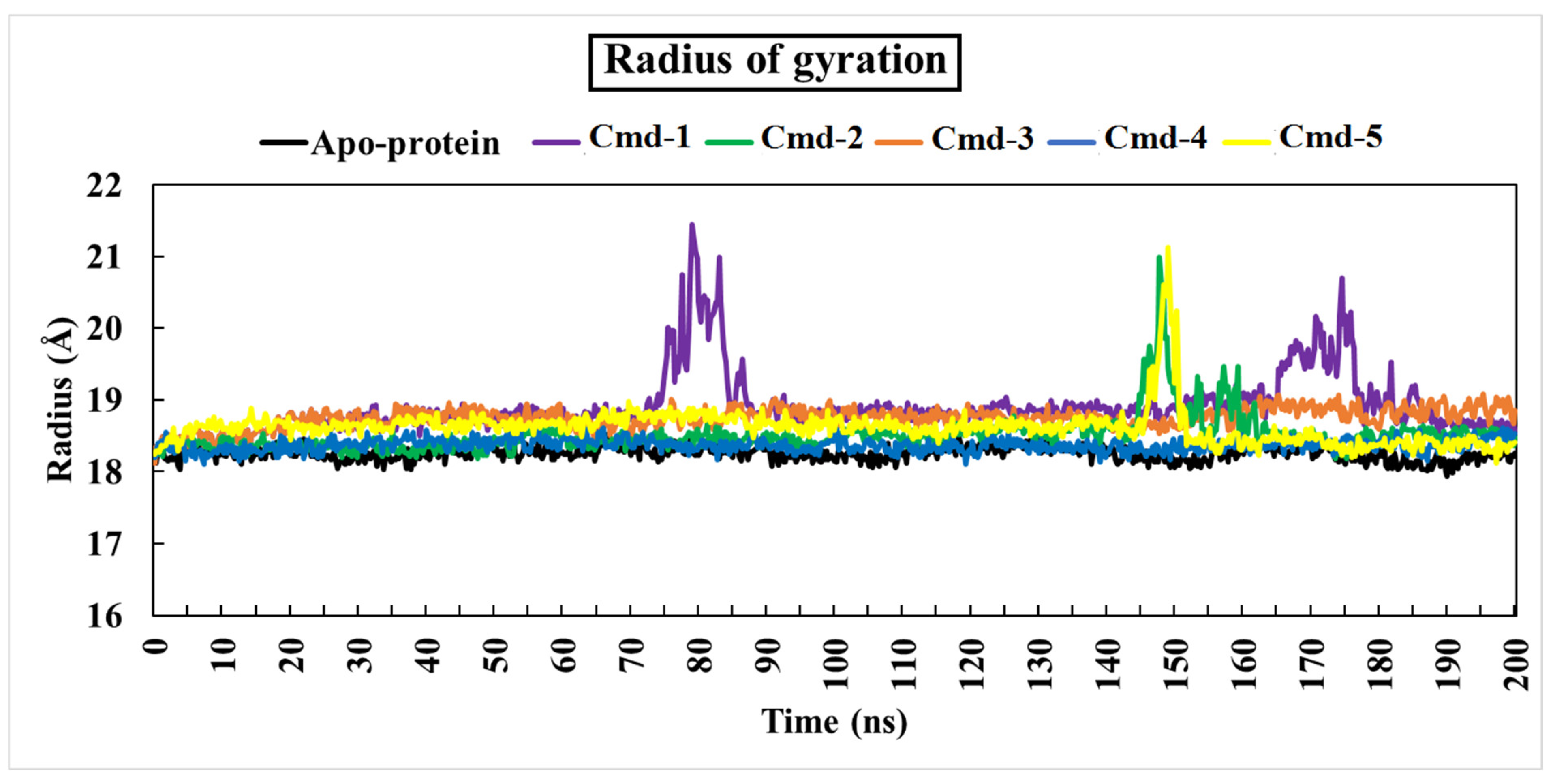
3.4.3. The Radius of Gyration (Rg)
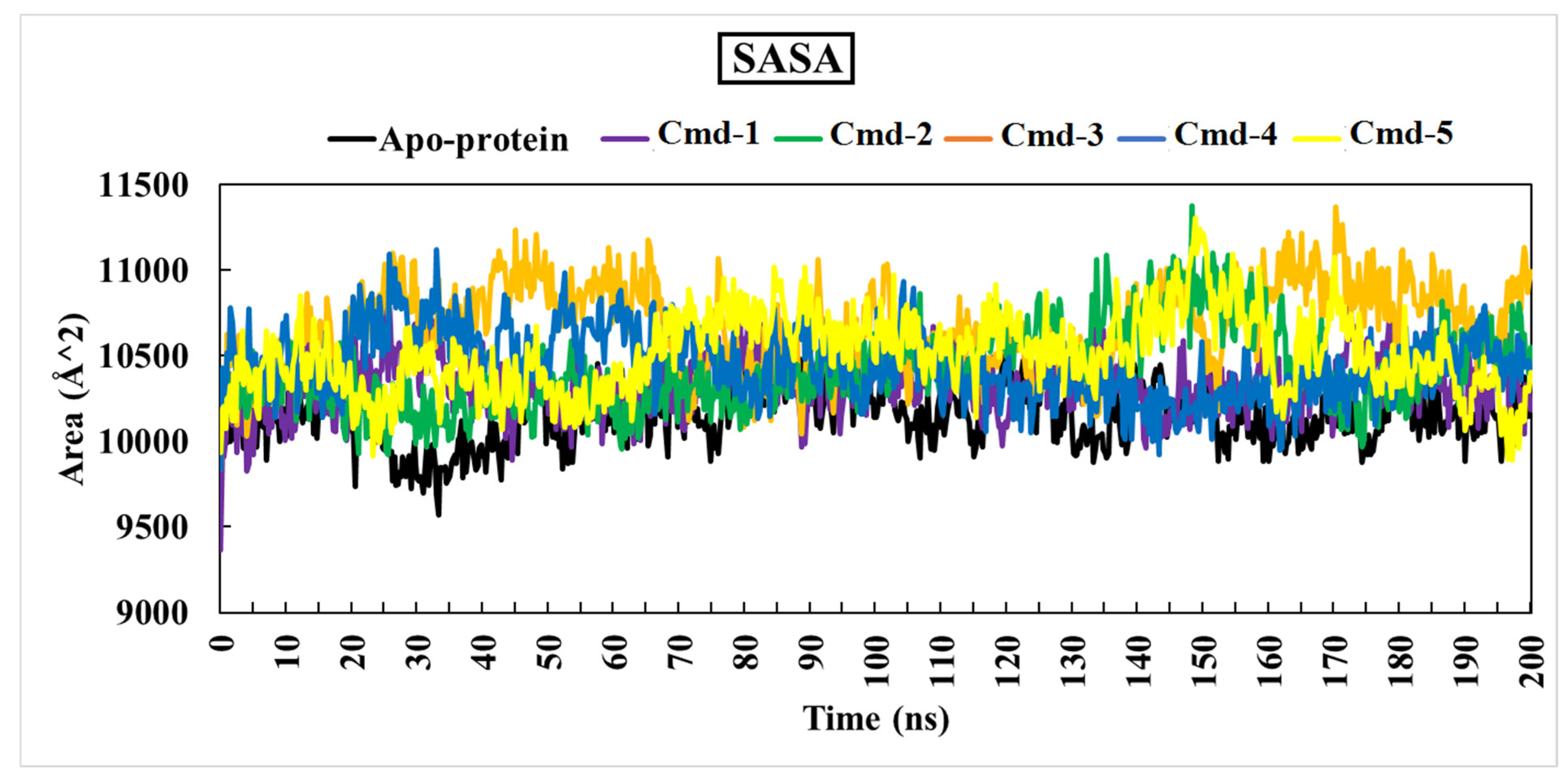
3.4.4. The Solvent-Accessible Surface area Analysis (SASA)
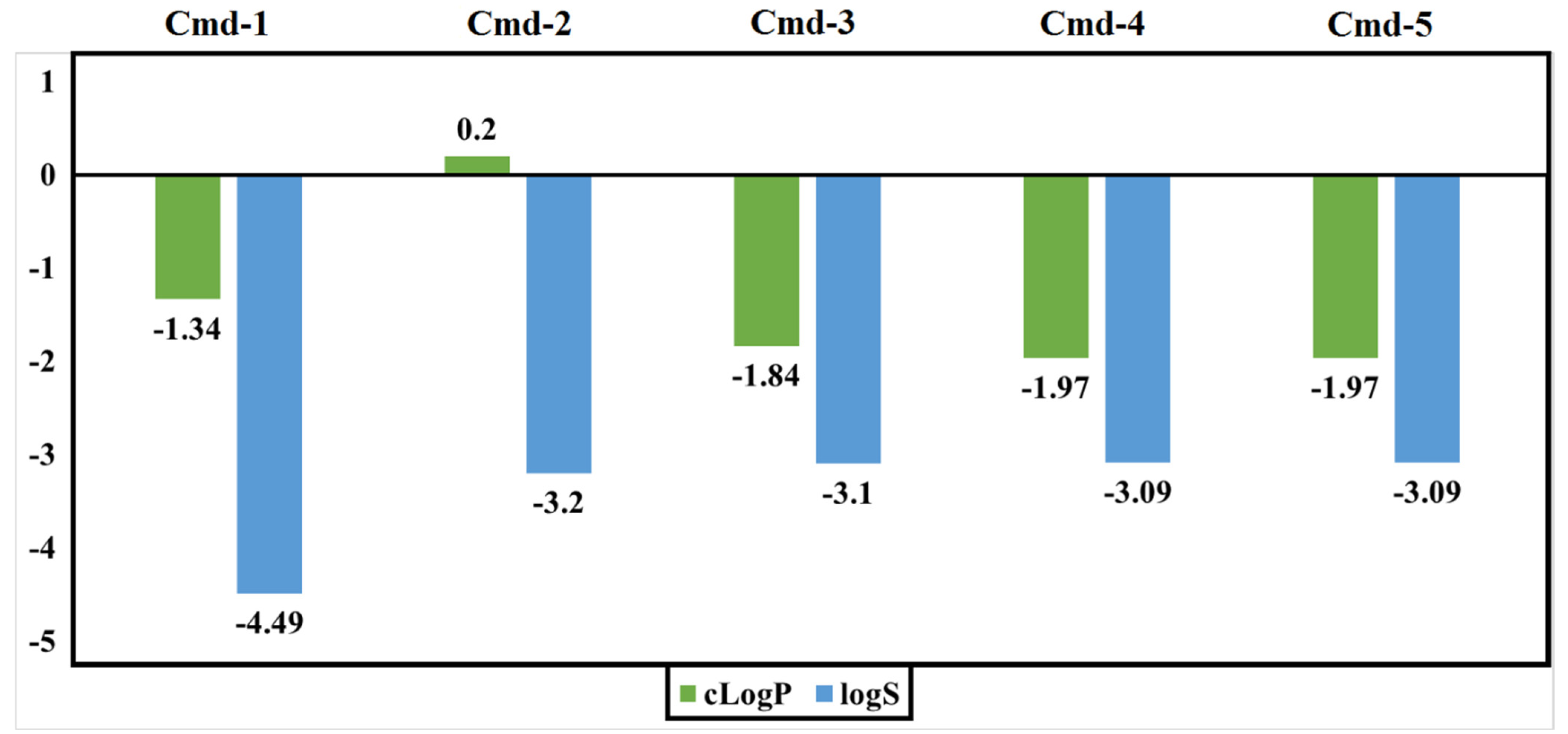
3.5. Drug-Relevant Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID-19’s next move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Krishnan, P.; Ng, D.Y.M.; Chang, L.D.J.; Liu, G.Y.Z.; Cheng, S.S.M.; Hui, M.M.Y.; Fan, M.C.Y.; Wan, J.H.L.; Lau, L.H.K.; et al. Probable Transmission of SARS-CoV-2 Omicron Variant in Quarantine Hotel, Hong Kong, China, November 2021. Emerg. Infect. Dis. 2022, 28, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; Kumar, S.; Ansari, S.; Paweska, J.T.; Maurya, V.K.; Tripathi, A.K.; Abdel-Moneim, A.S. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective. J. Med. Virol. 2022, 94, 1738–1744. [Google Scholar] [CrossRef]
- Maslo, C.; Friedland, R.; Toubkin, M.; Laubscher, A.; Akaloo, T.; Kama, B. Characteristics and Outcomes of Hospitalized Patients in South Africa During the COVID-19 Omicron Wave Compared With Previous Waves. JAMA 2022, 327, 583–584. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Peacock, T.P.; Brown, J.C.; Zhou, J.; Thakur, N.; Sukhova, K.; Newman, J.; Kugathasan, R.; Yan, A.W.C.; Furnon, W.; De Lorenzo, G.; et al. The altered entry pathway and antigenic distance of the SARS-CoV-2 Omicron variant map to separate domains of spike protein. bioRxiv 2022, 15, e0241955. [Google Scholar] [CrossRef]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: A data linkage study. Lancet 2022, 399, 437–446. [Google Scholar] [CrossRef]
