
Familial Hyperaldosteronism Type 3 with a Rapidly Growing Adrenal Tumor: An In Situ Aldosterone Imaging Study

 , ,
, , 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Case
3. Materials and Methods
3.1. DNA and RNA Isolation from Flash Frozen Tissues, Blood, and Hair Root
3.2. Whole Exome Sequencing
3.3. Microarray Analyses
3.4. Confirmation of CYP11B2 Expression
4. Result
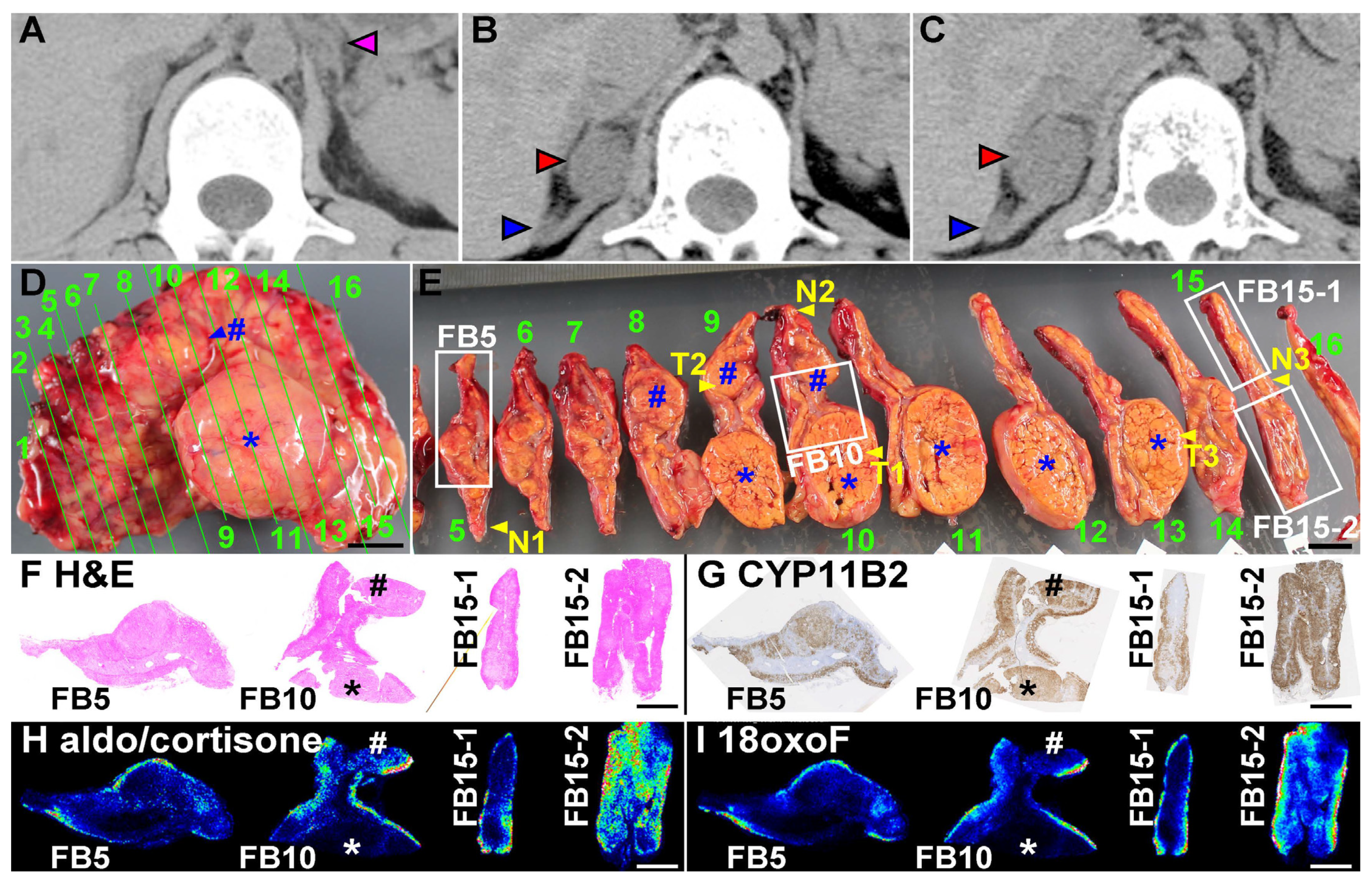
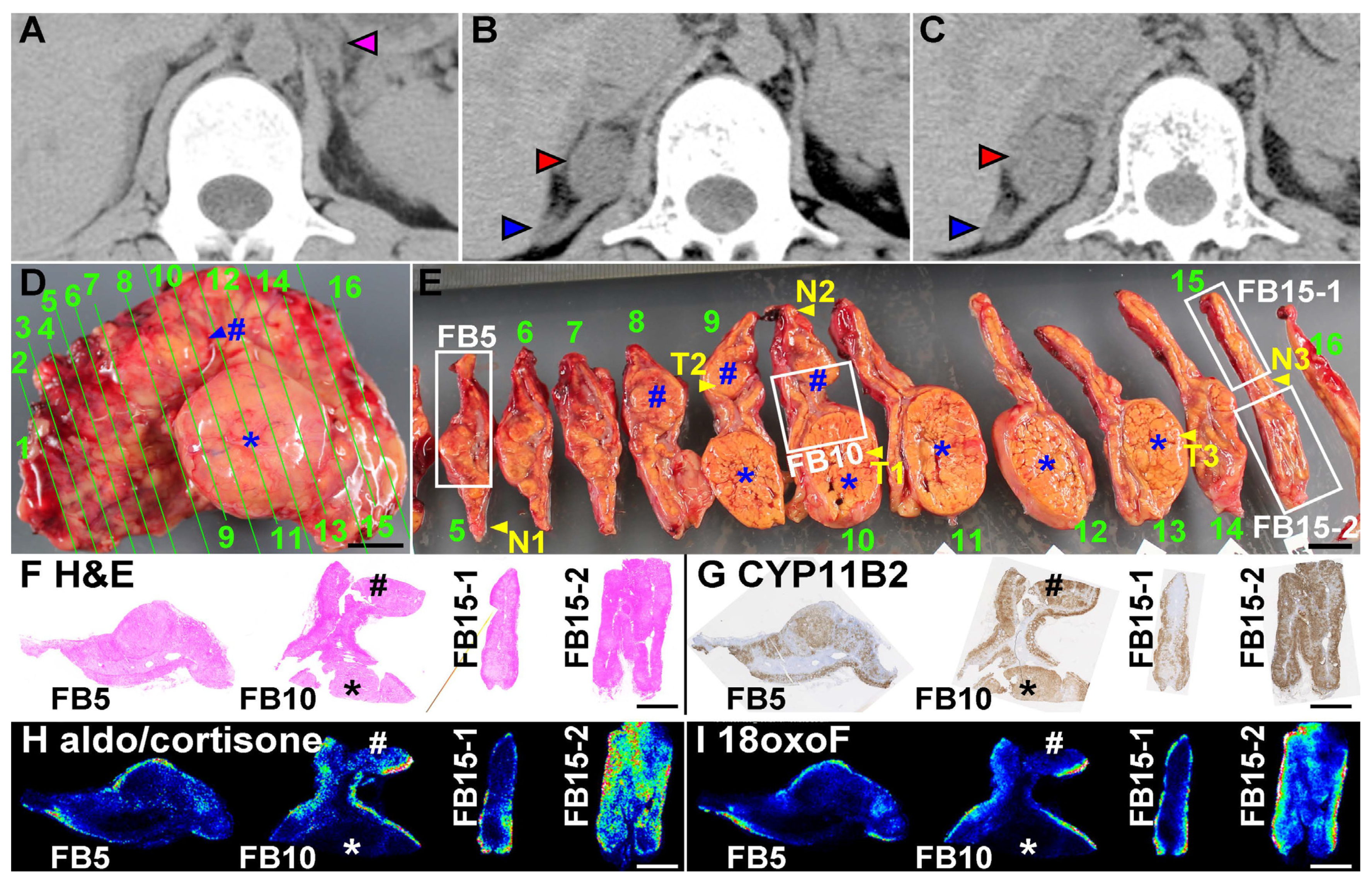
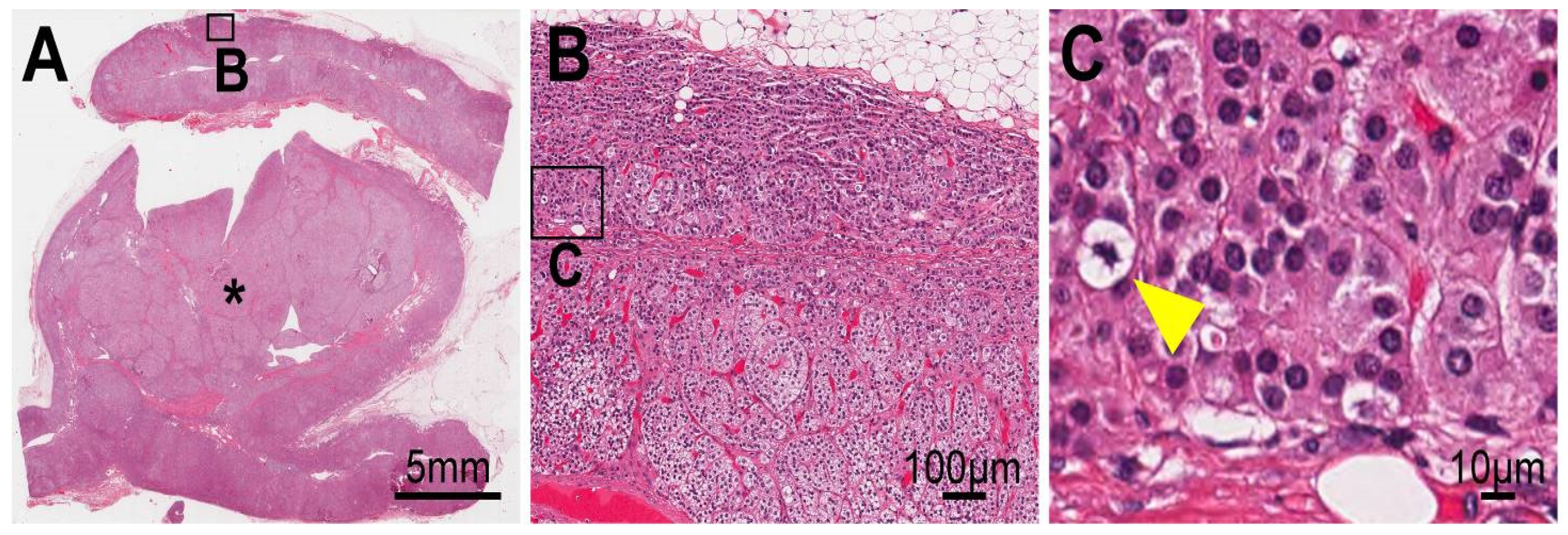
4.1. Analyses of the Surgically Removed Adrenal Gland
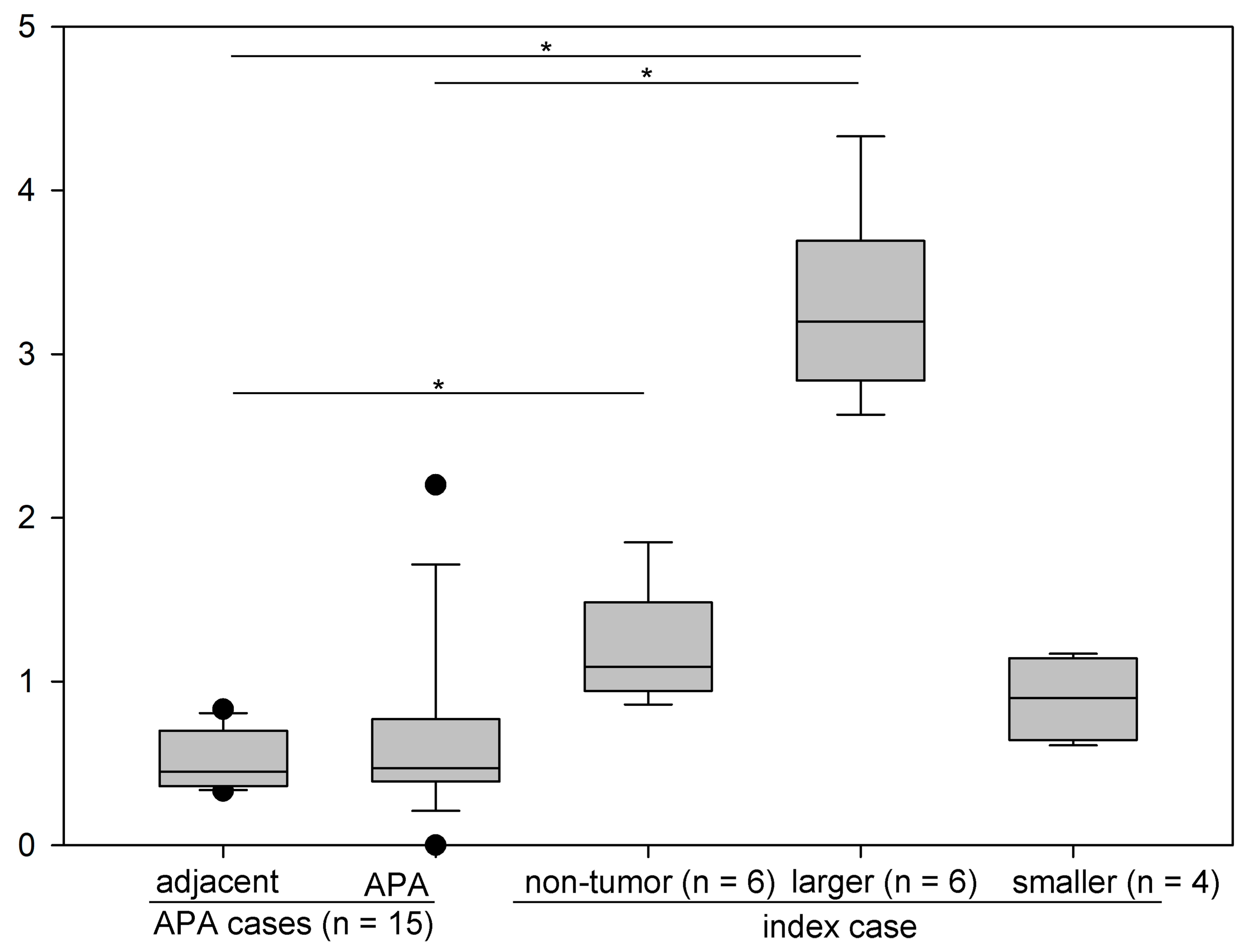
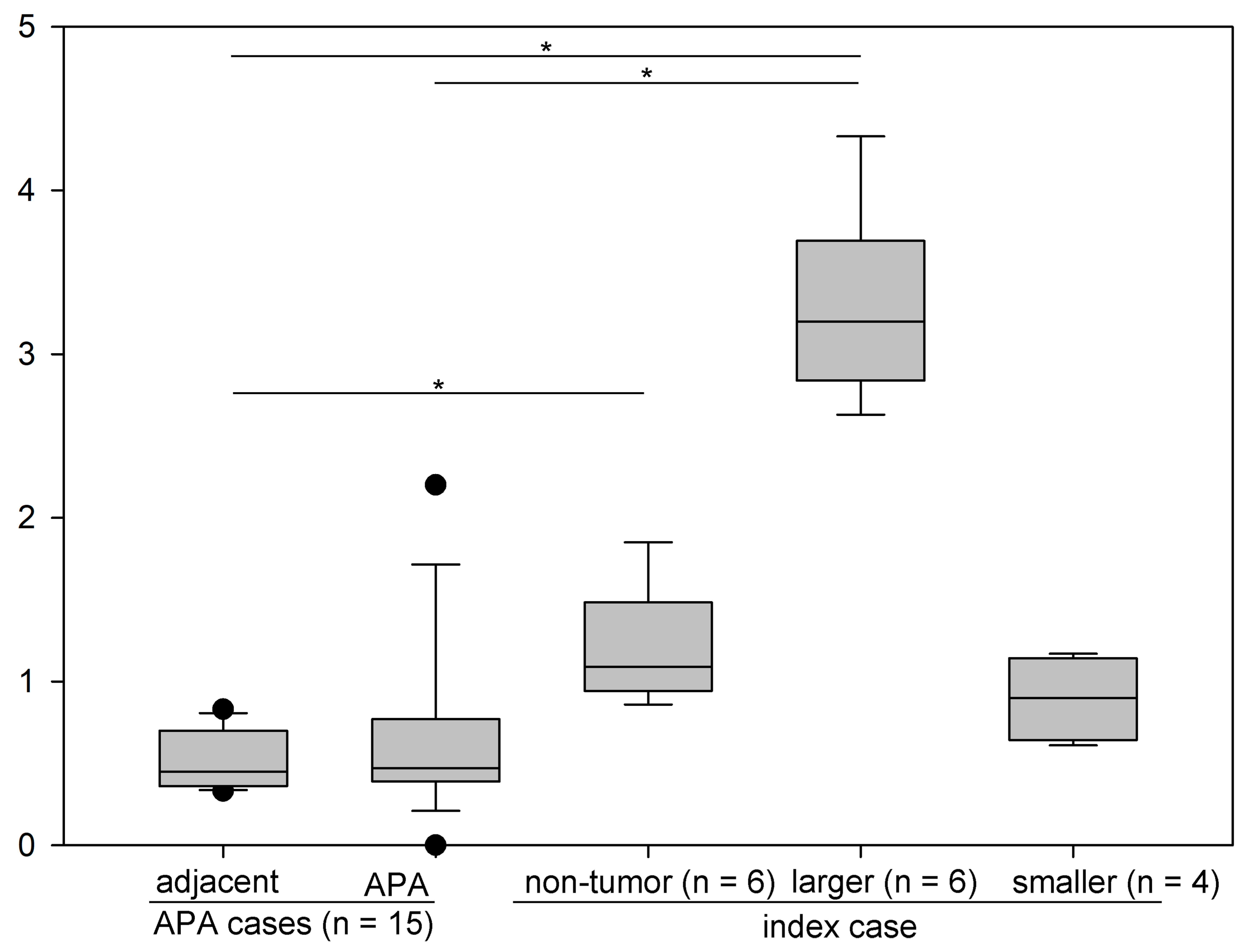
4.2. Production of Aldosterone in FH3 Adrenal
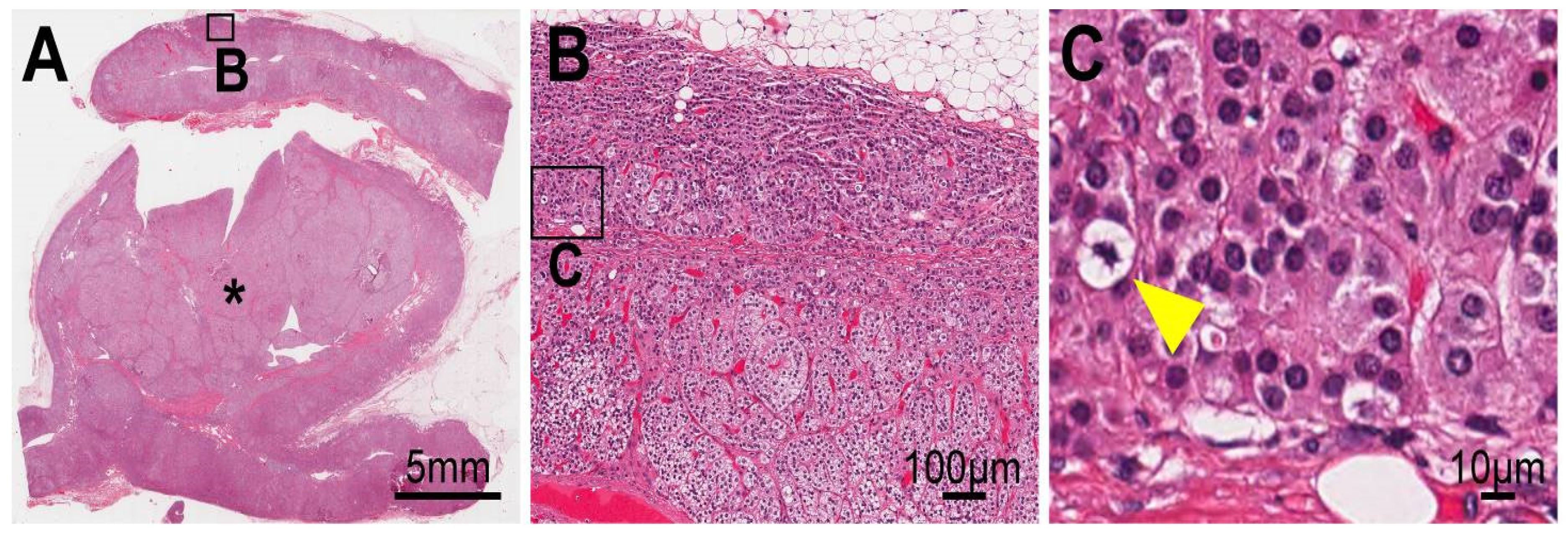
4.3. Cellular Progression in Non-Tumor and Tumor Portions of the Case
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef] [Green Version]
