1. Introduction
Coffee (Cf, family
Rubiaceae), as a drink, occupies an exceptionally respectable position that no other beverage has reached. This is owing to its attractive flavor and aroma, cognition stimulation in addition to its relative safety for human consumption (to a certain limit) [
1]. Therefore, it participates in different sectors of food, pharmaceutical and cosmetic industries. Moreover, Cf was anciently used in several cultures in their traditional medicine [
2], and scientifically proved thereafter to possess a wide variety of health effects (ranging from antioxidant, antibacterial, antifungal, anticancer, anti-inflammatory, neuroprotective, etc.) which arise from a huge collection of naturally occurring bioactive compounds of which polyphenols, phenolic acids (e.g., chlorogenic acids (CGAs), caffeic, coumaric and ferulic acids, etc.) and alkaloids (caffeine and trigonelline) have an evident existence and particular attention. These biologically active constituents vary in their ratios with respect to the bean roasting degrees (green, light, medium and dark roasted brews) [
3,
4], the plant species (
Coffee arabica and
Coffee canephora (known as Robusta)) [
3,
4] and the plant origins (Brazil, Vietnam, Colombia, Indonesia, etc.) [
5,
6,
7,
8]. In addition, it is worth mentioning that besides the natural bioactive components, there are other active substances that develop upon roasting the beans by binding to the existing phenolic compounds, called Maillard Reaction Products (MRPs, e.g., melanoidins), and were evidenced to possess certain biological effects [
9,
10,
11].
In light of our study, coffee was reported, as a whole plant extract and as isolated ingredients, to have well-established antioxidant [
8,
9,
10] and anticancer activities [
11,
12] which encouraged its use as a natural alternative to synthetic drugs or as an additive to improve the potentiality of other drugs in terms of delivery enhancement, dose reduction and side effect minimization. Usually, a remarkable antioxidant activity potentially confers further biological activity enhancement such as the anticancer and anti-rheumatic effects since an antioxidant neutralizes the free radicals and oxidative products (reactive oxygen species (ROS) and reactive nitrogen species (RNS) generated and accumulated in the human cells and eventually leads to cellular function impairment and the evolution of degenerative diseases such as cancer [
13,
14].
Nevertheless, the health benefits of the polyphenolics were found largely restricted due to multiple intrinsic properties such as the unpleasant oral taste, low water solubility, poor intestinal absorption (low bioavailability), and the overall in vivo and in vitro instability. Therefore, nanotechnology is one of the recently evidenced state-of-the-art solutions that provided superior pharmaceutical rewards to overcome these limitations [
15].
Particularly, nano-encapsulation into polymeric NPs is a highly rich field of experimental research since the polymeric drug delivery nano-systems possess high structural integrity and high stability during storage and when ingested in addition to their easy design, preparation and functionalization and their capability of enhancing water solubility and controlling the release of the contained material [
16]. In addition, incorporation of plant-based active compounds into polymeric NPs and their exploitation as adjuvant and/or synergistic therapy to existing drugs has reported promising achievements in providing treatments with low drug dosage to ensure safety and sustain immunity [
17].
Polymeric NPs made from biodegradable and biocompatible polymers (biopolymers) have been applied to encapsulate a huge variety of molecules. PLGA is one of the highly evidenced biopolymers in carrying and protecting a drug, targeting its delivery, and controlling its release. It is readily hydrolyzed in the body into its original building blocks (lactic and glycolic acids) which are already endogenous monomers and normally metabolized in the human body with insignificant systemic toxicity. Accordingly, it has been approved by the FDA as a biomaterial for human use in a wide range of applications in the pharmaceutical, diagnostic and therapeutic field [
18]. As reviewed in previous literature, the most implemented method for fabricating the PLGA-based NPs involved the single emulsion-solvent evaporation (SE-SE) which is best suited for encapsulating the hydrophobic drugs in the form of oil/water (o/w) emulsions. This procedure is derived from the generalized and basic technique: the emulsification-solvent evaporation (ESE) [
19]. Furthermore, polyvinyl alcohol (PVA) is a very common emulsifier/stabilizer that is extensively used in formulating PLGA-based nano-systems, due to its capability of formulating stable and homogeneously distributed small-sized NPs [
20]. There are several attractive features that made PVA an ideal candidate, such as its high water-solubility, biocompatibility, biodegradability, relative safety and high bioavailability upon oral [
21], topical [
22] and intravenous administration [
23,
24].
Considering our study, it is important to mention that there has not yet been any attempt to encapsulate extracts of Cf beans in a PLGA-based nano-system. However, it was first encapsulated in 2016 [
25] as a Cf residue extract into oxidized tapioca starch through coacervation but the obtained size was beyond the nano-range, reaching up to 1699.3 µm. Another group of researchers published a study in July 2019 [
26] that involved formulating Cf extract into solid lipid nanoparticles (SLNs) via melt emulsification-high pressure homogenization method which were intended to be taken up by the lymphatic route. Another research group published a study that comprised encapsulating green Cf beans extract within maltodextrin through spray drying technique, applying different polymer-to-drug ratios (
g/
g) at different temperatures to investigate their effects on four different responses: the moisture percentage of NPs, product yield, EE%, TPC and antioxidant activity (by ABTS assay) [
27]. The most recent experimental study was held early in 2020 [
28] to investigate the potentiality of Arabica Cf grounds to be used as an active pharmaceutical ingredient, achieved via ball milling combined with ultrasonication. The produced NPs were poly-dispersed with a size of 396.0 nm and the particles were found as irregular granules under the SEM [
28]. Moreover, the volatile oil from pressed roasted beans was experimented on by a group of researchers who attempted to stabilize its flavoring compounds by encapsulation through the ESE method [
29].
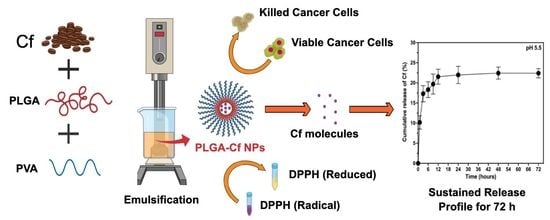
Accordingly, we attempted here to evidence the hypothesis of extract-encapsulated NPs that could improve the physicochemical properties and enhance the bioactivity of the Cf extract in terms of the antioxidant and anticancer activities. Therefore, we designed biopolymeric nanoparticulate Cf carriers made of PLGA with controlled diameter, size distribution, surface charge, EE and loading capacity (LC).
2. Materials and Methods
2.1. Materials
PLGA (Mw 38,000–54,000), Polyvinyl alcohol (PVA) (30,000–70,000 Mw), and 2,2-Diphenyl-1-picryl-hydrazyl (DPPH) were purchased from Sigma Aldrich, Hamburg, Germany. Dichloromethane (DCM) and Folin–Ciocalteau (F-C) were purchased from Fisher Chemical, Hampton, Ireland. The medium-roasted powdered Colombian Cf was purchased from international brand, Starbucks, (Cairo), Egypt. Gallic acid was purchased from (Merck KGaA, Darmstadt, Germany). For the cytotoxicity assay, all the cell lines were obtained from VACSERA Tissue Culture Unit, Cairo, Egypt.
2.2. Methods
First of all, this study comprised three main experimental stages, starting with the extraction of Cf, followed by the preparation of PLGA-Cf NPs (with the aid of BBD), characterization and optimization of the preparation conditions where the optimally obtained NPs of this stage were subjected to a final optimization stage where the initial extract amount was varied. Lastly, the in vitro evaluation (of antioxidant and cytotoxic activities) was carried out on the PLGA-Cf NPs obtained from the third stage, having the most desirable characteristics.
