
Microwave-Assisted Synthesis of Modified Glycidyl Methacrylate–Ethyl Methacrylate Oligomers, Their Physico-Chemical and Biological Characteristics

 , ,
, ,  , , and
, , and 
Abstract
:
1. Introduction
2. Results and Discussion
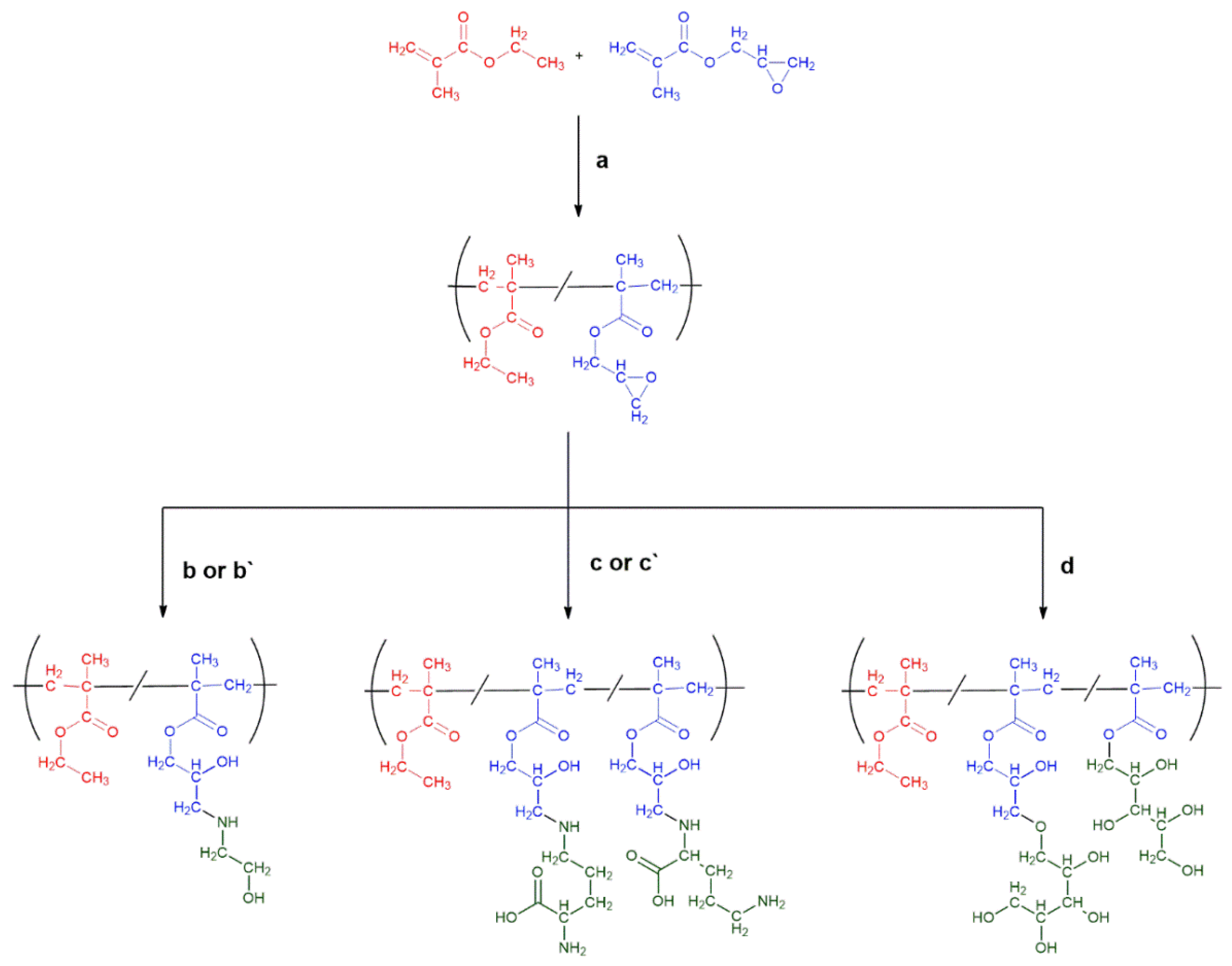
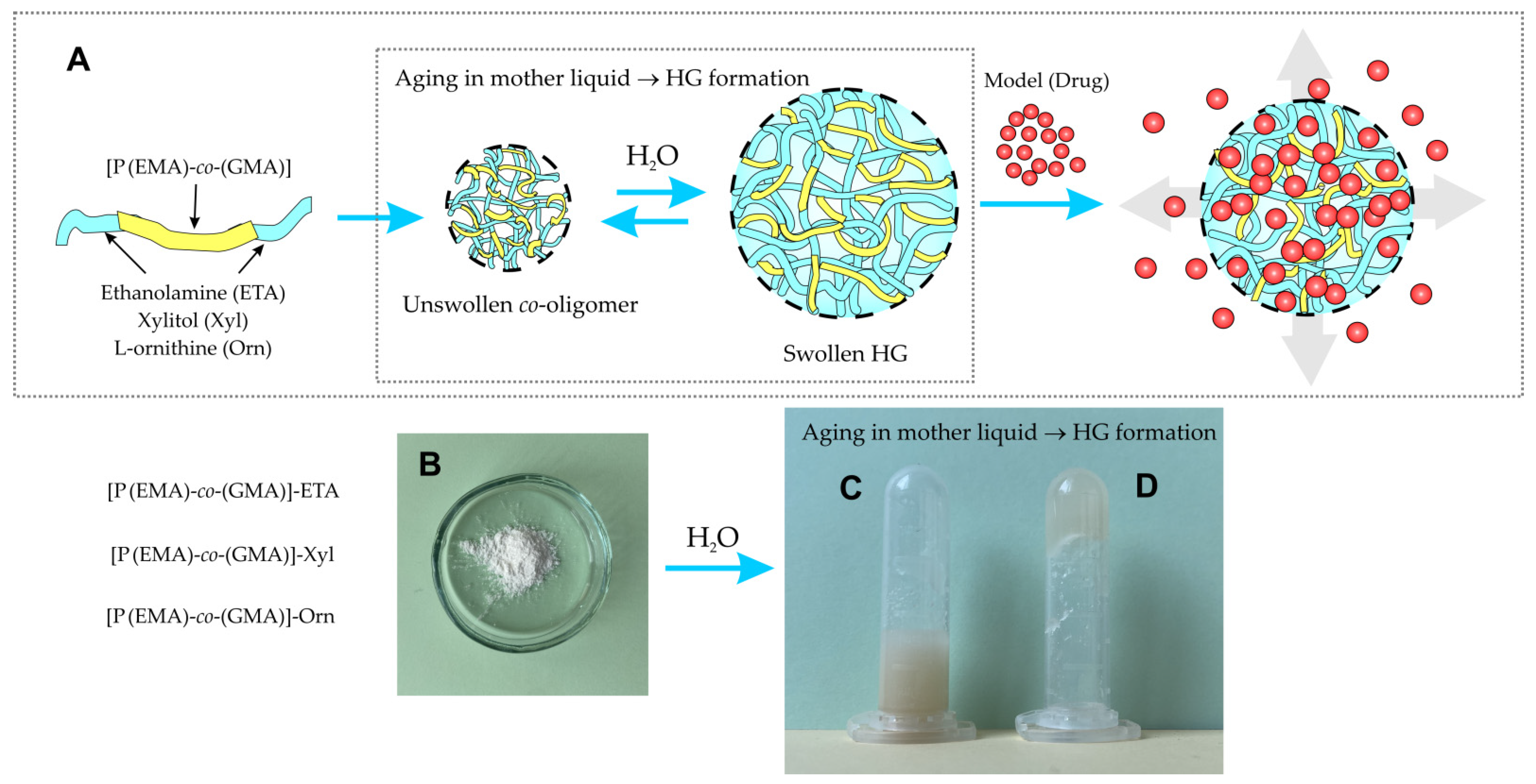
2.1. HGs Preparation: Synthesis of [P(EMA)-co-(GMA)] and Their Covalent Functionalization with Hydrophilic Moieties
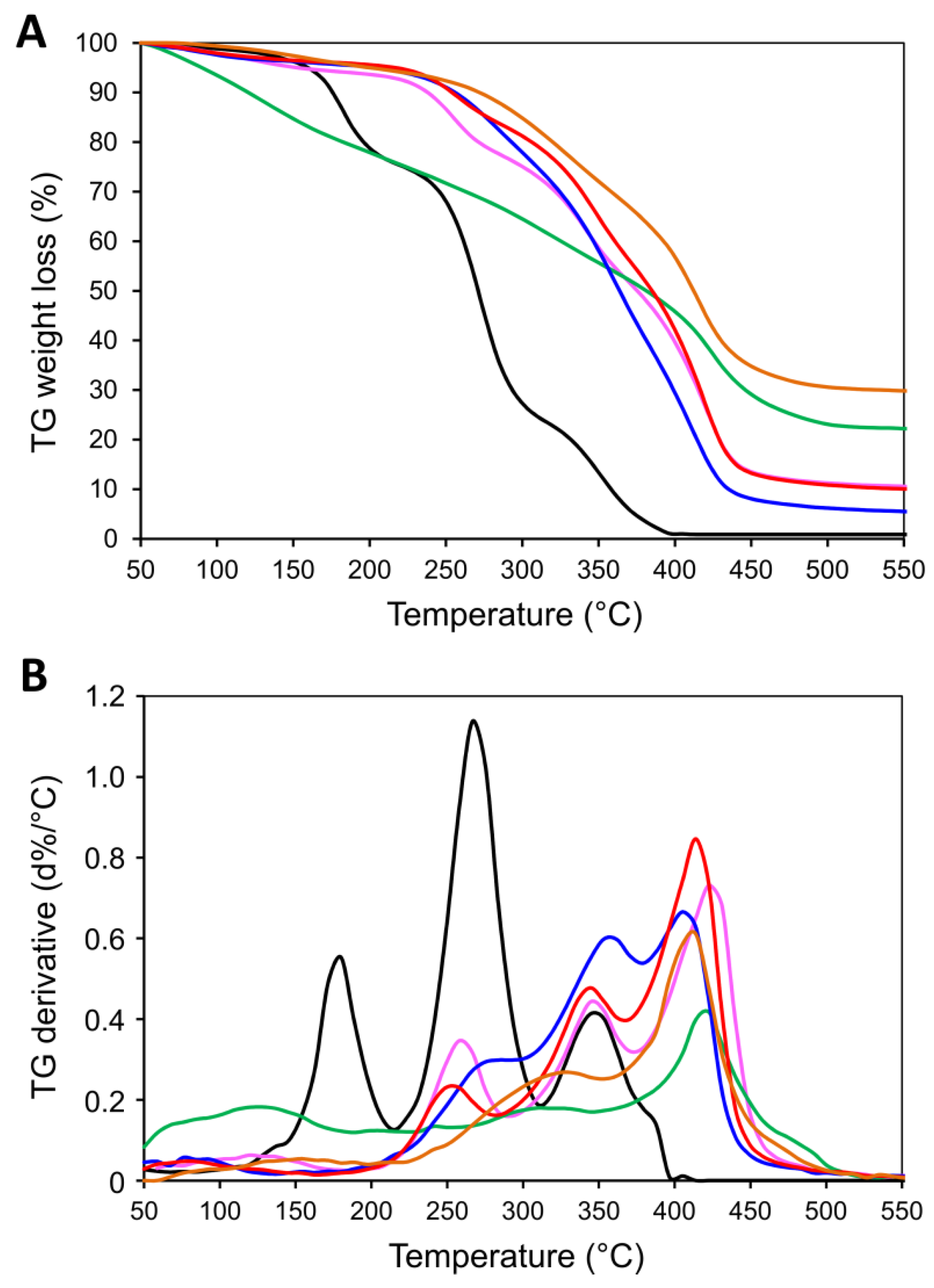
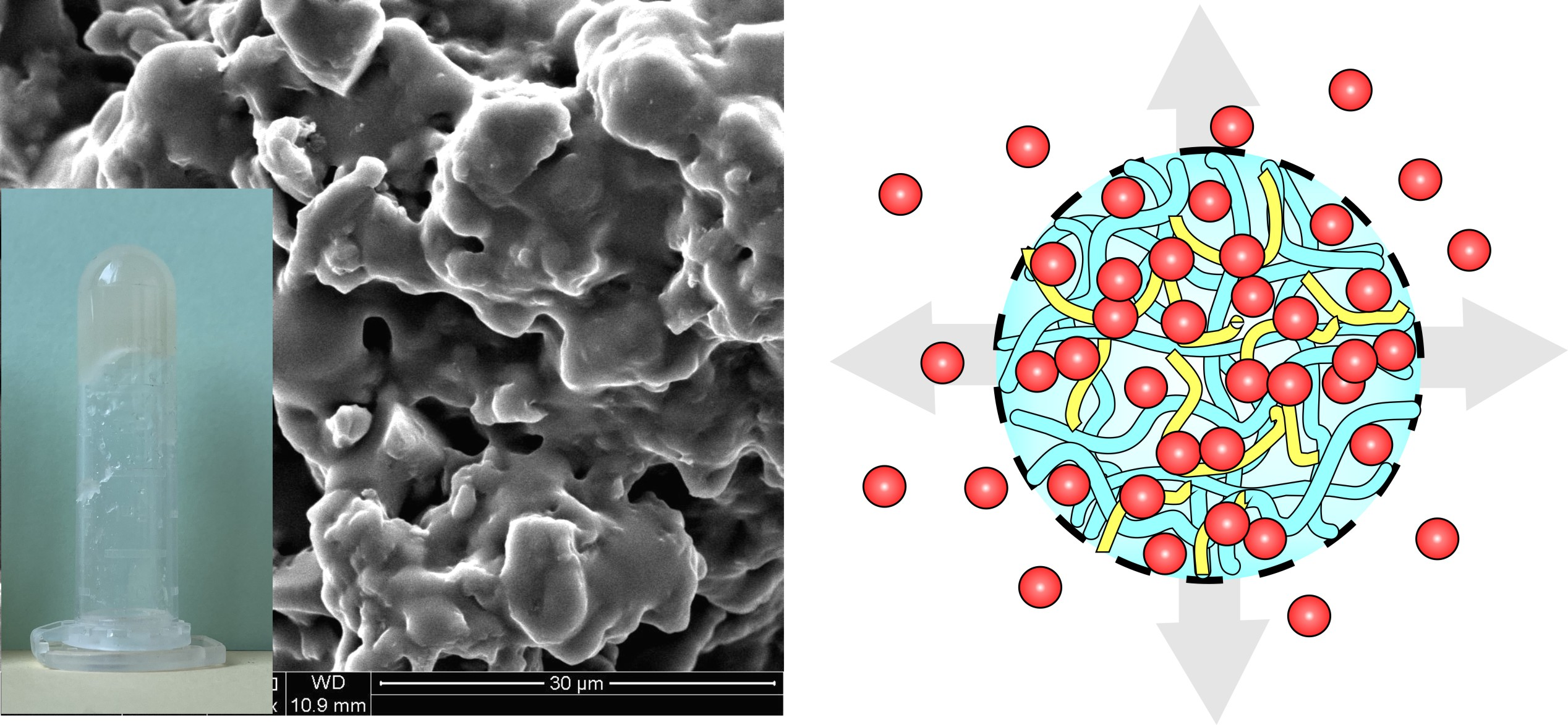
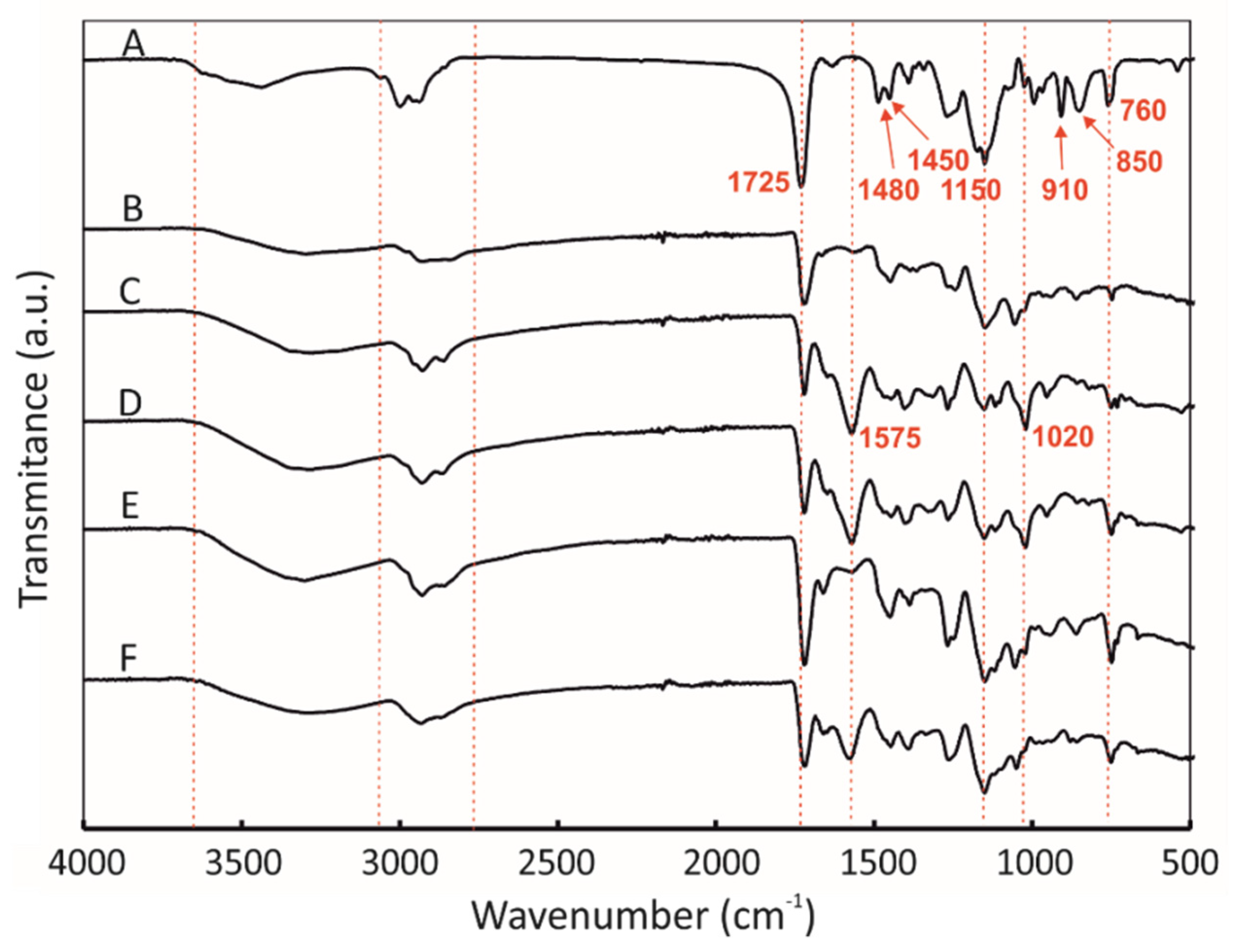
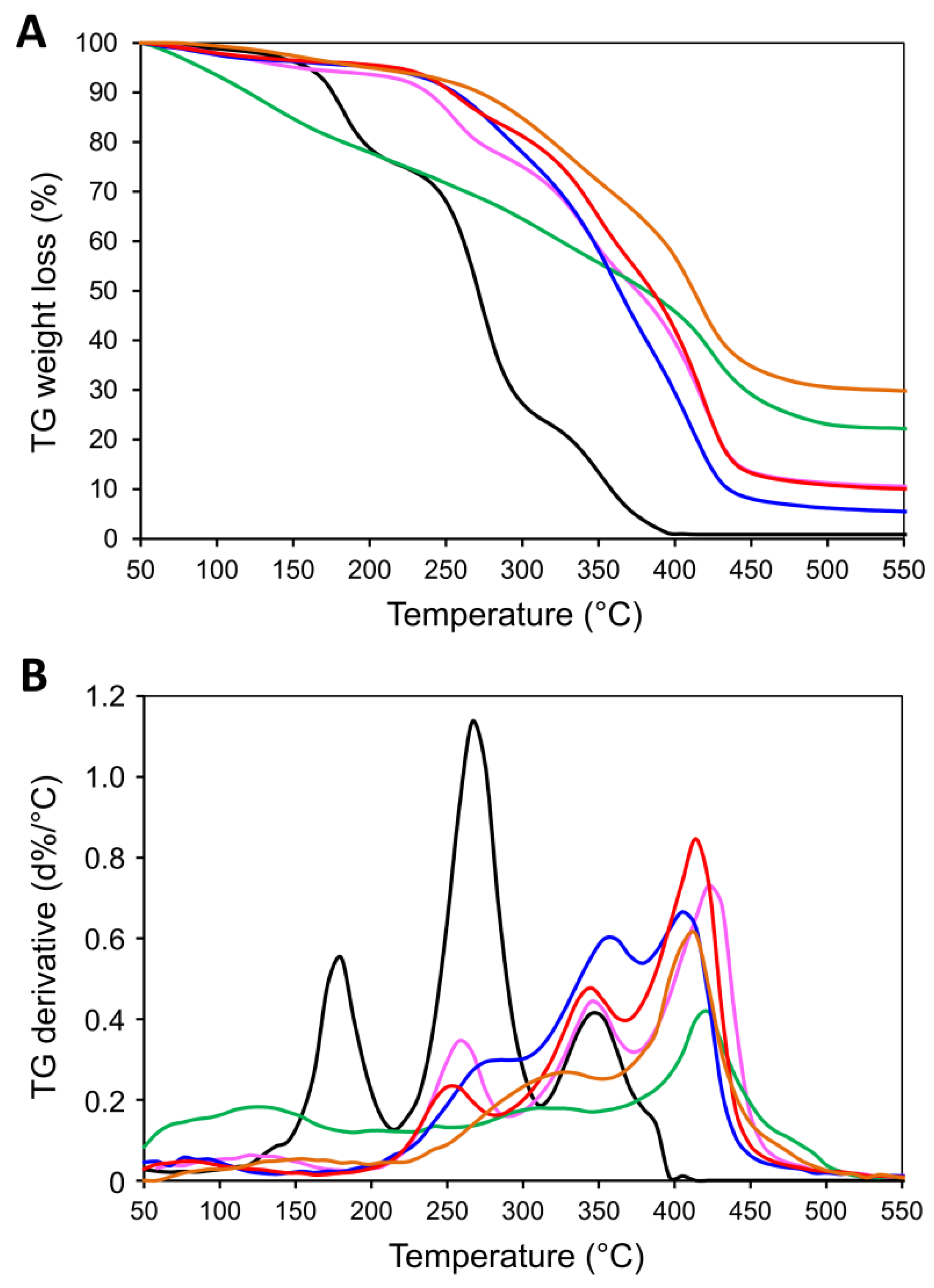
2.2. Physico-Chemical Characteristics of [P(EMA)-co-(GMA)] and Their Derivatives
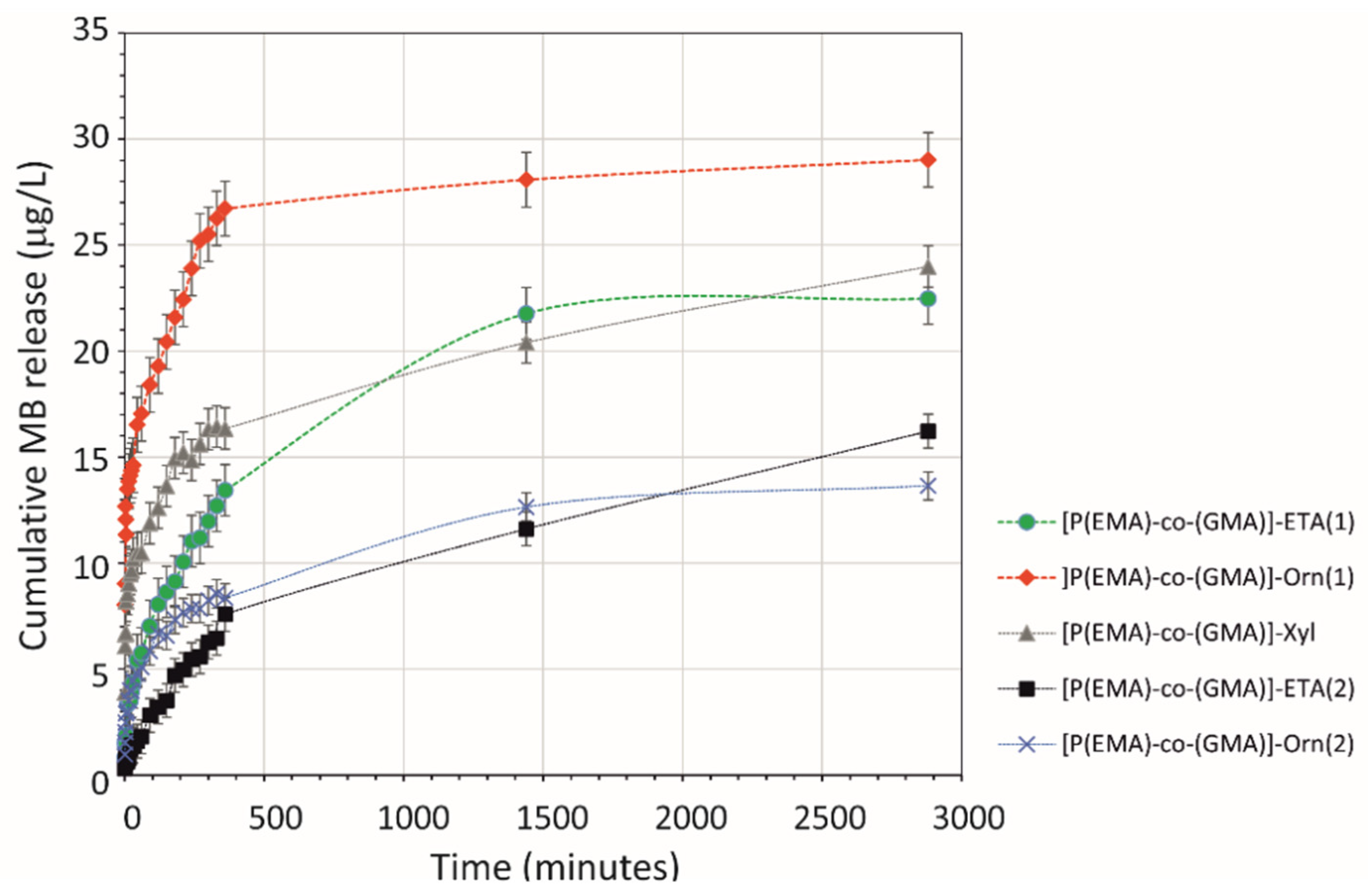
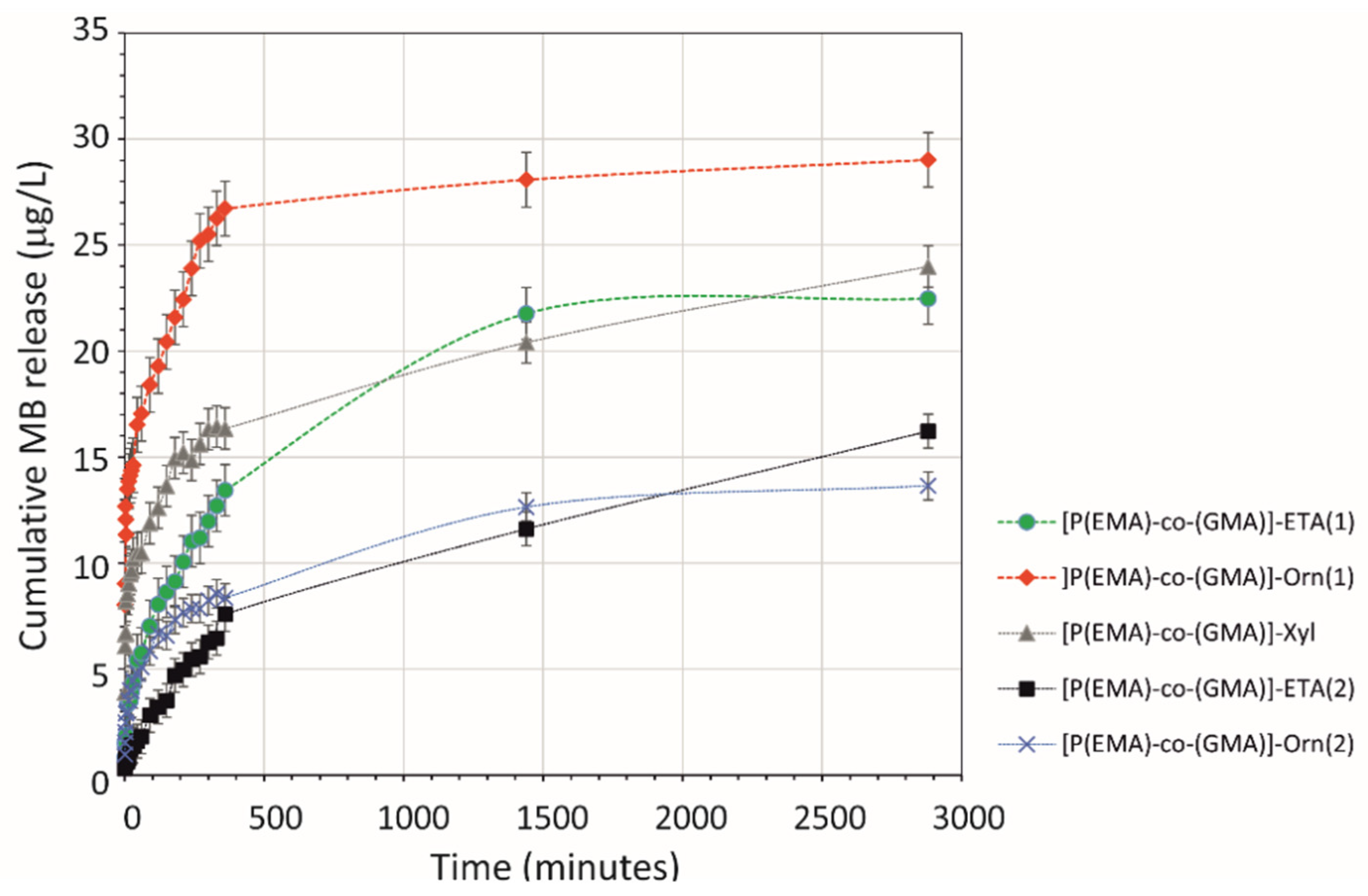
2.3. Release of Model Compounds from HGs Network
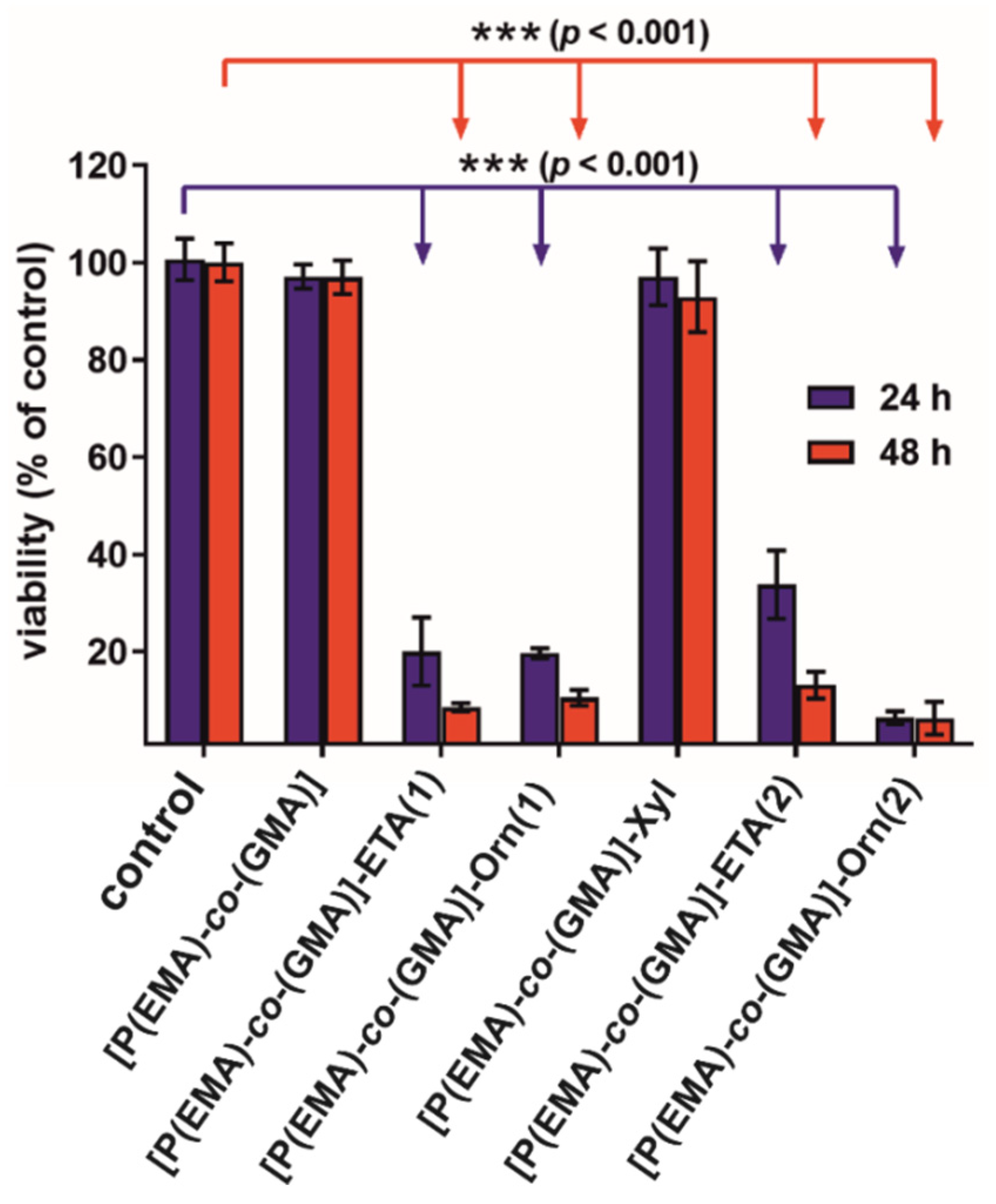
2.4. Biological Studies
3. Materials and Methods
3.1. Materials
3.1.1. Synthesis of Poly(ethyl methacrylate)-co-poly(glycidyl methacrylate) [P(EMA)-co-(GMA)]
3.1.2. Synthesis of [P(EMA)-co-(GMA)] with ETA {[P(EMA)-co-(GMA)]-ETA(1)}
3.1.3. Synthesis of [P(EMA)-co-(GMA)] with ETA in DMF {[P(EMA)-co-(GMA)]-ETA(2)}
3.1.4. Synthesis of [P(EMA)-co-(GMA)] with Xyl {[P(EMA)-co-(GMA)]-Xyl}
3.1.5. Synthesis of [P(EMA)-co-(GMA)] with Orn {[P(EMA)-co-(GMA)]-Orn(1) and [P(EMA)-co-(GMA)]-Orn(2)}
3.2. Methods
3.3. Determination of Epoxy Groups for [P(EMA)-co-(GMA)]
3.4. In Vitro Release Studies
3.5. Preparation of Oligomeric Extracts
3.6. Cytotoxicity Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, Characterization, and Applications: A Review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, A.; Ajazuddin; Khan, J.; Saraf, S.; Saraf, S. Poly(Ethylene Glycol)–Poly(Lactic-Co-Glycolic Acid) Based Thermosensitive Injectable Hydrogels for Biomedical Applications. J. Control. Release 2013, 172, 715–729. [Google Scholar] [CrossRef]
- Bakaic, E.; Smeets, N.M.B.; Hoare, T. Injectable Hydrogels Based on Poly(Ethylene Glycol) and Derivatives as Functional Biomaterials. RSC Adv. 2015, 5, 35469–35486. [Google Scholar] [CrossRef]
- Basso, J.; Miranda, A.; Nunes, S.; Cova, T.; Sousa, J.; Vitorino, C.; Pais, A. Hydrogel-Based Drug Delivery Nanosystems for the Treatment of Brain Tumors. Gels 2018, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Bayat, M.; Nasri, S. Injectable Microgel–Hydrogel Composites “Plum Pudding Gels”: New System for Prolonged Drug Delivery. In Nanomaterials for Drug Delivery and Therapy; Elsevier: Amsterdam, The Netherlands, 2019; pp. 343–372. ISBN 978-0-12-816505-8. [Google Scholar]