- Perez-Rivas, L.G.; Williams, T.A.; Reincke, M. Inherited Forms of Primary Hyperaldosteronism: New Genes, New Phenotypes and Proposition of a New Classification. Exp. Clin. Endocrinol. Diabetes 2019, 127, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Maria, A.G.; Suzuki, M.; Berthon, A.; Kamilaris, C.; Demidowich, A.; Lack, J.; Zilbermint, M.; Hannah-Shmouni, F.; Faucz, F.R.; Stratakis, C.A. Mosaicism for KCNJ5 causing early-onset primary aldosteronism due to bilateral adrenocortical hyperplasia. Am. J. Hypertens. 2020, 33, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Tamura, A.; Nishimoto, K.; Seki, T.; Matsuzawa, Y.; Saito, J.; Omura, M.; Gomez-Sanchez, C.E.; Makita, K.; Matsui, S.; Moriya, N.; et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol. Cell. Endocrinol. 2017, 441, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Nakagawa, K.; Li, D.; Kosaka, T.; Oya, M.; Mikami, S.; Shibata, H.; Itoh, H.; Mitani, F.; Yamazaki, T.; et al. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab. 2010, 95, 2296–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, K.; Seki, T.; Hayashi, Y.; Mikami, S.; Al-Eyd, G.; Nakagawa, K.; Morita, S.; Kosaka, T.; Oya, M.; Mitani, F.; et al. Human Adrenocortical Remodeling Leading to Aldosterone-Producing Cell Cluster Generation. Int. J. Endocrinol. 2016, 2016, 7834356. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, K.; Tomlins, S.A.; Kuick, R.; Cani, A.K.; Giordano, T.J.; Hovelson, D.H.; Liu, C.J.; Sanjanwala, A.R.; Edwards, M.A.; Gomez-Sanchez, C.E.; et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl. Acad. Sci. USA 2015, 112, E4591–E4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sanchez, C.E.; Qi, X.; Gomez-Sanchez, E.P.; Sasano, H.; Bohlen, M.O.; Wisgerhof, M. Disordered zonal and cellular CYP11B2 enzyme expression in familial hyperaldosteronism type 3. Mol. Cell. Endocrinol. 2017, 439, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, Y.; Takeo, E.; Shimma, S.; Yokota, M.; Higashi, T.; Seki, T.; Mizuno, Y.; Oya, M.; Kosaka, T.; Omura, M.; et al. Aldosterone and 