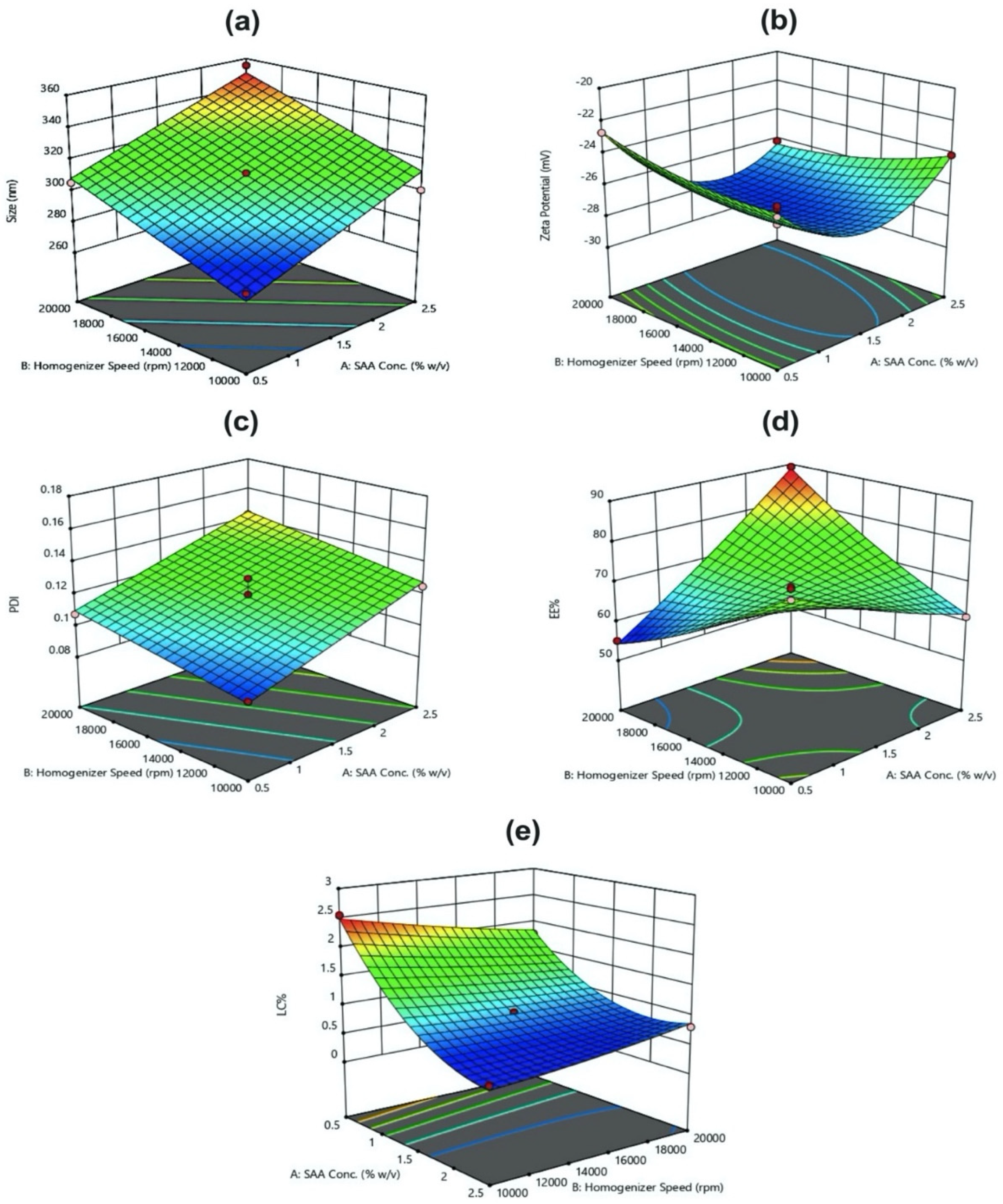
Three-factor, three-level BBD was implemented for optimization of PLGA-Cf NPs. The BBD includes 15 experimental runs with three replicated center points. The independent factors were PVA concentration (X
1), homogenization speed (X
2), and homogenization time (X
3). The three variables were varied at three levels: low (coded as −1), middle (coded as 0) and high (coded as 1). The ratio of Cf extract to PLGA was kept constant (0.1:1
w/
w). The responses or dependent variables studied were particle size (Y
1), Zeta Potential (ZP) (Y
2), poly disperse index (PDI) (Y
3), encapsulation efficiency (EE%) (Y
4), and loading capacity percentage (LC%) (Y
5). Selected ranges are shown in
Table 1.
Generally, we employed the BBD since it is commonly used to construct higher-order response surfaces applying minimal number of runs compared to a normal factorial technique. Altogether with the central composite technique, they suppress certain runs in order to maintain the higher-order surface prospects.
2.3. Optimization of PLGA-Cf NPs by Multiple Response
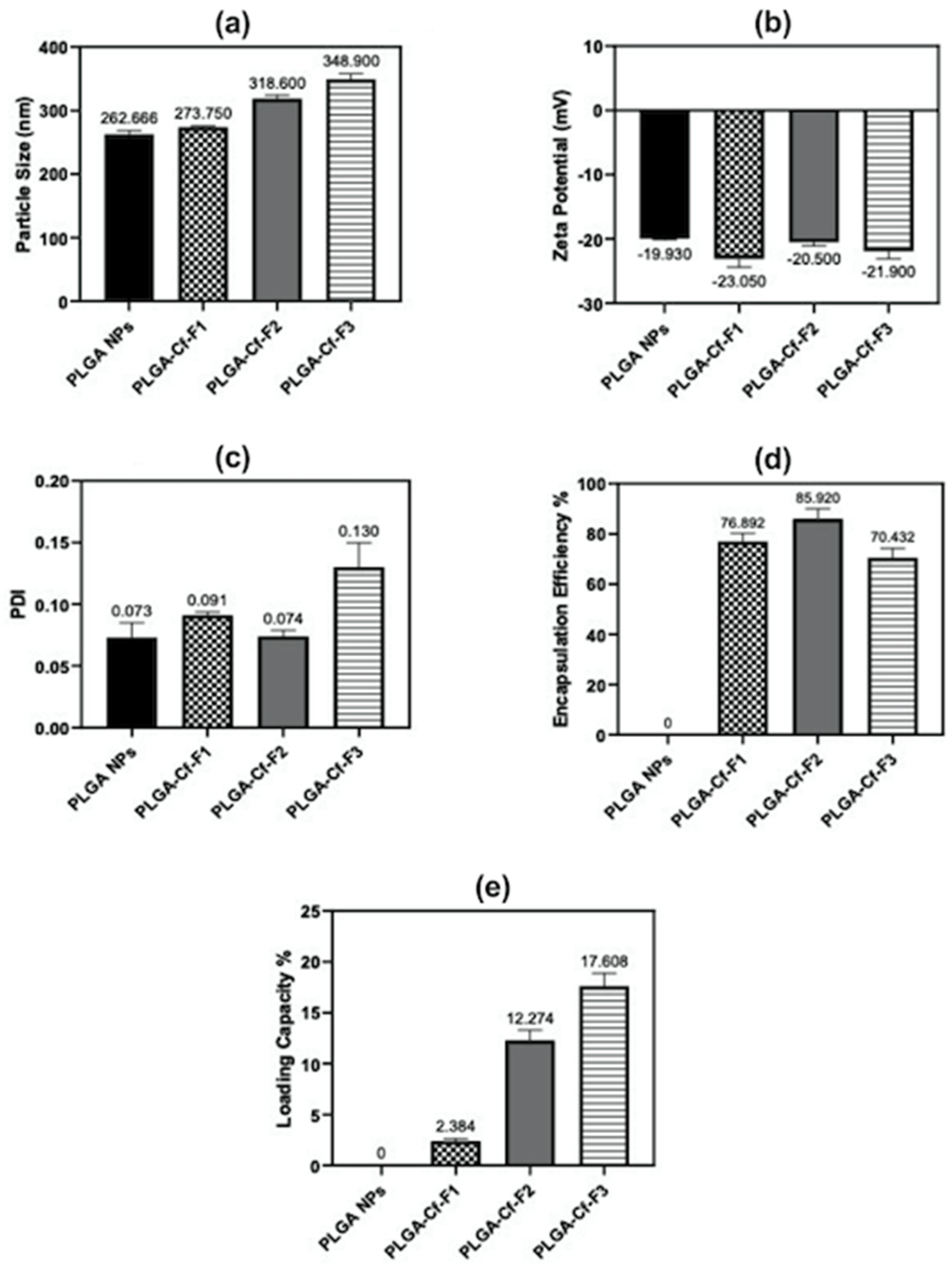
In this study, all three responses were simultaneously optimized by using the general linear scale desirability function. The optimum formulation was selected based on the criteria of attaining the minimum particle size and PDI, whereas maximum ZP, EE%, and LC% as shown in (
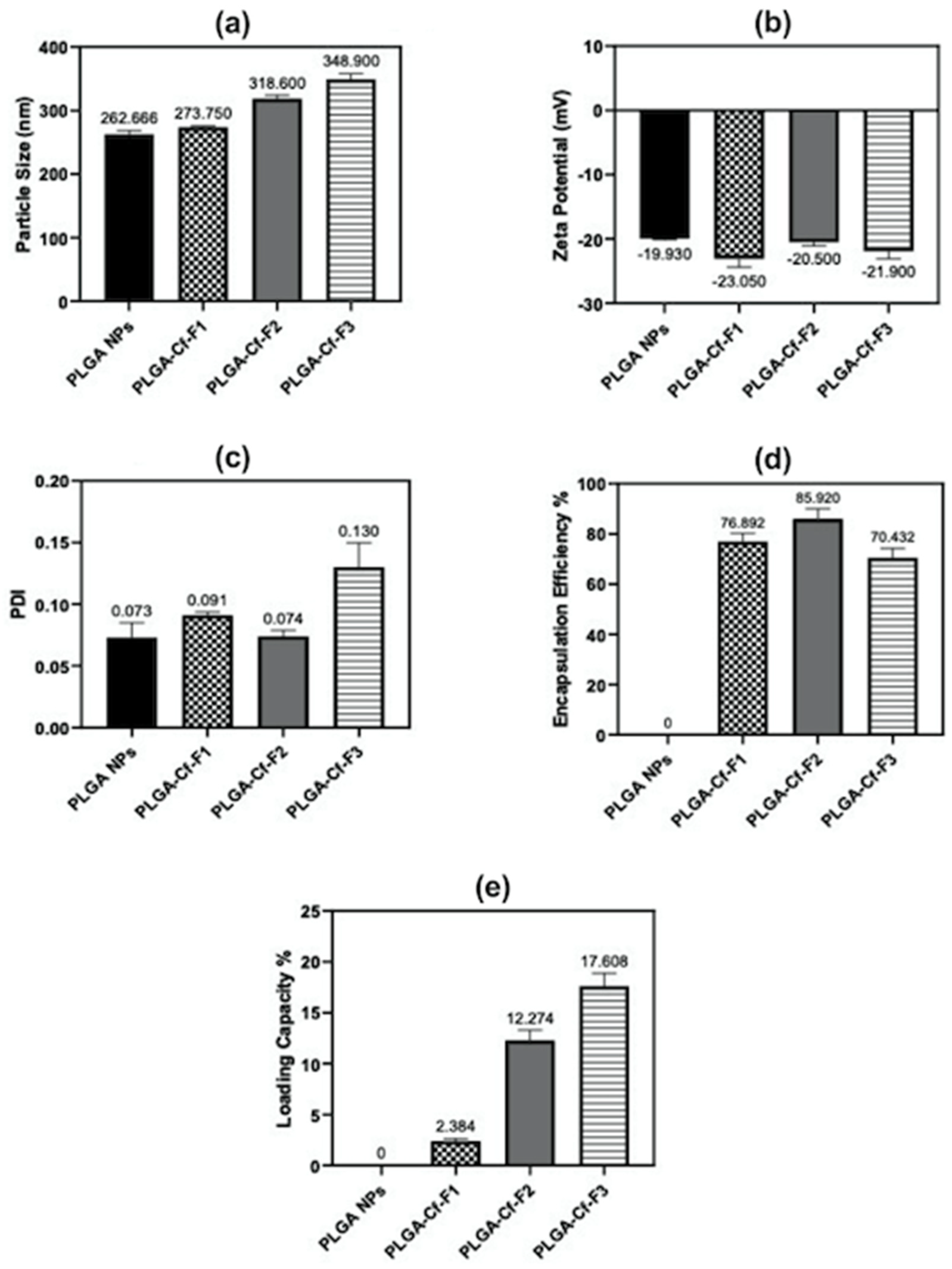
Table 1). New formulations were prepared according to the optimized independent variables. The Cf extract-to-PLGA polymer ratio was changed to study its impact on the responses that were previously studied in phase one. Accordingly, the resulted samples were denoted as PLGA-Cf-F