- Biondi, M.; Borzacchiello, A.; Mayol, L.; Ambrosio, L. Nanoparticle-Integrated Hydrogels as Multifunctional Composite Materials for Biomedical Applications. Gels 2015, 1, 162–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, N.; Kumar, N.; Kumar, M.N.V.R. Hydrogels for Pharmaceutical and Biomedical Applications. Crit. Rev. Ther. Drug Carr. Syst. 2005, 22, 107–150. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, M.; Betts, D.; Suh, A.; Bui, K.; Kim, L.; Cho, H. Hydrogel-Based Drug Delivery Systems for Poorly Water-Soluble Drugs. Molecules 2015, 20, 20397–20408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochis, A.; Bonetti, L.; Sorrentino, R.; Contessi Negrini, N.; Grassi, F.; Leigheb, M.; Rimondini, L.; Farè, S. 3D Printing of Thermo-Responsive Methylcellulose Hydrogels for Cell-Sheet Engineering. Materials 2018, 11, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guvendiren, M.; Lu, H.D.; Burdick, J.A. Shear-Thinning Hydrogels for Biomedical Applications. Soft Matter 2012, 8, 260–272. [Google Scholar] [CrossRef]
- Jia, X.; Kiick, K.L. Hybrid Multicomponent Hydrogels for Tissue Engineering. Macromol. Biosci. 2009, 9, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, J.; Deng, C.; Suuronen, E.J.; Zhong, Z. Click Hydrogels, Microgels and Nanogels: Emerging Platforms for Drug Delivery and Tissue Engineering. Biomaterials 2014, 35, 4969–4985. [Google Scholar] [CrossRef]
- Chatterjee, S.; Hui, P.; Kan, C. Thermoresponsive Hydrogels and Their Biomedical Applications: Special Insight into Their Applications in Textile Based Transdermal Therapy. Polymers 2018, 10, 480. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Long, H.; Zeuschner, D.; Räder, A.F.B.; Polacheck, W.J.; Kessler, H.; Sorokin, L.; Trappmann, B. Synthetic Extracellular Matrices with Tailored Adhesiveness and Degradability Support Lumen Formation during Angiogenic Sprouting. Nat. Commun. 2021, 12, 3402. [Google Scholar] [CrossRef]
