
Identification of Gut Microbiota Affecting Fiber Digestibility in Pigs

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection in Animals
2.2. Chemical Analysis
2.3. 16S rRNA Sequencing and Bioinformatics Analysis
2.4. Statistical Analysis
3. Results
3.1. Variation of Apparent NDF and ADF Digestibility within Pigs
3.2. Comparison of Apparent NDF and ADF Digestibility between High- and Low-AD Pigs
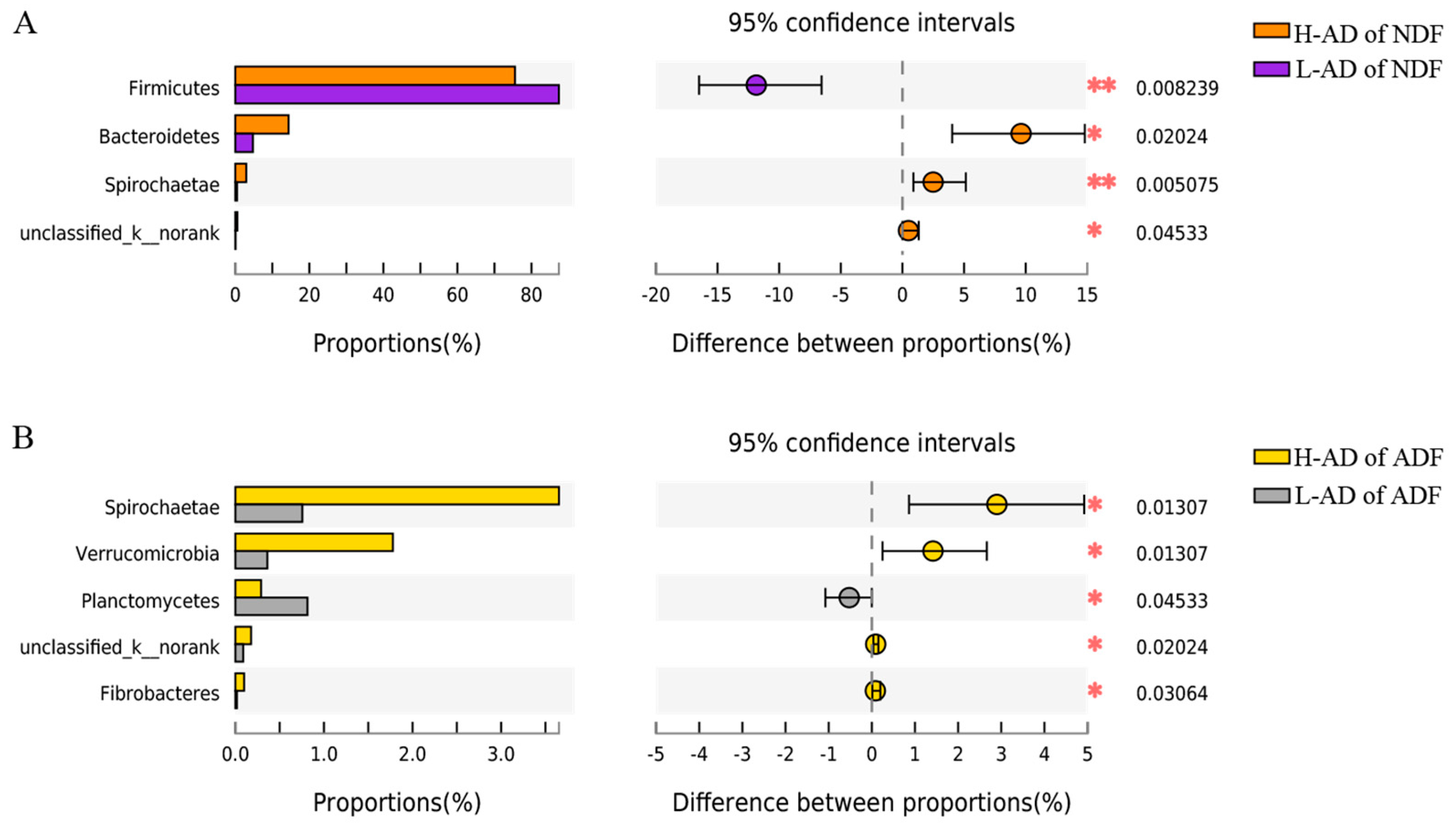
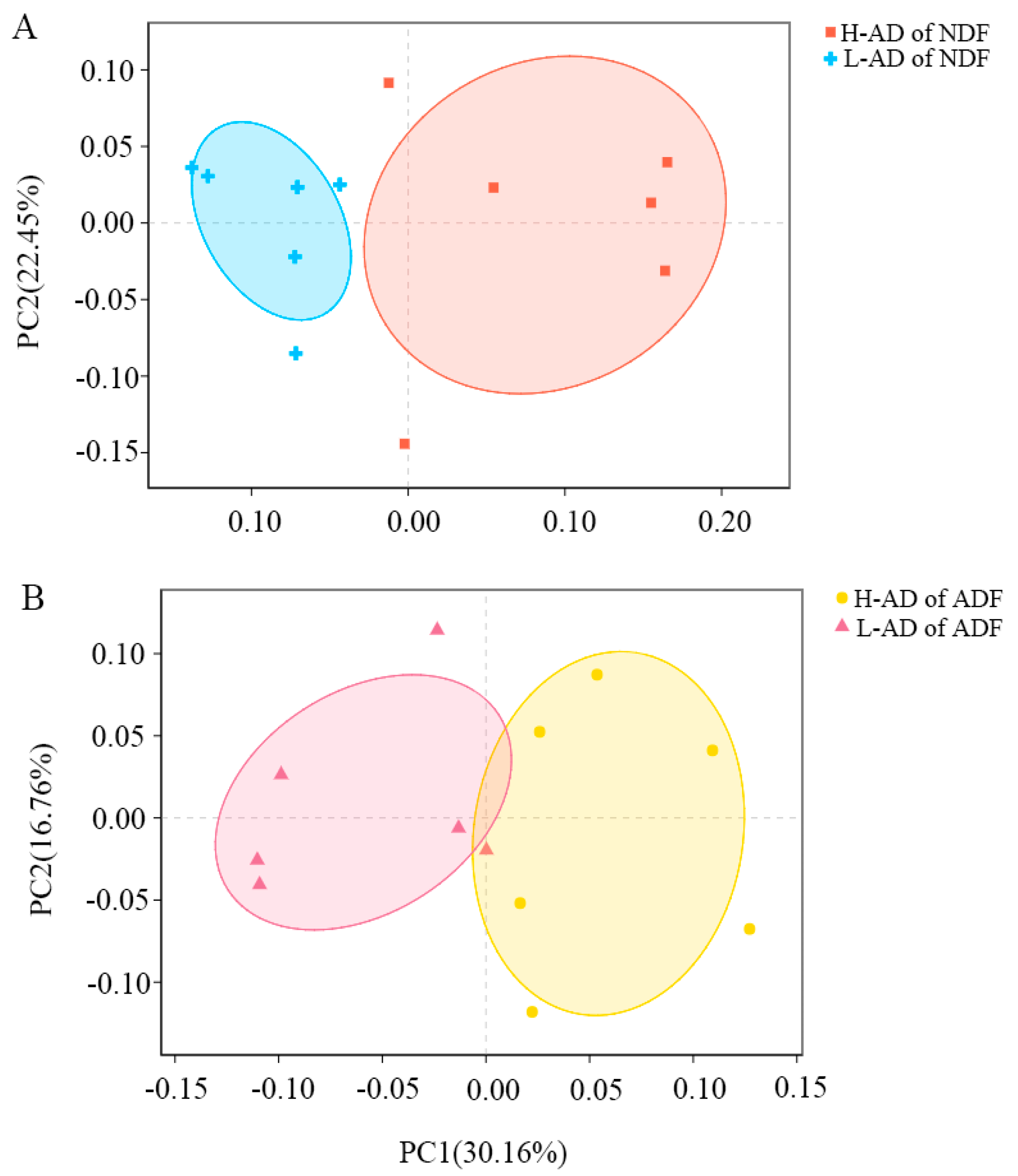
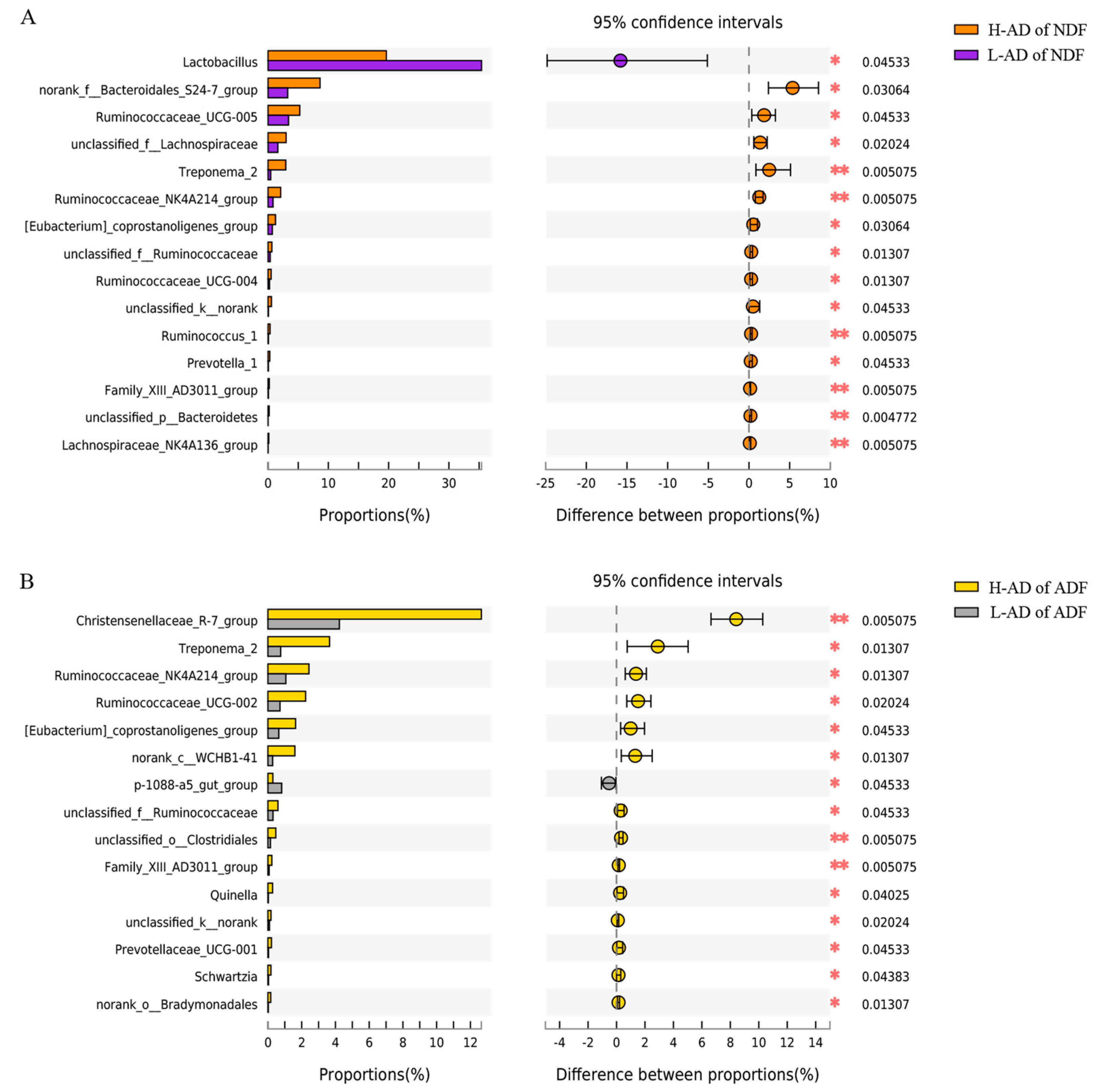
3.3. DNA Sequence Data and Bacterial Community Structure of the ATTD of NDF and of ADF Samples between H- and L-AD Pigs
3.4. Prediction Functions of Microbial Metabolism
4. Discussion
4.1. Chinese Indigenous Pig Showed Better Fiber Tolerance Characteristics as Compared with Foreign Varieties
4.2. Bacterial Community Structures Were Significantly Different between High and Low Fiber Digestibility Groups
4.3. Carbohydrate Transport and Metabolism and Carbohydrate Metabolism Pathway Were One of the Most Important Functions and Pathways of the Potential Biomarkers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiofalo, V.; Liotta, L.; Spanò, G.; D’Alessandro, E.; Chiofalo, B. Dietary neutral detergent fibre level on performance traits and meat quality in nero siciliano pigs. J. Nutr. Ecol. Food Res. 2013, 1, 207–212. [Google Scholar] [CrossRef]
- Mackowiak, K.; Torlinska-Walkowiak, N.; Torlinska, B. Dietary fibre as an important constituent of the diet. Postepy Hig. Med. Dosw. 2016, 70, 104–109. [Google Scholar] [CrossRef]
- Waldern, D.E. A rapid micro-digestion procedure for neutral and acid detergent fiber. Can. J. Anim. Sci. 1971, 51, 67–69. [Google Scholar] [CrossRef]
- Sommer, F.; Backhed, F. The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Isaacson, R.; Kim, H.B. The intestinal microbiome of the pig. Anim. Health Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Mikkelsen, D.; Flanagan, B.M.; Gidley, M.J. “Dietary fibre”: Moving beyond the “soluble/insoluble” classification for monogastric nutrition, with an emphasis on humans and pigs. J. Anim. Sci. Biotechnol. 2019, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Holscher, H.D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef]