X where F
1 stands for “Formula (1)” of 0.1:1.0
w/
w ratio and similarly for F
2 and F
3 which refer to 0.5:1.0 and 1.0:1.0
w/
w ratios, respectively.
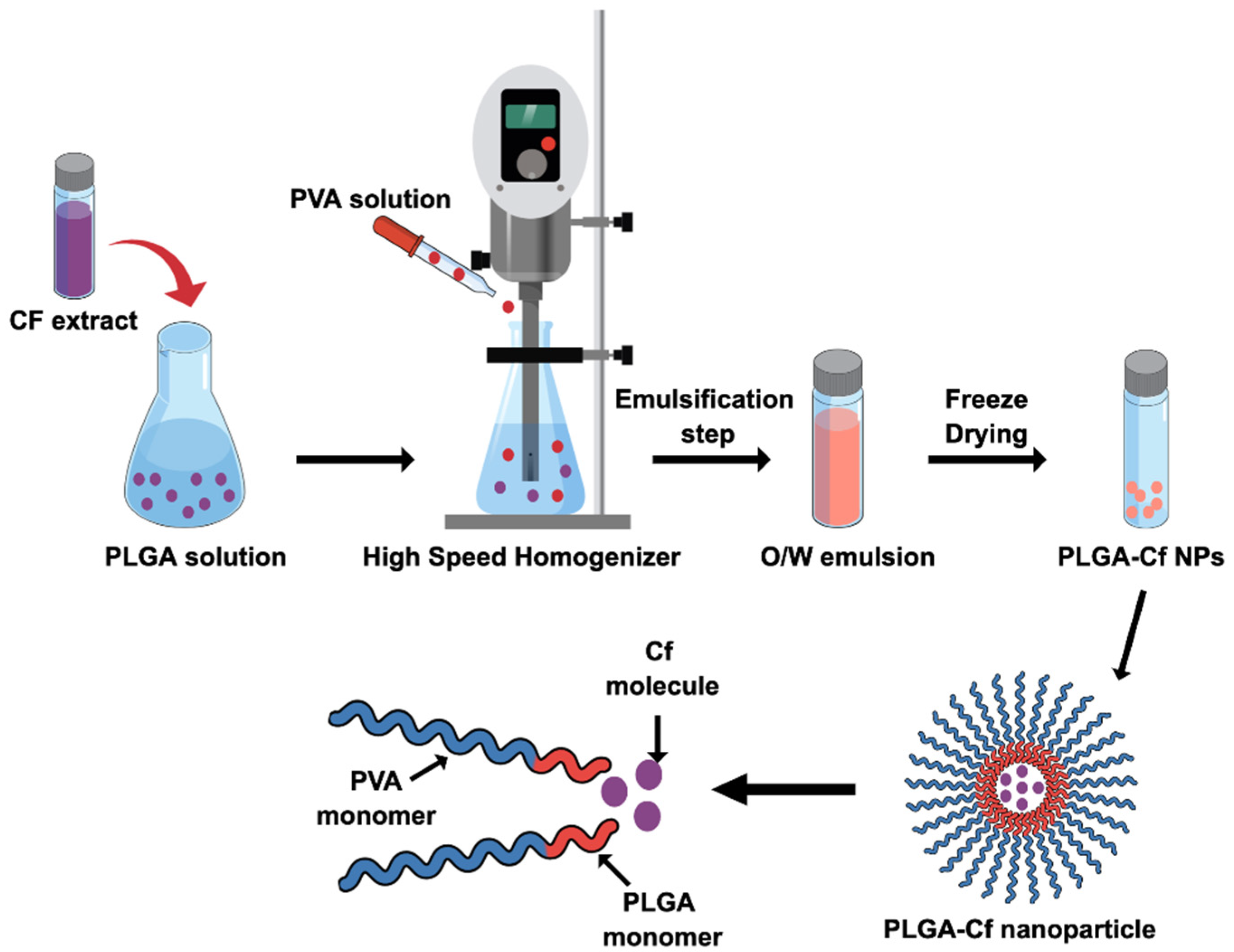
2.4. Synthesis of the PLGA-Cf NPs
PLGA-Cf NPs were synthesized based on SE-SE technique according to previous literature with minute specifications [
19]. First, the Cf extract was prepared according to the Solid-Liquid Extraction method (SLE) as described in literature [
30] (see
Appendix A [
8,
30,
31,
32,
33,
34,
35,
36,
37],
Table A1,
Table A2 and
Table A3 and
Figure A1). For the preparation of PLGA-Cf NPs, PLGA was dissolved in DCM in a concentration of 50 mg/mL then mixed with an equal volume of methanol-based solution of Cf extract, achieving a ratio of 0.1:1.0 (
w/
w) of Cf-to-PLGA. The organic solution was added dropwise to a 20 mL aqueous solution of PVA during homogenization then transferred to ultrasonication (in ice bath). The resulted
o/
w nano-emulsion was subjected to magnetic stirring overnight to evaporate the organic phase. The NPs were collected by centrifugation and washed twice. The final sediment was freeze dried. The produced NPs were characterized and subjected to a second phase of optimization involving the study of impact of varying the initial drug amount in the NP on the same abovementioned responses. Accordingly, other two Cf-to-PLGA ratios were selected, 0.5:1.0 and 1.0:1.0
w/
w, formulated under the identified optimum conditions and characterized.