- Hudson, S.P.; Langer, R.; Fink, G.R.; Kohane, D.S. Injectable in Situ Cross-Linking Hydrogels for Local Antifungal Therapy. Biomaterials 2010, 31, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- López-Noriega, A.; Hastings, C.L.; Ozbakir, B.; O’Donnell, K.E.; O’Brien, F.J.; Storm, G.; Hennink, W.E.; Duffy, G.P.; Ruiz-Hernández, E. Hyperthermia-Induced Drug Delivery from Thermosensitive Liposomes Encapsulated in an Injectable Hydrogel for Local Chemotherapy. Adv. Healthc. Mater. 2014, 3, 854–859. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Mohan, A.M. Novel PH Switchable Gelatin Based Hydrogel for the Controlled Delivery of the Anti Cancer Drug 5-Fluorouracil. RSC Adv. 2014, 4, 12109. [Google Scholar] [CrossRef]
- Tomczykowa, M.; Plonska-Brzezinska, M. Conducting Polymers, Hydrogels and Their Composites: Preparation, Properties and Bioapplications. Polymers 2019, 11, 350. [Google Scholar] [CrossRef] [Green Version]
- Chyzy, A.; Tomczykowa, M.; Plonska-Brzezinska, M.E. Hydrogels as Potential Nano-, Micro- and Macro-Scale Systems for Controlled Drug Delivery. Materials 2020, 13, 188. [Google Scholar] [CrossRef] [Green Version]
- Razavi, M. (Ed.) Hydrogels: Types, Structure, Properties, and Applications. In Frontiers in Biomaterials; Bentham Science Publishers Ltd.: Sharjah, United Arab Emirates, 2017; Volume 4, pp. 143–169. ISBN 978-1-68108-536-4. [Google Scholar]
- Basu, A.; Kunduru, K.R.; Doppalapudi, S.; Domb, A.J.; Khan, W. Poly(Lactic Acid) Based Hydrogels. Adv. Drug Deliv. Rev. 2016, 107, 192–205. [Google Scholar] [CrossRef]
- Li, C.; Ren, S.; Dai, Y.; Tian, F.; Wang, X.; Zhou, S.; Deng, S.; Liu, Q.; Zhao, J.; Chen, X. Efficacy, Pharmacokinetics, and Biodistribution of Thermosensitive Chitosan/β-Glycerophosphate Hydrogel Loaded with Docetaxel. AAPS PharmSciTech 2014, 15, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Kong, X.; Zhang, J.; Wang, Y.; Wang, Y.; Shi, S.; Guo, G.; Luo, F.; Zhao, X.; Wei, Y.; et al. Pharmaceutical Nanotechnology: A Novel Composite Hydrogel Based on Chitosan and Inorganic Phosphate for Local Drug Delivery of Camptothecin Nanocolloids. J. Pharm. Sci. 2011, 100, 232–241. [Google Scholar] [CrossRef]
- Bauri, K.; Nandi, M.; De, P. Amino Acid-Derived Stimuli-Responsive Polymers and Their Applications. Polym. Chem. 2018, 9, 1257–1287. [Google Scholar] [CrossRef]
- Chatterjee, S.; Chi-leung Hui, P. Stimuli-Responsive Hydrogels: An Interdisciplinary Overview. In Hydrogels—Smart Materials for Biomedical Applications; Popa, L., Violeta Ghica, M., Dinu-Pîrvu, C.-E., Eds.; IntechOpen: London, UK, 2019; ISBN 978-1-78985-875-4. [Google Scholar]