- Yushun, W.; Jian, S.; Rachel, G.; Ralph, S.B.; Fang, L.; Tom, G. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.M.S.; Mok, C.K.P.; Leung, Y.W.Y.; Ng, S.S.; Chan, K.C.K.; Ko, F.W.; Chen, C.; Yiu, K.; Lam, B.H.S.; Lau, E.H.Y.; et al. Neutralizing antibodies against the SARS-CoV-2 Omicron variant BA.1 following homologous and heterologous CoronaVac or BNT162b2 vaccination. Nat. Med. 2022, 28, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Quandt, J.; Muik, A.; Salisch, N.; Lui, B.G.; Lutz, S.; Krüger, K.; Wallisch, A.-K.; Adams-Quack, P.; Bacher, M.; Finlayson, A.; et al. Omicron BA.1 breakthrough infection drives cross-variant neutralization and memory B cell formation against conserved epitopes. Sci. Immunol. 2022, 7, eabq2427. [Google Scholar] [CrossRef]
- Qibin, G.; Ke, S.; Gang, Y.; Wei, Z.; Hideki, A.; Fang, L.; Tom, G. Structural Basis for Human Receptor Recognition by SARS-CoV-2 Omicron Variant BA.1. J. Virol. 2022, 96, e00249-22. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (80-) 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Zhang, L.; Mann, M.; Syed, Z.A.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2109905118. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and sub-variants (BA.1.1, BA.2, and BA.3) of SARS-CoV-2 spike infectivity and pathogenicity: A comparative sequence and structural-based computational assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar] [CrossRef] [PubMed]
- Tufts Center for the Study of Drug Development. Cost to develop and win marketing approval for a new drug is $2.6 billion. News of the Tufts Center for the Study of Drug Development, 14 November 2014. [Google Scholar]
- Young, D. Computational Drug Design: A Guide for Computational and Medicinal Chemists; John Wiley & Sons: Birmingham, AL, USA, 2009; ISBN 978-0470126851. [Google Scholar]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Mohamed, E.A.R.; Abdelrahman, A.H.M.; Allemailem, K.S.; Moustafa, M.F.; Shawky, A.M.; Mahzari, A.; Hakami, A.R.; Abdeljawaad, K.A.A.; Atia, M.A.M. Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: In silico drug discovery study. J. Mol. Graph. Model. 2021, 105, 107904. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.R.; Sanad, I.M.; Allam, A.E.; Abouelela, M.E.; Sayed, A.M.; Emam, S.S.; El-Kousy, S.M.; Shimizu, K. Chemical constituents from Limonium tubiflorum and their in silico evaluation as potential antiviral agents against SARS-CoV-2. RSC Adv. 2021, 11, 32346–32357. [Google Scholar] [CrossRef]
- Hassan, H.A.; Hassan, A.R.; Mohamed, E.A.R.; Al-Khdhairawi, A.; Karkashan, A.; Attar, R.; Allemailem, K.S.; Al Abdulmonem, W.; Shimizu, K.; Abdel-Rahman, I.A.M.; et al. Conducting the RBD of SARS-CoV-2 Omicron Variant with Phytoconstituents from Euphorbia dendroides to Repudiate the Binding of Spike Glycoprotein Using Computational Molecular Search and Simulation Approach. Molecules 2022, 27, 2929. [Google Scholar] [CrossRef]
- El-Tantawy, H.M.; Hassan, A.R.; Taha, H.E. Antioxidant potential and LC/MS metabolic profile of anticancer fractions from Echium angustifolium Mill. aerial parts. J. Appl. Pharm. Sci. 2021, 11, 200–208. [Google Scholar] [CrossRef]
- Allam, A.E.; Amen, Y.; Ashour, A.; Assaf, H.K.; Hassan, H.A.; Abdel-Rahman, I.M.; Sayed, A.M.; Shimizu, K. In silico study of natural compounds from sesame against COVID-19 by targeting Mpro, PLpro and RdRp. RSC Adv. 2021, 11, 22398–22408. [Google Scholar] [CrossRef]
- Allam, A.E.; Assaf, H.K.; Hassan, H.A.; Shimizu, K.; Elshaier, Y.A.M.M. An in silico perception for newly isolated flavonoids from peach fruit as privileged avenue for a countermeasure outbreak of COVID-19. RSC Adv. 2020, 10, 29983–29998. [Google Scholar] [CrossRef]
- Zakaryan, H.; Arabyan, E.; Oo, A.; Zandi, K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017, 162, 2539–2551. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Lu, Y.; Li, J.; Song, Y.; Nyamgerelt, M.; Wang, X. Diagnosis and treatment of novel coronavirus pneumonia based on the theory of traditional Chinese medicine. J. Integr. Med. 2020, 18, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sadawe, I.A.; Gbaj, A.A.; Algheryane, A.O.; Meiqal, N.H.; Bensaber, S.M.; Alshoushan, A.A.A.; Gbaj, H.A.; Maamar, M.S.; Hermann, A.; Gbaj, A.M. Evaluation of aerial parts of Echium angustifolium on ciguatoxins toxicity using molecular modeling and albino mice models. MOJ Anat. Physiol. 2020, 7, 134–145. [Google Scholar]
- Popović, B.M.; Blagojević, B.; Kucharska, A.Z.; Agić, D.; Magazin, N.; Milović, M.; Serra, A.T. Exploring fruits from genus Prunus as a source of potential pharmaceutical agents—In vitro and in silico study. Food Chem. 2021, 358, 129812. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.R. Chemical profile and cytotoxic activity of a polyphenolic-rich fraction from Euphorbia dendroides aerial parts. S. Afr. J. Bot. 2022, 147, 332–339. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p Ka s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- BIOVIA, D.S. BIOVIA discovery studio visualizer. Softw. Version 2016, 20, 779. [Google Scholar]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A Gen. Phys. 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Reher, R.; Kühl, T.; Annala, S.; Benkel, T.; Kaufmann, D.; Nubbemeyer, B.; Odhiambo, J.P.; Heimer, P.; Bäuml, C.A.; Kehraus, S.; et al. Deciphering Specificity Determinants for FR900359-Derived Gqα Inhibitors Based on Computational and Structure–Activity Studies. ChemMedChem 2018, 13, 1634–1643. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- El-Rokh, A.R.; Negm, A.; El-Shamy, M.; El-Gindy, M.; Abdel-Mogib, M. Sucrose diester of aryldihydronaphthalene-type lignans from Echium angustifolium Mill. and their antitumor activity. Phytochemistry 2018, 149, 155–160. [Google Scholar] [CrossRef]
- Agalar, H.G.D.; Ciftci, G.A.; Goger, F.; Kirimer, N. Activity Guided Fractionation of Arum italicum Miller Tubers and the LC/MS-MS Profiles. Rec. Nat. Prod. 2018, 12, 64–75. [Google Scholar] [CrossRef]