2.5. Characterization of the Prepared PLGA-Cf NPs
The hydrodynamic particle size, PDI and ZP measurements were obtained using the particle size analyzer (DLS: Dynamic Light Scattering, Malvern, NANO-series, Zetasizer, Worcestershire, United Kingdom). FT-IR Spectroscopy analysis (FT-IR: Fourier Transform-Infrared Spectroscopy, using Thermo Scientific Nicolet 8700, Waltham, MA, USA) was carried out to examine the chemical composition of the unprocessed Cf extract and the formed NPs (with and without the extract) working over a range of wavenumber of 4000–400 cm−1. The method adopted here relied on the use of KBr-based solid pellet technique. The particle morphology and microscopic size were imaged by the FE-SEM (FE-SEM: Field Emission-Scanning Electron Microscopy, Gemini Sigma, New Philadelphia, OH, USA). Typical sample preparation required initial surface coating by sputtering with a gold film.
2.6. Determination of Encapsulation Efficiency (EE%) and Loading Capacity (LC%)
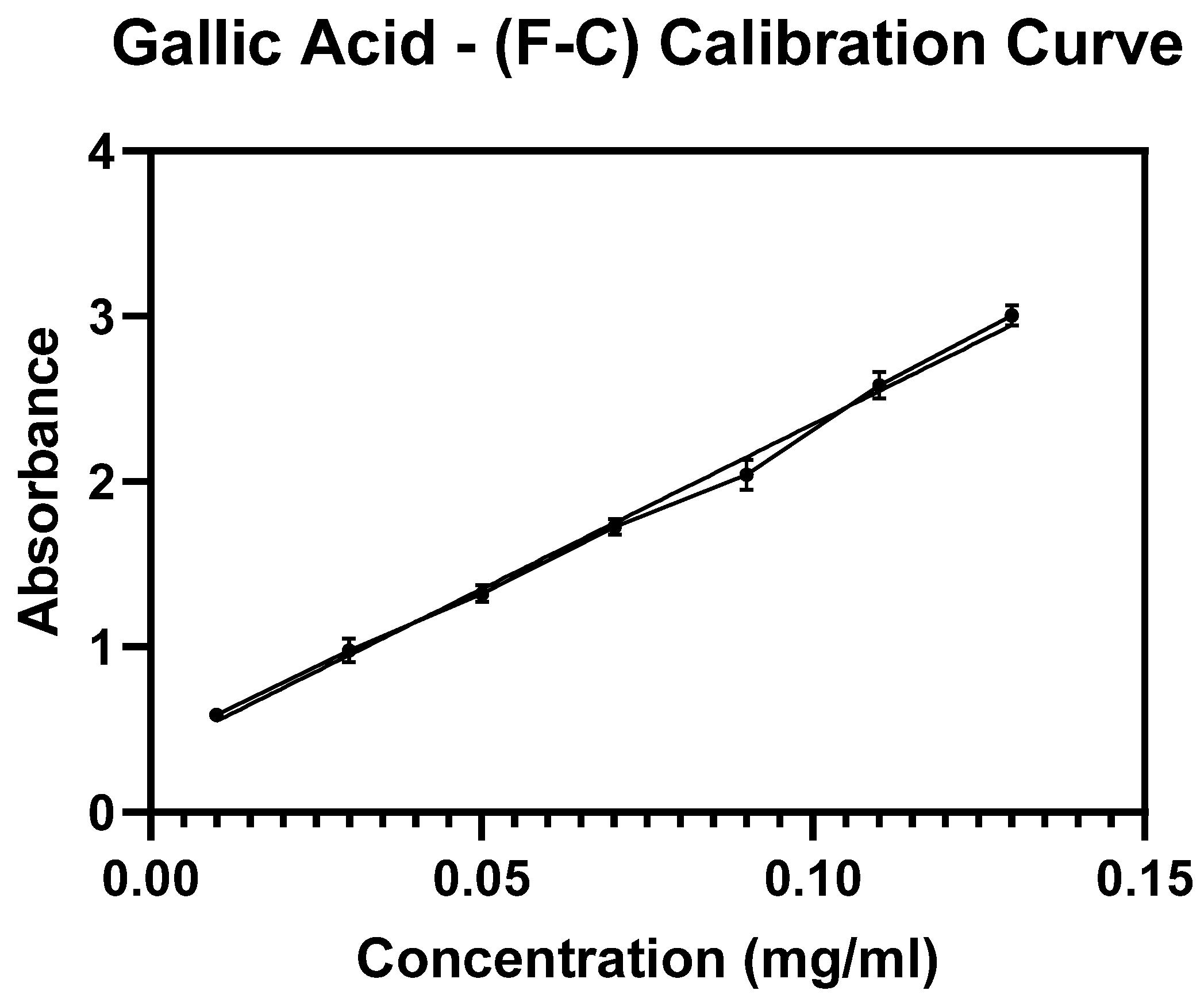
The amount of entrapped Cf extract within the NPs was determined indirectly based on constructing a standard calibration curve of serial concentrations of the Cf extract where the absorbance readings were detected at a maximum wavelength (
= 273 nm). Therefore, the collected supernatants were subjected to Ultraviolet-Visible spectroscopy (UV-Vis, Jenway 74 series, Staffordshire, UK) (R
2 of the calibration curve was 0.9979) for absorbance detection at the same
. The entrapped mass of the extract was determined using Equation (1), thereby the EE% value was calculated through Equation (2). Finally, the LC% was determined from Equation (3). This method was performed according to previously applied protocols [
18,
19,
38,
39,
40,
41,
42].
2.7. Stability of TPC of Cf Extract in PLGA NPs
F-C assay was applied on the formulated NPs in order to indicate the stability of the TPC after encapsulation [
8,
31,
32,
38]. Briefly, specific equal weights of pure Cf extract, optimally prepared plain PLGA NPs and the PLGA-Cf NPs (0.1:1, 0.5:1 and 1:1) were added to 2.5 mL of 10% (
v/
v) aqueous F-C reagent, followed by 2 mL of 7.5% (
w/
v) sodium carbonate. All the prepared tubes were left in the dark for 90 min followed by centrifugation for 15 min at 20,000 rpm. Finally, supernatants were measured by UV-Vis at a wavelength of 765 nm. Gallic acid (GA) was used as a standard reference phenolic compound to which a material known for its TPC (Total Phenolic Content) is compared. Similarly, F-C assay was applied to establish a standard calibration curve using a series of known concentrations of GA. The TPC was expressed as the number of milligrams of GA that was equivalent to a gram of the extract (mg GAE/g dry extract).