- Indermun, S.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Modi, G.; Luttge, R.; Pillay, V. An Interfacially Plasticized Electro-Responsive Hydrogel for Transdermal Electro-Activated and Modulated (TEAM) Drug Delivery. Int. J. Pharm. 2014, 462, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Ariffin, A.; Musa, M.S.; Othman, M.B.H.; Razali, M.A.A.; Yunus, F. Effects of Various Fillers on Anionic Polyacrylamide Systems for Treating Kaolin Suspensions. Colloids Surf. Physicochem. Eng. Asp. 2014, 441, 306–311. [Google Scholar] [CrossRef]
- Brzonova, I.; Kozliak, E.I.; Andrianova, A.A.; LaVallie, A.; Kubátová, A.; Ji, Y. Production of Lignin Based Insoluble Polymers (Anionic Hydrogels) by C. Versicolor. Sci. Rep. 2017, 7, 17507. [Google Scholar] [CrossRef]
- Liu, S.; Oderinde, O.; Hussain, I.; Yao, F.; Fu, G. Dual Ionic Cross-Linked Double Network Hydrogel with Self-Healing, Conductive, and Force Sensitive Properties. Polymer 2018, 144, 111–120. [Google Scholar] [CrossRef]
- Refojo, M.F. Glyceryl Methacrylate Hydrogels. J. Appl. Polym. Sci. 1965, 9, 3161–3170. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, S.-H.; Nair, S.; Moore, E. Reactive Electrospinning of Cross-Linked Poly(2-Hydroxyethyl Methacrylate) Nanofibers and Elastic Properties of Individual Hydrogel Nanofibers in Aqueous Solutions. Macromolecules 2005, 38, 3719–3723. [Google Scholar] [CrossRef]
- Sivakumaran, D.; Maitland, D.; Hoare, T. Injectable Microgel-Hydrogel Composites for Prolonged Small-Molecule Drug Delivery. Biomacromolecules 2011, 12, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.S.; Desale, S.S.; Bronich, T.K. Nanogels: An Overview of Properties, Biomedical Applications and Obstacles to Clinical Translation. J. Control. Release 2016, 240, 109–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosnik, A.; Seremeta, K. Polymeric Hydrogels as Technology Platform for Drug Delivery Applications. Gels 2017, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-Laden Microengineered Gelatin Methacrylate Hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, T.C.; Yu, J.; Tsai, W.B. Gelatin Methacrylate/Carboxybetaine Methacrylate Hydrogels with Tunable Crosslinking for Controlled Drug Release. J. Mater. Chem. B 2016, 4, 2304–2313. [Google Scholar] [CrossRef]