18-Oxocortisol Coaccumulation in Aldosterone-Producing Lesions. Hypertension 2018, 72, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Isozaki, Y.; Hirose, Y.; Higashino, H.; Noda, Y.; Kobayashi, Y. Long-term follow-up of a girl with primary aldosteronism: Effect of potassium supplement. Acta Paediatr. 2005, 94, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Seki, T.; Kurihara, I.; Yokota, K.; Omura, M.; Nishikawa, T.; Shibata, H.; Kosaka, T.; Oya, M.; Suematsu, M.; et al. Case Report: Nodule Development from Subcapsular Aldosterone-Producing Cell Clusters Causes Hyperaldosteronism. J. Clin. Endocrinol. Metab. 2016, 101, 6–9. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Higasa, K.; Miyake, N.; Yoshimura, J.; Okamura, K.; Niihori, T.; Saitsu, H.; Doi, K.; Shimizu, M.; Nakabayashi, K.; Aoki, Y.; et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J. Hum. Genet. 2016, 61, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.X.; Nishimoto, K.; Nanba, K.; Rainey, W.E. Potassium channels related to primary aldosteronism: Expression similarities and differences between human and rat adrenals. Mol. Cell. Endocrinol. 2015, 417, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Klöppel, G.; La Rosa, S. Correction to: Ki67 labeling index: Assessment and prognostic role in gastroenteropancreatic neuroendocrine neoplasms. Virchows Arch. Int. J. Pathol. 2018, 472, 515. [Google Scholar] [CrossRef] [Green Version]
- Berthon, A.; Sahut-Barnola, I.; Lambert-Langlais, S.; de Joussineau, C.; Damon-Soubeyrand, C.; Louiset, E.; Taketo, M.M.; Tissier, F.; Bertherat, J.; Lefrancois-Martinez, A.M.; et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet. 2010, 19, 1561–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, D.S.; Zhang, J.; Wisgerhof, M.V.; Shackleton, C.; Kashgarian, M.; Lifton, R.P. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J. Clin. Endocrinol. Metab. 