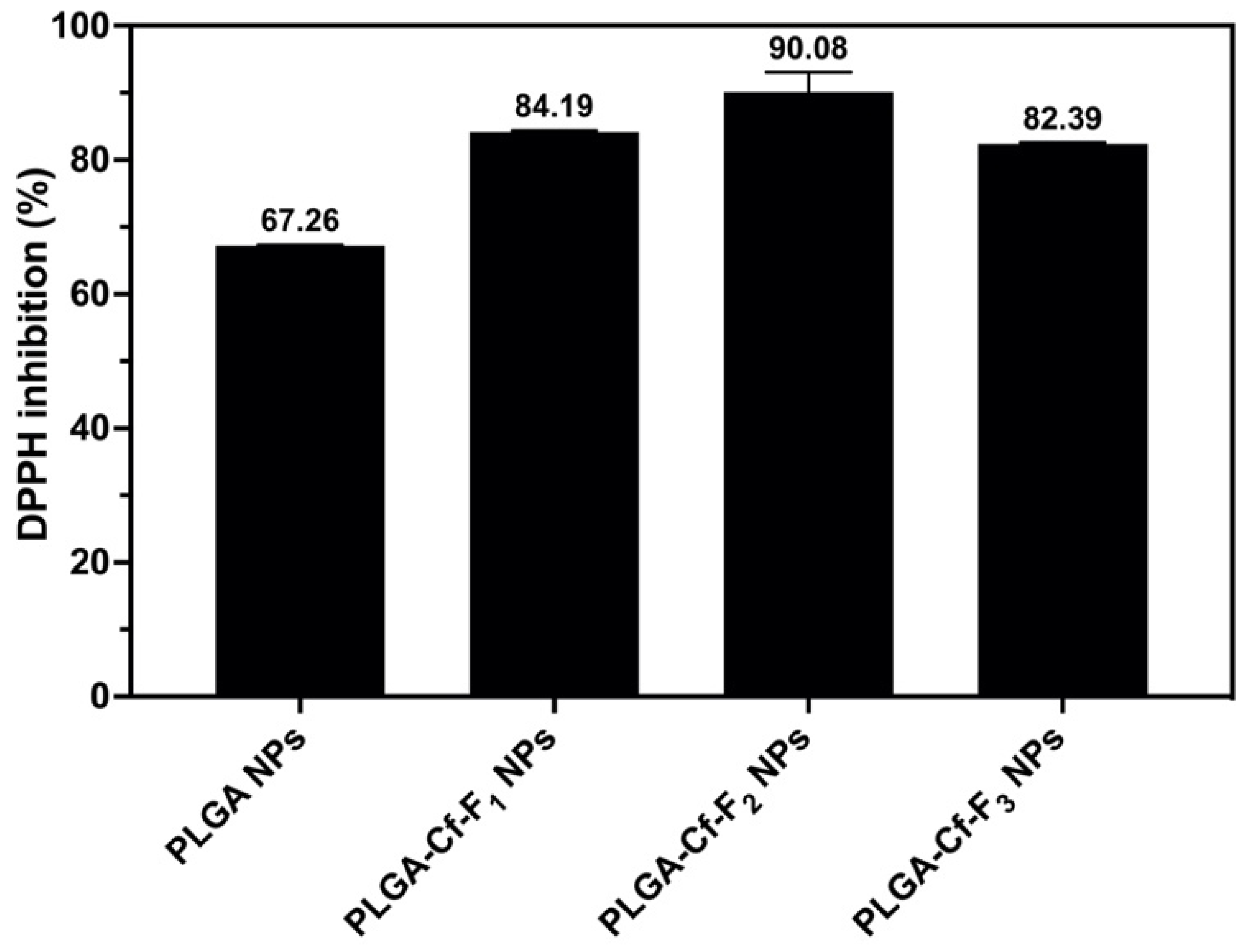
2.8. Evaluating the Antioxidant Activity
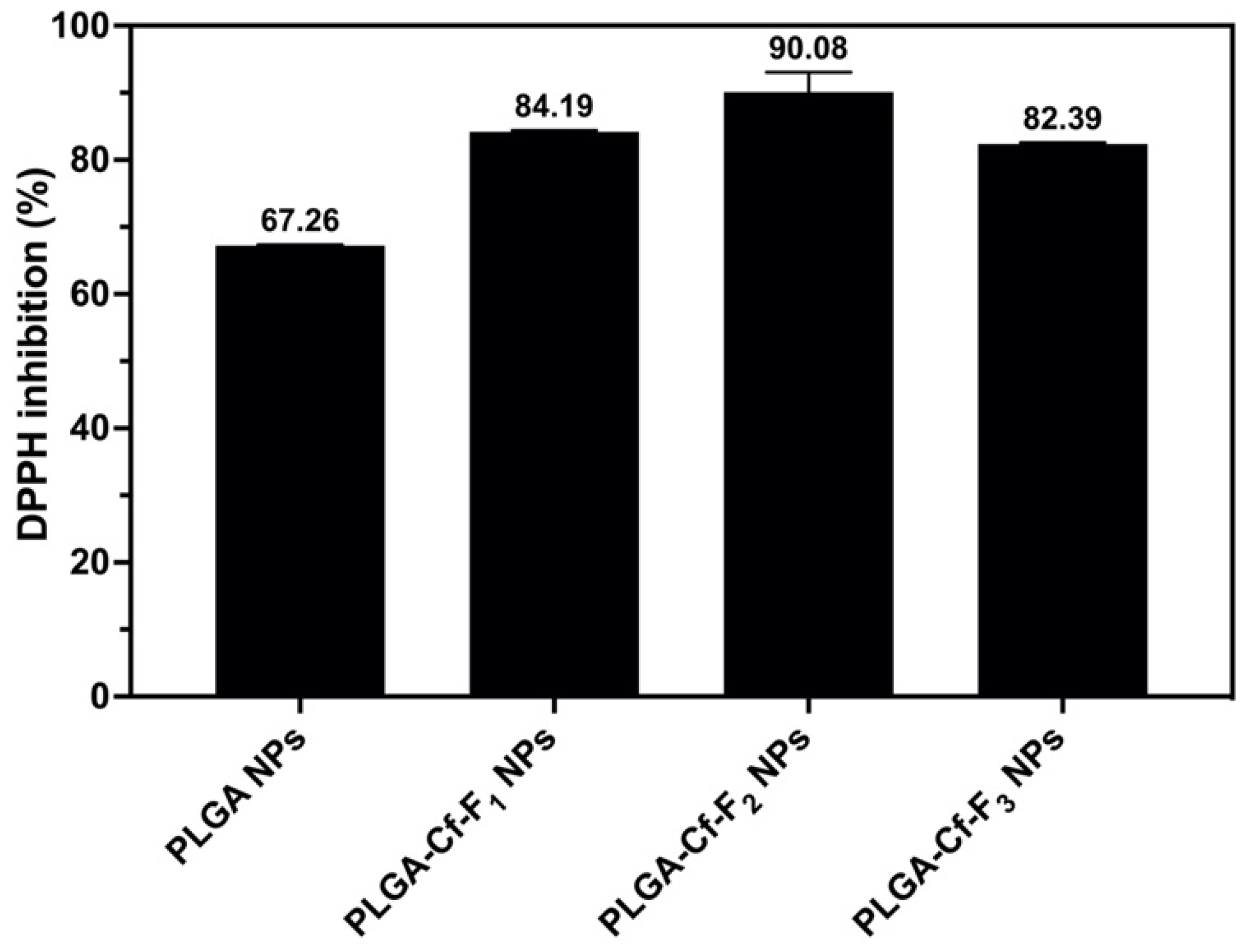
The DPPH assay protocol was applied as elaborated previously [
38] using equal predetermined weights of the NPs samples. The samples containing NPs were centrifuged and the supernatants were separated for further UV-Vis spectrometry at 517 nm. The % inhibition values were calculated using Equation (4). The NP sample that showed the highest % inhibition value among others was selected for further analyses to obtain the 50% DPPH inhibition (IC
50) value through preparing a series of known concentrations to construct a calibration curve.
where
is the absorbance of the DPPH reagent,
is the absorbance of the prepared samples.