- Ermis, M. Photo-Crosslinked Gelatin Methacrylate Hydrogels with Mesenchymal Stem Cell and Endothelial Cell Spheroids as Soft Tissue Substitutes. J. Mater. Res. 2021, 36, 176–190. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Y.; Ferracci, G.; Zheng, J.; Cho, N.-J.; Lee, B.H. Gelatin Methacryloyl and Its Hydrogels with an Exceptional Degree of Controllability and Batch-to-Batch Consistency. Sci. Rep. 2019, 9, 6863. [Google Scholar] [CrossRef] [Green Version]
- Woerly, S.; Maghami, G.; Duncan, R. Poly(Glyceryl Methacrylate) Hydrogels—Effect of Composition and Crosslinking Density on Structure and Release of Dextran as a Model Macromolecule. J. Bioact. Compat. Polym. 1992, 7, 305–323. [Google Scholar] [CrossRef]
- Parhi, R. Cross-Linked Hydrogel for Pharmaceutical Applications: A Review. Adv. Pharm. Bull. 2017, 7, 515–530. [Google Scholar] [CrossRef]
- Palmese, L.L.; Thapa, R.K.; Sullivan, M.O.; Kiick, K.L. Hybrid Hydrogels for Biomedical Applications. Curr. Opin. Chem. Eng. 2019, 24, 143–157. [Google Scholar] [CrossRef]
- Caccavo, D.; Cascone, S.; Lamberti, G.; Barba, A.A.; Larsson, A. Swellable Hydrogel-Based Systems for Controlled Drug Delivery. In Smart Drug Delivery System; Sezer, A.D., Ed.; InTech: London, UK, 2016; ISBN 978-953-51-2247-0. [Google Scholar]
- Narayanaswamy, R.; Torchilin, V.P. Hydrogels and Their Applications in Targeted Drug Delivery. Molecules 2019, 24, 603. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Li, J.; Zhang, J.; Ma, S.; Wang, L.; Gao, F.; Guan, F. Dual-Enzymatically Cross-Linked Gelatin Hydrogel Enhances Neural Differentiation of Human Umbilical Cord Mesenchymal Stem Cells and Functional Recovery in Experimental Murine Spinal Cord Injury. J. Mater. Chem. B 2021, 9, 440–452. [Google Scholar] [CrossRef]
- Lin, F.-S.; Lee, J.-J.; Lee, A.K.-X.; Ho, C.-C.; Liu, Y.-T.; Shie, M.-Y. Calcium Silicate-Activated Gelatin Methacrylate Hydrogel for Accelerating Human Dermal Fibroblast Proliferation and Differentiation. Polymers 2020, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Lin, R.-Z.; Qi, H.; Yang, Y.; Bae, H.; Melero-Martin, J.M.; Khademhosseini, A. Functional Human Vascular Network Generated in Photocrosslinkable Gelatin Methacrylate Hydrogels. Adv. Funct. Mater. 