- Ashida, H.; Ogawa, M.; Kim, M.; Mimuro, H.; Sasakawa, C. Bacteria and host interactions in the gut epithelial barrier. Nat. Chem Biol. 2011, 8, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Bouter, K.E.; van Raalte, D.H.; Groen, A.K.; Nieuwdorp, M. Role of the gut microbiome in the pathogenesis of obesity and obesity-related metabolic dysfunction. Gastroenterology 2017, 152, 1671–1678. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Pu, G.; Li, P.; Du, T.; Niu, Q.; Fan, L.; Wang, H.; Liu, H.; Li, K.; Niu, P.; Wu, C.; et al. Adding appropriate fiber in diet increases diversity and metabolic capacity of distal gut microbiota without altering fiber digestibility and growth rate of finishing pig. Front. Microbiol. 2020, 11, 533. [Google Scholar] [CrossRef] [PubMed]
- Varel, V.H.; Pond, W.G.; Pekas, J.C.; Yen, J.T. Influence of high-fibre diet on bacterial populations in gastrointestinal tracts of obese- and lean-genotype pigs. Appl. Environ. Microbiol. 1982, 44, 107–112. [Google Scholar] [PubMed]
- Niu, Q.; Li, P.; Hao, S.; Zhang, Y.; Kim, S.W.; Li, H.; Ma, X.; Gao, S.; He, L.; Wu, W.; et al. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 2015, 5, 9938. [Google Scholar] [CrossRef] [PubMed]
- Kemp, B.; den Hartog, L.A.; Klok, J.J.; Zandstra, T. The digestibility of nutrients, energy and nitrogen in the meishan and dutch landrace pig. J. Anim. Physiol. Anim. Nutr. 1991, 65, 263–266. [Google Scholar] [CrossRef]
- Fevrier, C.; Bourdon, D.; Aumaitre, A. Effects of level of dietary fibre from wheat bran on digestibility of nutrients, digestive enzymes and performance in the european large white and chinese mei shan pig. J. Anim. Physiol. Anim. Nutr. 1992, 68, 60–72. [Google Scholar] [CrossRef]