- Veit, M.; Pauli, G.F. Major Flavonoids from Arabidopsis thaliana Leaves. J. Nat. Prod. 1999, 62, 1301–1303. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-R.; Zhou, P.; Zhi, Y.-E.; Wu, J.; Zhang, S. Three new flavonol triglycosides from Derris trifoliata. J. Asian Nat. Prod. Res. 2009, 11, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Ablajan, K.; Abliz, Z.; Shang, X.-Y.; He, J.-M.; Zhang, R.-P.; Shi, J.-G. Structural characterization of flavonol 3,7-di-O-glycosides and determination of the glycosylation position by using negative ion electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2006, 41, 352–360. [Google Scholar] [CrossRef]
- Zhou, J.; Xie, G.; Yan, X. Encyclopedia of Traditional Chinese Medicines—Molecular Structures, Pharmacological Activities, Natural Sources and Applications; Springer: Berlin/Heidelberg, Germany, 2011; ISBN 978-3-642-16740-9. [Google Scholar]
- Halabalaki, M.; Urbain, A.; Paschali, A.; Mitakou, S.; Tillequin, F.; Skaltsounis, A.-L. Quercetin and Kaempferol 3-O-[α-l-Rhamnopyranosyl-(1→2)-α-l-arabinopyranoside]-7-O-α-l-rhamnopyranosides from Anthyllis hermanniae: Structure Determination and Conformational Studies. J. Nat. Prod. 2011, 74, 1939–1945. [Google Scholar] [CrossRef]
- Manguro, L.O.A.; Ugi, I.; Lemmen, P.; Hermann, R. Flavonol glycosides of Warburgia ugandensis leaves. Phytochemistry 2003, 64, 891–896. [Google Scholar] [CrossRef]
- Petpiroon, N.; Suktap, C.; Pongsamart, S.; Chanvorachote, P.; Sukrong, S. Kaempferol-3-O-rutinoside from Afgekia mahidoliae promotes keratinocyte migration through FAK and Rac1 activation. J. Nat. Med. 2015, 69, 340–348. [Google Scholar] [CrossRef]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; Pérez-Guerrero, C.; López-Lázaro, M. A review on the dietary flavonoid kaempferol. Mini Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef]