2008, 93, 3117–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholl, U.I.; Nelson-Williams, C.; Yue, P.; Grekin, R.; Wyatt, R.J.; Dillon, M.J.; Couch, R.; Hammer, L.K.; Harley, F.L.; Farhi, A.; et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl. Acad. Sci. USA 2012, 109, 2533–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamid, S.; Muller-Lobeck, H.; Pahl, S.; Remberger, K.; Bonhof, J.A.; Walb, D.; Rockel, A. Prevalence of adrenal and extra-adrenal Conn syndrome in hypertensive patients. Arch. Intern. Med. 1996, 156, 1190–1195. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takizawa, N.; Tanaka, S.; Nishimoto, K.; Sugiura, Y.; Suematsu, M.; Ohe, C.; Ohsugi, H.; Mizuno, Y.; Mukai, K.; Seki, T.; et al. Familial Hyperaldosteronism Type 3 with a Rapidly Growing Adrenal Tumor: An In Situ Aldosterone Imaging Study. Curr. Issues Mol. Biol. 2022, 44, 128-138. https://doi.org/10.3390/cimb44010010
Takizawa N, Tanaka S, Nishimoto K, Sugiura Y, Suematsu M, Ohe C, Ohsugi H, Mizuno Y, Mukai K, Seki T, et al. Familial Hyperaldosteronism Type 3 with a Rapidly Growing Adrenal Tumor: An In Situ Aldosterone Imaging Study. Current Issues in Molecular Biology. 2022; 44(1):128-138. https://doi.org/10.3390/cimb44010010
Chicago/Turabian StyleTakizawa, Nae, Susumu Tanaka, Koshiro Nishimoto, Yuki Sugiura, Makoto Suematsu, Chisato Ohe, Haruyuki Ohsugi, Yosuke Mizuno, Kuniaki Mukai, Tsugio Seki, and et al. 2022. "Familial Hyperaldosteronism Type 3 with a Rapidly Growing Adrenal Tumor: An In Situ Aldosterone Imaging Study" Current Issues in Molecular Biology 44, no. 1: 128-138. https://doi.org/10.3390/cimb44010010
APA StyleTakizawa, N., Tanaka, S., Nishimoto, K., Sugiura, Y., Suematsu, M., Ohe, C., Ohsugi, H., Mizuno, Y., Mukai, K., Seki, T., Oki, K., Gomez-Sanchez, C. E., & Matsuda, T. (2022). Familial Hyperaldosteronism Type 3 with a Rapidly Growing Adrenal Tumor: An In Situ Aldosterone Imaging Study. Current Issues in Molecular Biology, 44(1), 128-138. https://doi.org/10.3390/cimb44010010