- Janssen, A.W.; Kersten, S. The role of the gut microbiota in metabolic health. FASEB J. 2015, 29, 3111–3123. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Ngoc, T.T.; Len, N.T.; Lindberg, J.E. Impact of fibre intake and fibre source on digestibility, gut development, retention time and growth performance of indigenous and exotic pigs. Animal 2013, 7, 736–745. [Google Scholar] [CrossRef]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The ribosomal database project: Improved alignments and new tools for rrna analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rrna sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, D.; Ruan, Y.; Xue, C.; Zhang, J.; Li, R.; Shen, Q. Deep 16s rrna pyrosequencing reveals a bacterial community associated with banana fusarium wilt disease suppression induced by bio-organic fertilizer application. PLoS ONE 2014, 9, e98420. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xiao, X.H.; Zhang, Q.; Mao, L.L.; Yu, M.; Xu, J.P.; Wang, T. Correlation of placental microbiota with fetal macrosomia and clinical characteristics in mothers and newborns. Oncotarget 2017, 8, 82314–82325. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Hamady, M.; Knight, R. Unifrac—An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform. 2006, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. Emperor: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16s rrna marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Riva, A.; Kuzyk, O.; Forsberg, E.; Siuzdak, G.; Pfann, C.; Herbold, C.; Daims, H.; Loy, A.; Warth, B.; Berry, D. A fiber-deprived diet disturbs the fine-scale spatial architecture of the murine colon microbiome. Nat. Commun. 2019, 10, 4366. [Google Scholar] [CrossRef]
- Le Sciellour, M.; Labussiere, E.; Zemb, O.; Renaudeau, D. Effect of dietary fiber content on nutrient digestibility and fecal microbiota composition in growing-finishing pigs. PLoS ONE 2018, 13, e0206159. [Google Scholar] [CrossRef] [PubMed]
- Magwira, C.A.; Taylor, M.B. Composition of gut microbiota and its influence on the immunogenicity of oral rotavirus vaccines. Vaccine 2018, 36, 3427–3433. [Google Scholar] [CrossRef]