2012, 22, 2027–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikkhah, M.; Eshak, N.; Zorlutuna, P.; Annabi, N.; Castello, M.; Kim, K.; Dolatshahi-Pirouz, A.; Edalat, F.; Bae, H.; Yang, Y.; et al. Directed Endothelial Cell Morphogenesis in Micropatterned Gelatin Methacrylate Hydrogels. Biomaterials 2012, 33, 9009–9018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basara, G.; Yue, X.; Zorlutuna, P. Dual Crosslinked Gelatin Methacryloyl Hydrogels for Photolithography and 3D Printing. Gels 2019, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Hutson, C.B.; Nichol, J.W.; Aubin, H.; Bae, H.; Yamanlar, S.; Al-Haque, S.; Koshy, S.T.; Khademhosseini, A. Synthesis and Characterization of Tunable Poly(Ethylene Glycol): Gelatin Methacrylate Composite Hydrogels. Tissue Eng. Part A 2011, 17, 1713–1723. [Google Scholar] [CrossRef]
- Yin, M.; Wan, S.; Ren, X.; Chu, C.-C. Development of Inherently Antibacterial, Biodegradable, and Biologically Active Chitosan/Pseudo-Protein Hybrid Hydrogels as Biofunctional Wound Dressings. ACS Appl. Mater. Interfaces 2021, 13, 14688–14699. [Google Scholar] [CrossRef]
- Abdollahi, H.; Najafi, V.; Amiri, F. Determination of Monomer Reactivity Ratios and Thermal Properties of Poly(GMA-Co-MMA) Copolymers. Polym. Bull. 2021, 78, 493–511. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Ito, K. Dispersion Polymerization. In Polymer Particles; Okubo, M., Ed.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2005; Volume 175, pp. 299–328. ISBN 978-3-540-22923-0. [Google Scholar]
- Dou, X.B.; Chai, M.Y.; Zhu, Y.; Yang, W.T.; Xu, F.J. Aminated Poly(Glycidyl Methacrylate)s for Constructing Efficient Gene Carriers. ACS Appl. Mater. Interfaces 2013, 5, 3212–3218. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.J.; Patias, G.; Town, J.S.; Wemyss, A.M.; Eissa, A.M.; Shegiwal, A.; Haddleton, D.M. A Simple and Versatile Route to Amphiphilic Polymethacrylates: Catalytic Chain Transfer Polymerisation (CCTP) Coupled with Post-Polymerisation Modifications. Polym. Chem. 2019, 10, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Huerta, G.; Contreras-Ordoñez, G.; Alvarez-Toledano, C.; Santes, V.; Gómez, E.; Toscano, R.A. Facile Synthesis of Aminoalcohols by Ring Opening of Epoxides under Solvent Free Conditions. Synth. Commun. 2004, 34, 2393–2406. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Joseph, S.; Aluru, N.R. Effect of Cross-Linking on the Diffusion of Water, Ions, and Small Molecules in Hydrogels. J. Phys. Chem. B 2009, 113, 3512–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Renner, J.N.; Duval, C.E. A Lysine-Modified Polyethersulfone (PES) Membrane for the Recovery of Lanthanides. Front. Chem. 2020, 8, 512. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-Polymerization Modification Reactions of Poly(Glycidyl Methacrylate)s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef] [Green Version]