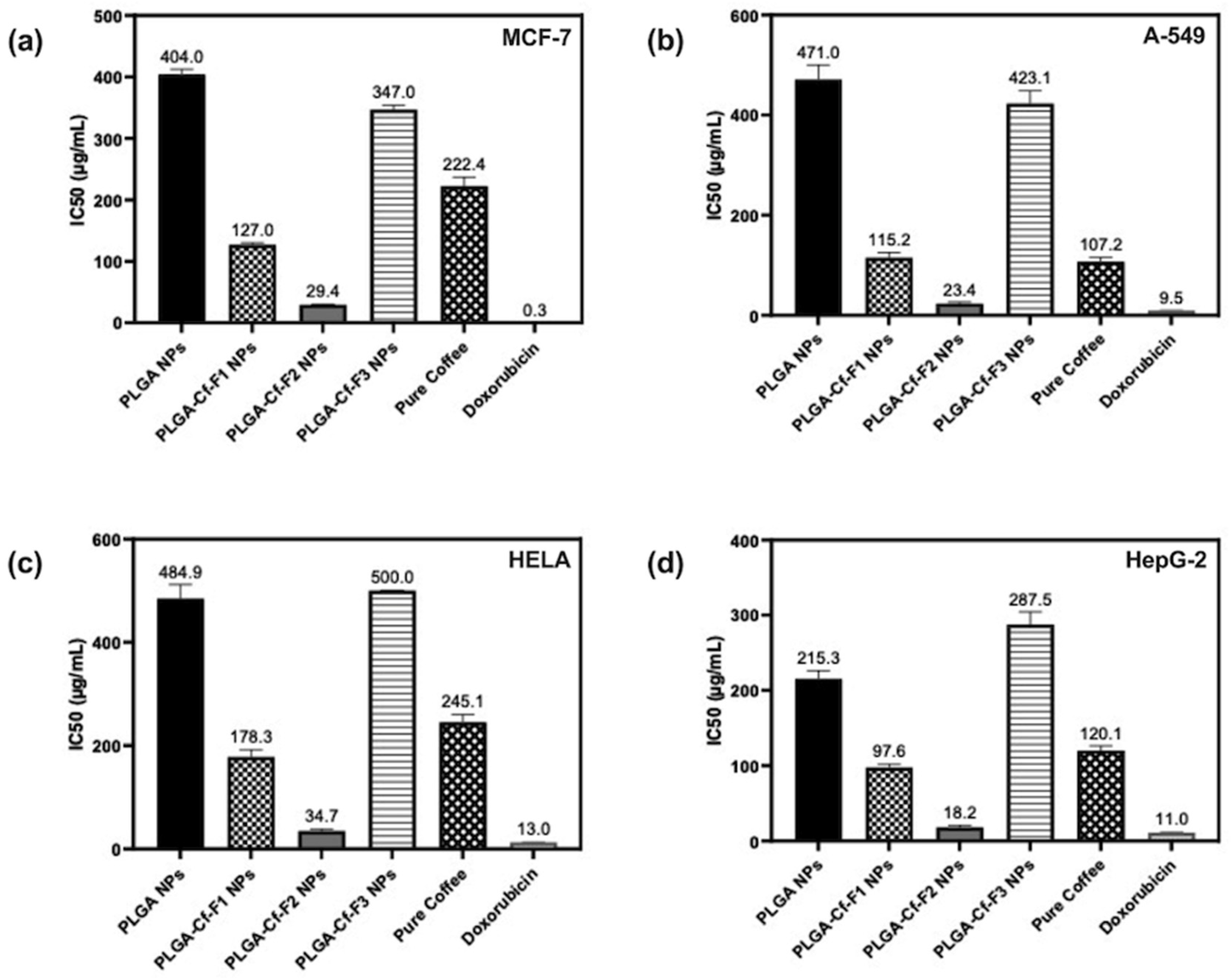
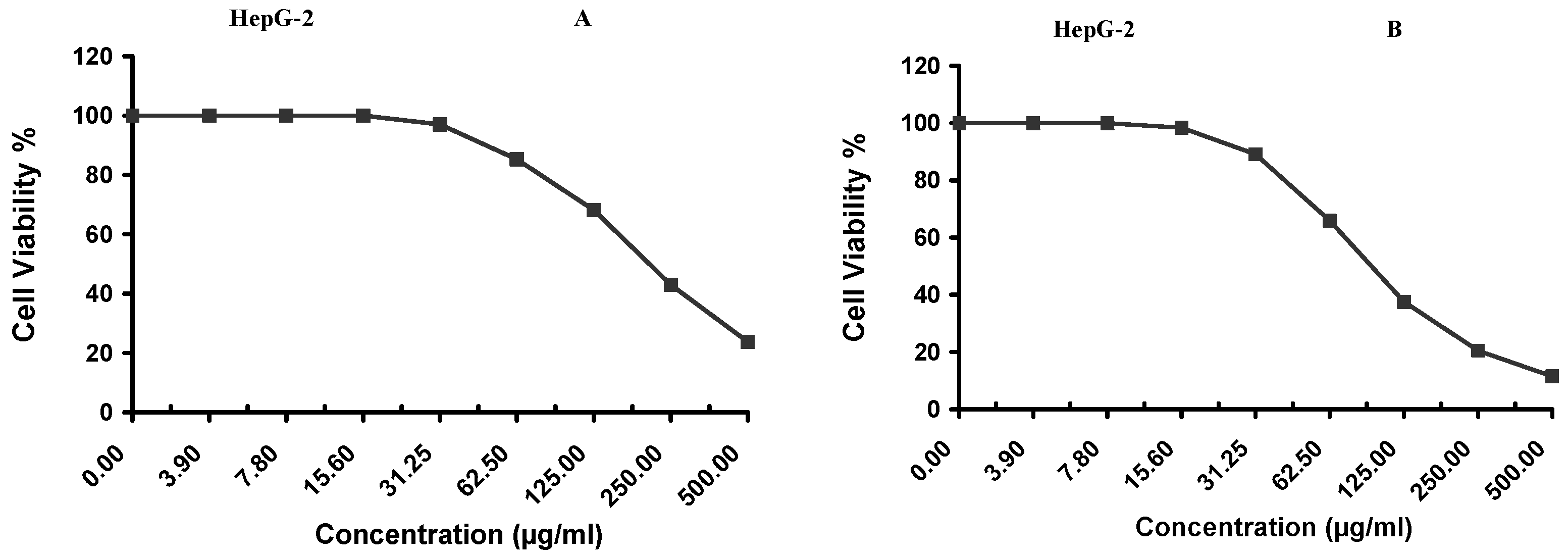
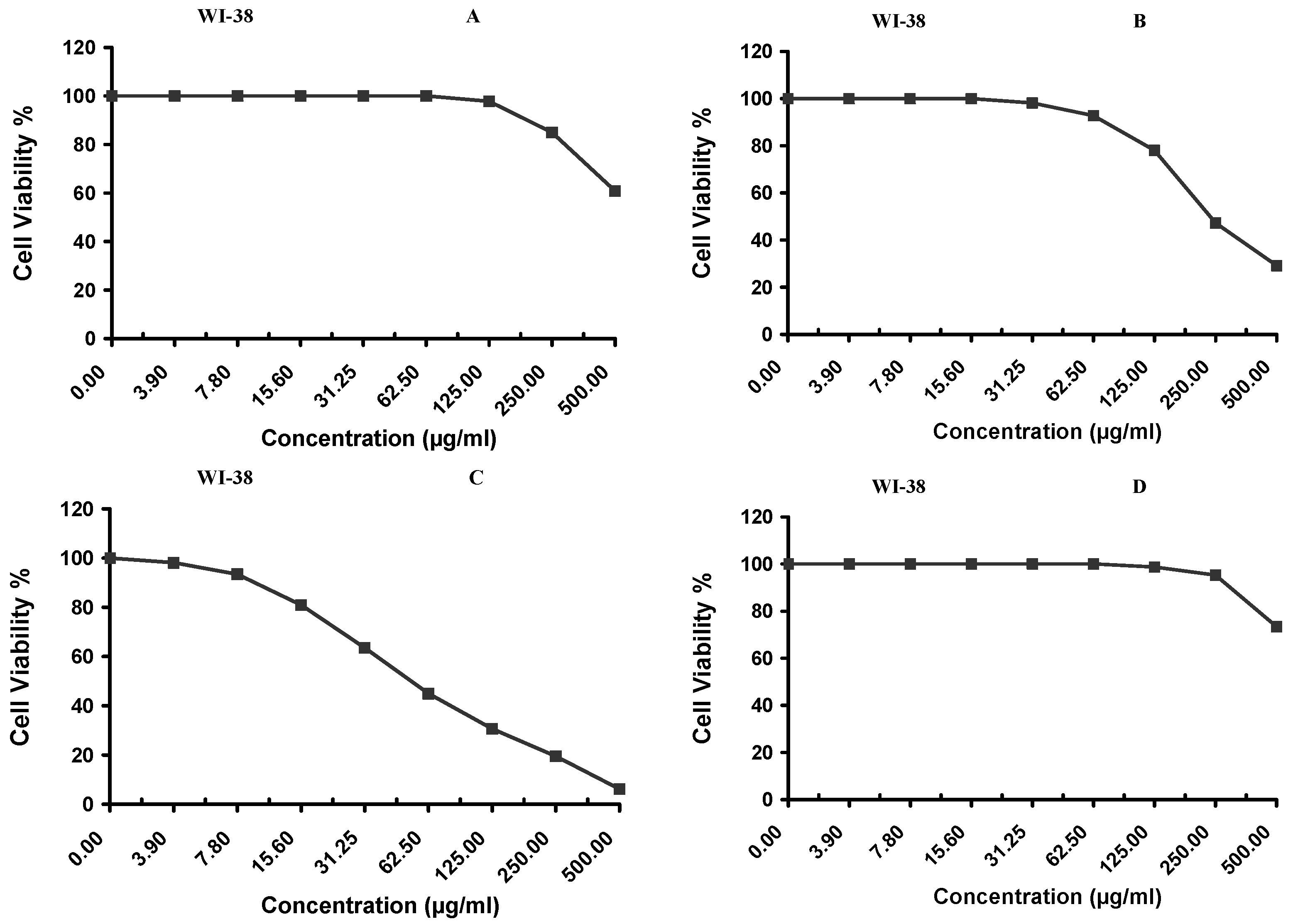