- Lu, X.M.; Lu, P.Z.; Zhang, H. Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16s rdna 454 pyrosequencing. Appl. Microbiol. Biotechnol. 2014, 98, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Pajarillo, E.A.; Chae, J.P.; Balolong, M.P.; Kim, H.B.; Seo, K.S.; Kang, D.K. Pyrosequencing-based analysis of fecal microbial communities in three purebred pig lines. J. Microbiol. 2014, 52, 646–651. [Google Scholar] [CrossRef]
- Kanengoni, A.T.; Chimonyo, M.; Tasara, T.; Cormican, P.; Chapwanya, A.; Ndimba, B.K.; Dzama, K. A comparison of faecal microbial populations of south african windsnyer-type indigenous pigs (sawips) and large white x landrace (lw x lr) crosses fed diets containing ensiled maize cobs. FEMS Microbiol. Lett. 2015, 362, fnv100. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Piazuelo, D.; Estelle, J.; Revilla, M.; Criado-Mesas, L.; Ramayo-Caldas, Y.; Ovilo, C.; Fernandez, A.I.; Ballester, M.; Folch, J.M. Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci. Rep. 2018, 8, 12727. [Google Scholar] [CrossRef]
- Morris, A.; Paulson, J.N.; Talukder, H.; Tipton, L.; Kling, H.; Cui, L.; Fitch, A.; Pop, M.; Norris, K.A.; Ghedin, E. Longitudinal analysis of the lung microbiota of cynomolgous macaques during long-term shiv infection. Microbiome 2016, 4, 38. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. The microbiota-gut-brain axis in obesity. Lancet Gastroenterol. Hepatol. 2017, 2, 747–756. [Google Scholar] [CrossRef]
- Simpson, H.L.; Campbell, B.J. Review article: Dietary fibre-microbiota interactions. Aliment. Pharmacol. Ther. 2015, 42, 158–179. [Google Scholar] [CrossRef]
- Makki, K.; Deehan, E.C.; Walter, J.; Backhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Heinritz, S.N.; Weiss, E.; Eklund, M.; Aumiller, T.; Louis, S.; Rings, A.; Messner, S.; Camarinha-Silva, A.; Seifert, J.; Bischoff, S.C.; et al. Intestinal microbiota and microbial metabolites are changed in a pig model fed a high-fat/low-fiber or a low-fat/high-fiber diet. PLoS ONE 2016, 11, e0154329. [Google Scholar] [CrossRef]
- Tan, Z.; Wang, Y.; Yang, T.; Ao, H.; Chen, S.; Xing, K.; Zhang, F.; Zhao, X.; Liu, J.; Wang, C. Differences in gut microbiota composition in finishing landrace pigs with low and high feed conversion ratios. Antonie Van Leeuwenhoek 2018, 111, 1673–1685. [Google Scholar] [CrossRef]