- Ding, Y.; Fang, Y.; Moreno, J.; Ramanujam, J.; Jarrell, M.; Brylinski, M. Assessing the similarity of ligand binding conformations with the Contact Mode Score. Comput. Biol. Chem. 2016, 64, 403–413. [Google Scholar] [CrossRef]
- Reva, B.A.; Finkelstein, A.V.; Skolnick, J. What is the probability of a chance prediction of a protein structure with an rmsd of 6 å? Fold. Des. 1998, 3, 141–147. [Google Scholar] [CrossRef]
- Azam, S.S.; Uddin, R.; Wadood, A. Structure and dynamics of alpha-glucosidase through molecular dynamics simulation studies. J. Mol. Liq. 2012, 174, 58–62. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spike Protein | Mutation Sites | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 339 | 371 | 373 | 375 | 417 | 440 | 446 | 477 | 478 | 484 | 493 | 496 | 498 | 501 | 505 | |
| Wuhan-Hu-1 (wild type) | GLY | SER | SER | SER | LYS | ASN | GLY | SER | THR | GLU | GLN | GLY | GLN | ASN | TYR |
| Omicron BA.1 (mutant type) | ASP | LEU | PRO | PHE | ASN | LYS | SER | ASN | LYS | ALA | ARG | SER | ARG | TYR | HIS |
| Molecule | 2D-Chemical Structure | Docking Score (Kcal/mol) | Binding Features (Hydrogen Bond Length in Å) |
|---|---|---|---|
| 1 |  | −9.7 | ASP405 (2.28 Å), ARG408 (2.80 Å), TYR453 (2.86, 3.13 Å), ARG498 (3.31 Å), TYR501 (3.10 Å) |
| 2 |  | −9.5 | LYS403 (2.99 Å), ASP406 (2.02 Å), GLN409 (3.08 Å), TYR453 (3.06 Å), SER494 (3.22 Å) |
| 3 |  | −9.2 | LYS403 (3.22 Å), GLN409 (2.83 Å), VAL417 (3.27 Å) |
| 4 |  | −9.0 | ASP406 (2.13 Å), ARG408 (2.92 Å), GLN409 (2.90 Å), TYR449 (2.45 Å), SER496 (3.01 Å), ARG498 (3.09 Å), TYR501 (2.75, 3.13 Å) |
| 5 |  | −9.0 | TYR453 (2.28, 2.80, 2.95 Å), TYR449 (2.14 Å), TYR501 (2.96 Å) |
| Toxicity Risk | Mu | Tu | Ir | Re |
|---|---|---|---|---|
| Cmd-1 | G | G | G | R |
| Cmd-2 | G | G | G | G |
| Cmd-3 | G | R | G | G |
| Cmd-4 | G | G | G | Y |