2.9. Evaluating the Cytotoxic Activity
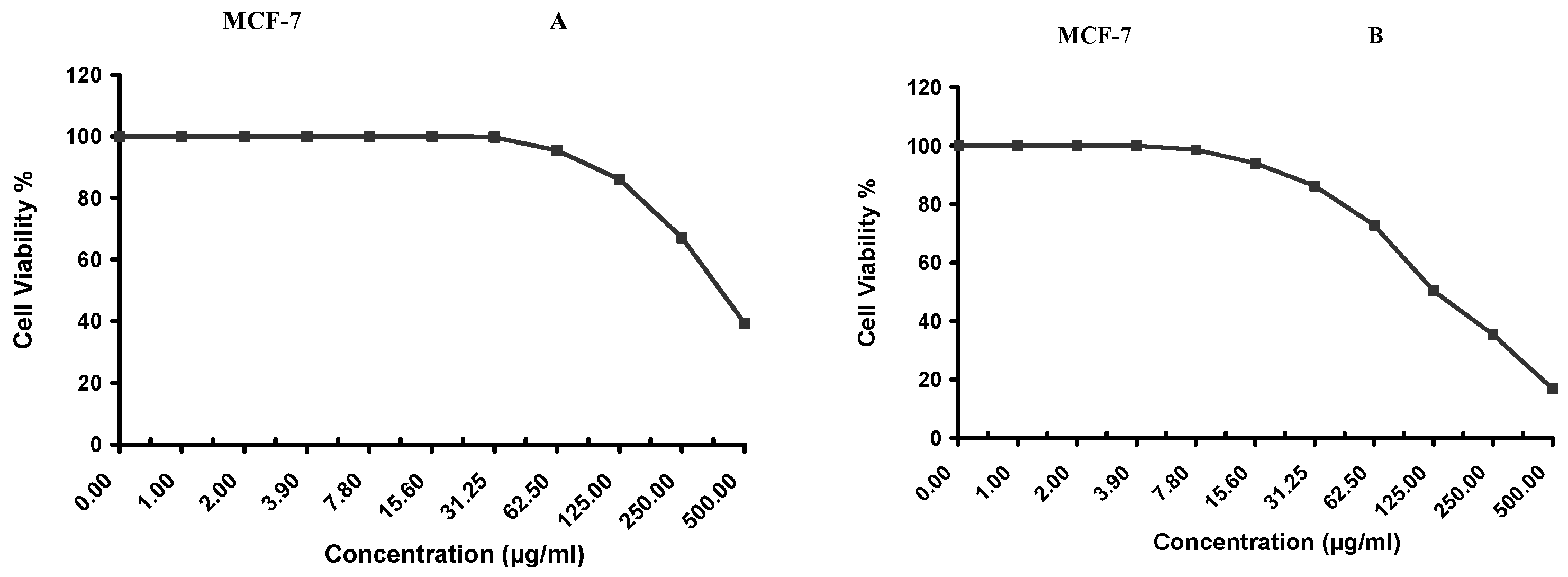