- Boand, G.; Houriet, R.; Gaumann, T. Gas-Phase Acidity of Aliphatic Alcohols. J. Am. Chem. Soc. 1983, 105, 2203–2206. [Google Scholar] [CrossRef]
- Deng, M.; Li, M.; Zhao, Y.; Jiang, Z.; Guo, X. A Novel One-Pot Strategy to Prepare β-Cyclodextrin Functionalized Capillary Monoliths for Enantioseparation of Basic Drugs. Talanta 2018, 189, 458–466. [Google Scholar] [CrossRef]
- Ahmadi, H.; Javanbakht, M.; Akbari-adergani, B.; Shabanian, M. Photo-grafting of Β-cyclodextrin onto the Polyethersulfone Microfiltration-membrane: Fast Surface Hydrophilicity Improvement and Continuous Phthalate Ester Removal. J. Appl. Polym. Sci. 2019, 136, 47632. [Google Scholar] [CrossRef]
- Plazek, D.J.; Frund, Z.N. Epoxy Resins (DGEBA): The Curing and Physical Aging Process. J. Polym. Sci. Part B Polym. Phys. 1990, 28, 431–448. [Google Scholar] [CrossRef]
- Liaw, D.-J.; Shen, W.-C. Curing of Acrylated Epoxy Resin Based on Bisphenol-S. Polym. Eng. Sci. 1994, 34, 1297–1303. [Google Scholar] [CrossRef]
- Fang, Q.; Zhou, X.; Deng, W.; Liu, Z. Hydroxyl-Containing Organic Molecule Induced Self-Assembly of Porous Graphene Monoliths with High Structural Stability and Recycle Performance for Heavy Metal Removal. Chem. Eng. J. 2017, 308, 1001–1009. [Google Scholar] [CrossRef]
- Zulfiqar, S.; Zulfiqar, M.; Nawaz, M.; McNeill, I.C.; Gorman, J.G. Thermal Degradation of Poly(Glycidyl Methacrylate). Polym. Degrad. Stab. 1990, 30, 195–203. [Google Scholar] [CrossRef]
- Fares, S. Influence of Gamma-Ray Irradiation on Optical and Thermal Degradation of Poly (Ethyl-Methacrylate) (PEMA) Polymer. Nat. Sci. 2012, 4, 499–507. [Google Scholar] [CrossRef]
- He, Z.; Wang, Y.; Zhao, T.; Ye, Z.; Huang, H. Ultrasonication-Assisted Rapid Determination of Epoxide Values in Polymer Mixtures Containing Epoxy Resin. Anal Methods 2014, 6, 4257–4261. [Google Scholar] [CrossRef]
- Zalfen, A.M.; Nizet, D.; Jérôme, C.; Jérôme, R.; Frankenne, F.; Foidart, J.M.; Maquet, V.; Lecomte, F.; Hubert, P.; Evrard, B. Controlled Release of Drugs from Multi-Component Biomaterials. Acta Biomater. 2008, 4, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Przekora, A.; Czechowska, J.; Pijocha, D.; Ślósarczyk, A.; Ginalska, G. Do Novel Cement-Type Biomaterials Reveal Ion Reactivity That Affects Cell Viability in Vitro? Open Life Sci. 2014, 9, 277–289. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ||||