| Cmd-5 | R | G | G | G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, H.A.; Hassan, A.R.; Mohamed, E.A.R.; Al-Khdhairawi, A.; Taha, H.E.; El-Tantawy, H.M.; Abdel-Rahman, I.A.M.; Raslan, A.E.; Allemailem, K.S.; Almatroudi, A.; et al. Targeting Natural Plant Metabolites for Hunting SARS-CoV-2 Omicron BA.1 Variant Inhibitors: Extraction, Molecular Docking, Molecular Dynamics, and Physicochemical Properties Study. Curr. Issues Mol. Biol. 2022, 44, 5028-5047. https://doi.org/10.3390/cimb44100342
Hassan HA, Hassan AR, Mohamed EAR, Al-Khdhairawi A, Taha HE, El-Tantawy HM, Abdel-Rahman IAM, Raslan AE, Allemailem KS, Almatroudi A, et al. Targeting Natural Plant Metabolites for Hunting SARS-CoV-2 Omicron BA.1 Variant Inhibitors: Extraction, Molecular Docking, Molecular Dynamics, and Physicochemical Properties Study. Current Issues in Molecular Biology. 2022; 44(10):5028-5047. https://doi.org/10.3390/cimb44100342
Chicago/Turabian StyleHassan, Heba Ali, Ahmed R. Hassan, Eslam A. R. Mohamed, Ahmad Al-Khdhairawi, Hala E. Taha, Hanan M. El-Tantawy, Iman A. M. Abdel-Rahman, Ali E. Raslan, Khaled S. Allemailem, Ahmad Almatroudi, and et al. 2022. "Targeting Natural Plant Metabolites for Hunting SARS-CoV-2 Omicron BA.1 Variant Inhibitors: Extraction, Molecular Docking, Molecular Dynamics, and Physicochemical Properties Study" Current Issues in Molecular Biology 44, no. 10: 5028-5047. https://doi.org/10.3390/cimb44100342
APA StyleHassan, H. A., Hassan, A. R., Mohamed, E. A. R., Al-Khdhairawi, A., Taha, H. E., El-Tantawy, H. M., Abdel-Rahman, I. A. M., Raslan, A. E., Allemailem, K. S., Almatroudi, A., Alrumaihi, F., Alshiekheid, M. A., Rehman, H. M., Abdelhamid, M. M., Abdel-Rahman, I. M., & Allam, A. E. (2022). Targeting Natural Plant Metabolites for Hunting SARS-CoV-2 Omicron BA.1 Variant Inhibitors: Extraction, Molecular Docking, Molecular Dynamics, and Physicochemical Properties Study. Current Issues in Molecular Biology, 44(10), 5028-5047. https://doi.org/10.3390/cimb44100342