- Metzler, B.U.; Mosenthin, R. A review of interactions between dietary fiber and the gastrointestinal microbiota and their consequences on intestinal phosphorus metabolism in growing pigs. Asian Austral. J. Anim. Sci. 2008, 21, 603–615. [Google Scholar] [CrossRef]
- Chen, H.; Mao, X.B.; Che, L.Q.; Yu, B.; He, J.; Yu, J.; Han, G.Q.; Huang, Z.Q.; Zheng, P.; Chen, D.W. Impact of fiber types on gut microbiota, gut environment and gut function in fattening pigs. Anim. Feed Sci. Technol. 2014, 195, 101–111. [Google Scholar] [CrossRef]
- Myhrstad, M.C.W.; Tunsjo, H.; Charnock, C.; Telle-Hansen, V.H. Dietary fiber, gut microbiota, and metabolic regulation-current status in human randomized trials. Nutrients 2020, 12, 859. [Google Scholar] [CrossRef] [PubMed]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Backhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef]
- Pu, G.; Hou, L.; Du, T.; Wang, B.; Liu, H.; Li, K.; Niu, P.; Zhou, W.; Huang, R.; Li, P. Effects of short-term feeding with high fiber diets on growth, utilization of dietary fiber, and microbiota in pigs. Front. Microbiol. 2022, 13, 963917. [Google Scholar] [CrossRef]
- Lamendella, R.; Domingo, J.W.; Ghosh, S.; Martinson, J.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ATTD | N | Range, % | Mean ± SE | CV, % |
|---|---|---|---|---|
| NDF | 274 | 44.6–88.3 | 70.4 ± 0.6 | 12.1 |
| ADF | 274 | 30.0–83.1 | 62. 9 ± 1.2 | 18.8 |
| Group | H-AD | L-AD | ||
|---|---|---|---|---|
| N | Mean ± SE, % | N | Mean ± SE, % | |
| NDF | 6 | 83.3 ± 5.2 A | 6 | 53.1 ± 5.1 B |
| ADF | 6 | 75.7 ± 4.8 A | 6 | 41.9 ± 8.5 B |
| Functions | H-AD of NDF∩H-AD of ADF | H-AD of NDF | H-AD of ADF |
|---|---|---|---|
| Carbohydrate transport and metabolism | 8.68 | 8.55 | 7.20 |
| General function prediction only | 8.01 | 7.95 | 8.25 |
| Transcription | 7.58 | 6.30 | 7.55 |
| Amino acid transport and metabolism | 7.42 | 7.94 | 8.71 |
| Translation, ribosomal structure, and biogenesis | 7.22 | 7.70 | 6.96 |