|---|---|---|---|---|
| Value | [P(EMA)-co-(GMA)] | [P(EMA)-co-(GMA)]-Xyl | [P(EMA)-co-(GMA)]-ETA(2) | [P(EMA)-co-(GMA)]-Orn(2) |
| Average molar mass (Mn) | 436.50 | 615.80 | 473.20 | 686.67 |
| Weighted average molar mass (Mw) | 478.70 | 721.78 | 543.91 | 783.37 |
| Dispersity index (Ð) | 1.10 | 1.17 | 1.14 | 1.14 |
| Sample | mdried (mg) | mswollen (mg) | SW |
|---|---|---|---|
| [P(EMA-co-GMA)]-ETA(1) | 1000 | 4687 | 3.70 |
| [P(EMA-co-GMA)]-Orn(1) | 1000 | 2257 | 1.25 |
| [P(EMA-co-GMA)]-Xyl | 1000 | 3475 | 2.47 |
| [P(EMA-co-GMA)]-ETA(2) | 1000 | 5149 | 4.15 |
| [P(EMA-co-GMA)]-Orn(2) | 1000 | 4584 | 3.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chyzy, A.; Pawelski, D.; Vivcharenko, V.; Przekora, A.; Bratychak, M.; Astakhova, O.; Breczko, J.; Drozdzal, P.; Plonska-Brzezinska, M.E. Microwave-Assisted Synthesis of Modified Glycidyl Methacrylate–Ethyl Methacrylate Oligomers, Their Physico-Chemical and Biological Characteristics. Molecules 2022, 27, 337. https://doi.org/10.3390/molecules27020337
Chyzy A, Pawelski D, Vivcharenko V, Przekora A, Bratychak M, Astakhova O, Breczko J, Drozdzal P, Plonska-Brzezinska ME. Microwave-Assisted Synthesis of Modified Glycidyl Methacrylate–Ethyl Methacrylate Oligomers, Their Physico-Chemical and Biological Characteristics. Molecules. 2022; 27(2):337. https://doi.org/10.3390/molecules27020337
Chicago/Turabian StyleChyzy, Adam, Damian Pawelski, Vladyslav Vivcharenko, Agata Przekora, Michael Bratychak, Olena Astakhova, Joanna Breczko, Pawel Drozdzal, and Marta E. Plonska-Brzezinska. 2022. "Microwave-Assisted Synthesis of Modified Glycidyl Methacrylate–Ethyl Methacrylate Oligomers, Their Physico-Chemical and Biological Characteristics" Molecules 27, no. 2: 337. https://doi.org/10.3390/molecules27020337
APA StyleChyzy, A., Pawelski, D., Vivcharenko, V., Przekora, A., Bratychak, M., Astakhova, O., Breczko, J., Drozdzal, P., & Plonska-Brzezinska, M. E. (2022). Microwave-Assisted Synthesis of Modified Glycidyl Methacrylate–Ethyl Methacrylate Oligomers, Their Physico-Chemical and Biological Characteristics. Molecules, 27(2), 337. https://doi.org/10.3390/molecules27020337