| Replication, recombination, and repair | 7.11 | 7.23 | 7.20 |
| Cell wall/membrane/envelope biogenesis | 7.00 | 9.25 | 6.04 |
| Signal transduction mechanisms | 6.26 | 4.50 | 6.50 |
| Energy production and conversion | 5.60 | 6.23 | 6.22 |
| Inorganic ion transport and metabolism | 5.00 | 5.91 | 5.24 |
| Pathways | H-AD of NDF∩H-AD of ADF | H-AD of NDF | H-AD of ADF |
|---|---|---|---|
| Membrane transport | 13.67 | 8.41 | 12.64 |
| Carbohydrate metabolism | 10.00 | 10.44 | 9.44 |
| Amino acid metabolism | 9.08 | 10.64 | 9.64 |
| Replication and repair | 8.79 | 9.70 | 8.78 |
| Translation | 6.23 | 6.33 | 5.88 |
| Cell motility | 5.47 | - | 4.45 |
| Energy metabolism | 5.26 | 6.69 | 5.65 |
| Poorly characterized | 5.01 | 4.74 | 4.49 |
| Nucleotide metabolism | 3.85 | 4.49 | 3.89 |
| Metabolism of cofactors and vitamins | 3.46 | 4.69 | 4.39 |
| Cellular processes and signaling | - | 3.80 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Q.; Pu, G.; Fan, L.; Gao, C.; Lan, T.; Liu, C.; Du, T.; Kim, S.W.; Niu, P.; Zhang, Z.; et al. Identification of Gut Microbiota Affecting Fiber Digestibility in Pigs. Curr. Issues Mol. Biol. 2022, 44, 4557-4569. https://doi.org/10.3390/cimb44100312
Niu Q, Pu G, Fan L, Gao C, Lan T, Liu C, Du T, Kim SW, Niu P, Zhang Z, et al. Identification of Gut Microbiota Affecting Fiber Digestibility in Pigs. Current Issues in Molecular Biology. 2022; 44(10):4557-4569. https://doi.org/10.3390/cimb44100312
Chicago/Turabian StyleNiu, Qing, Guang Pu, Lijuan Fan, Chen Gao, Tingxu Lan, Chenxi Liu, Taoran Du, Sung Woo Kim, Peipei Niu, Zongping Zhang, and et al. 2022. "Identification of Gut Microbiota Affecting Fiber Digestibility in Pigs" Current Issues in Molecular Biology 44, no. 10: 4557-4569. https://doi.org/10.3390/cimb44100312
APA StyleNiu, Q., Pu, G., Fan, L., Gao, C., Lan, T., Liu, C., Du, T., Kim, S. W., Niu, P., Zhang, Z., Li, P., & Huang, R. (2022). Identification of Gut Microbiota Affecting Fiber Digestibility in Pigs. Current Issues in Molecular Biology, 44(10), 4557-4569. https://doi.org/10.3390/cimb44100312