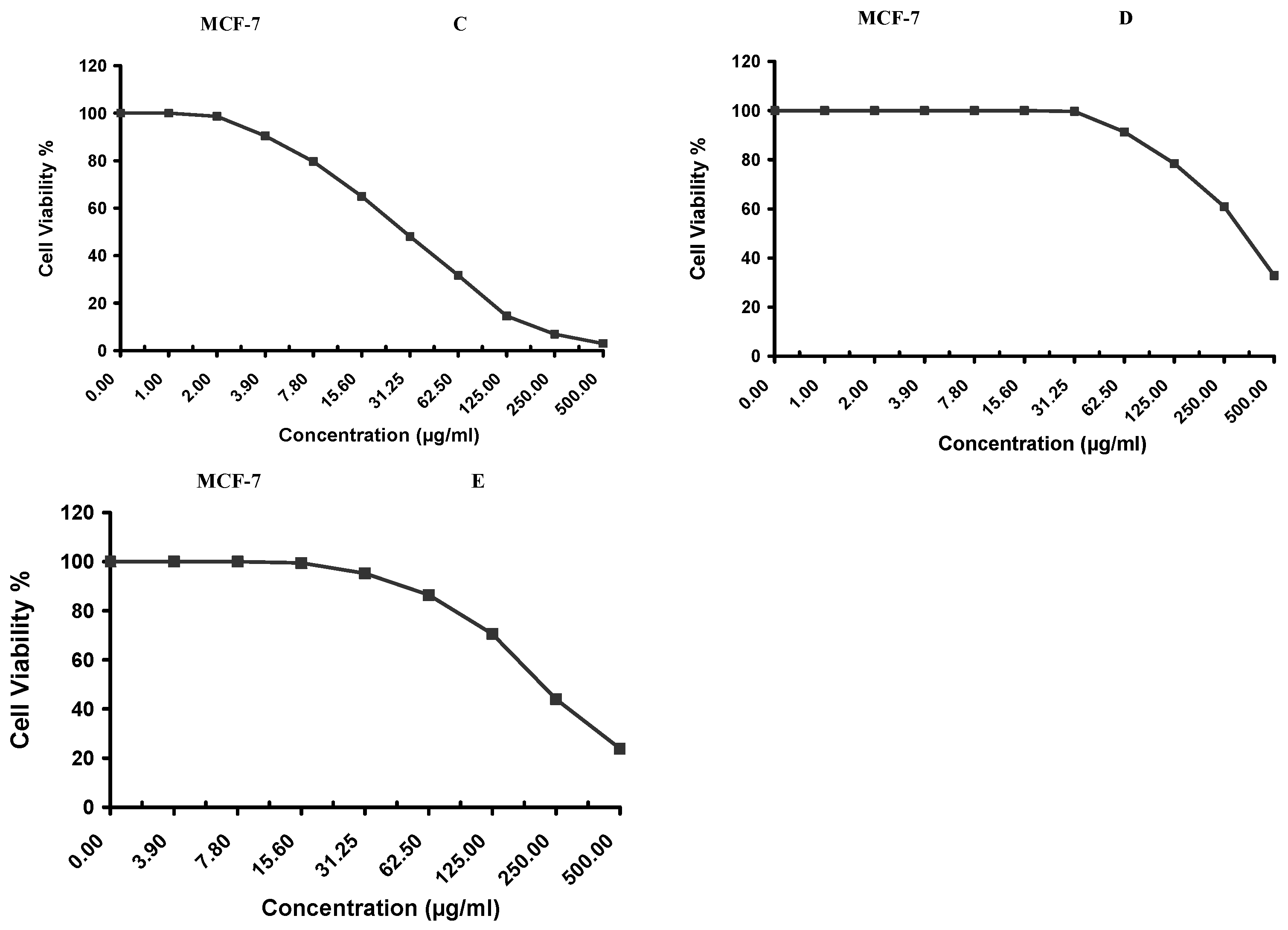
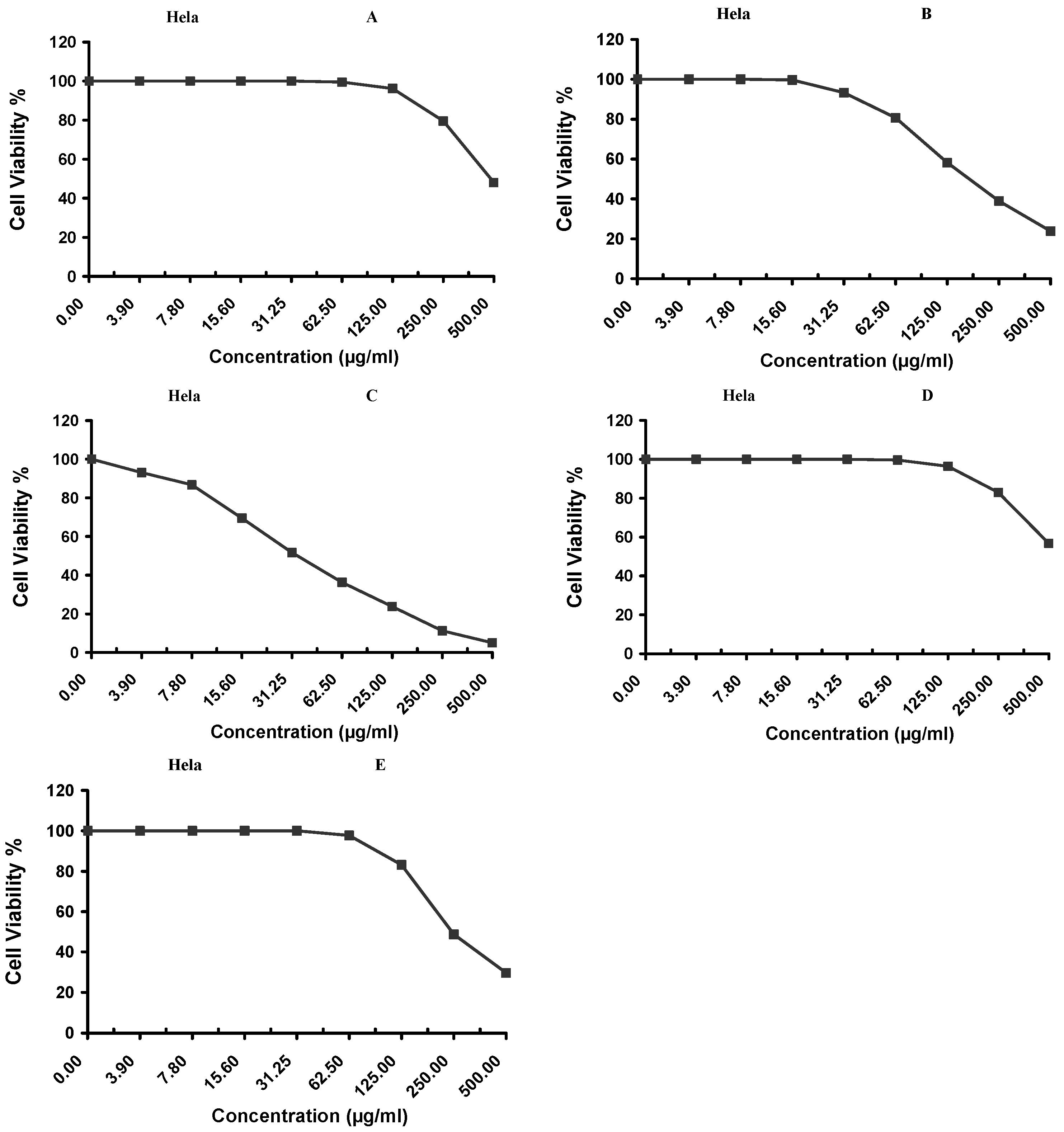
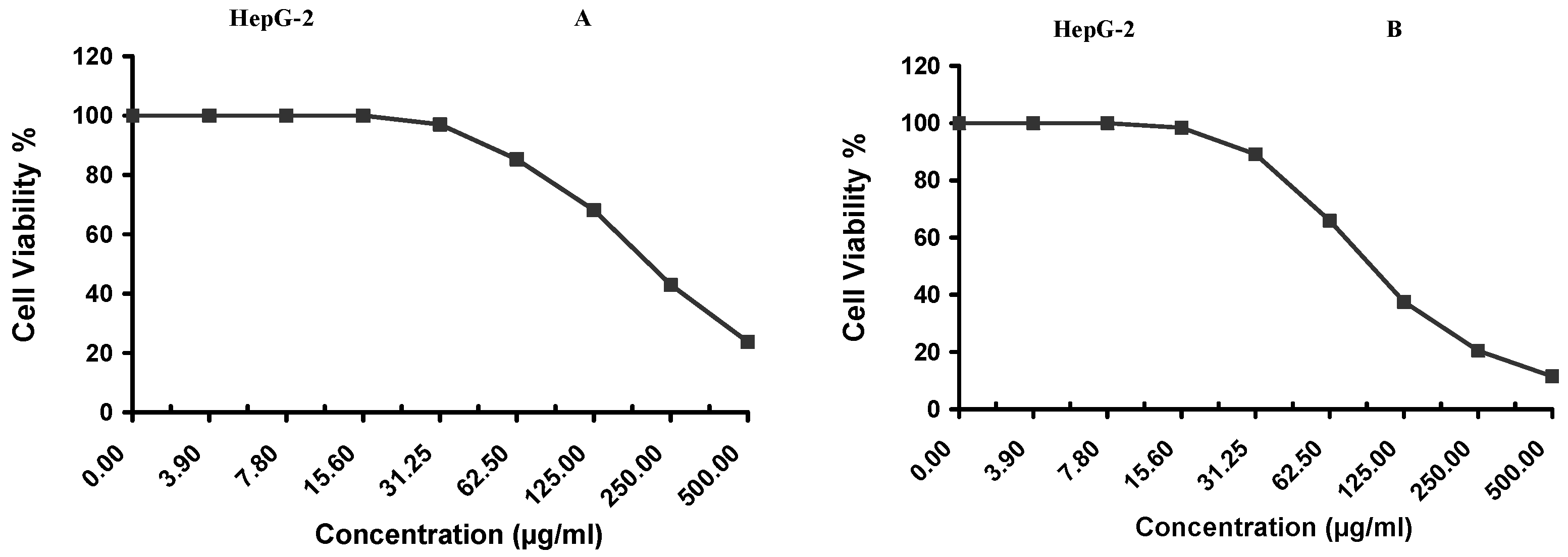
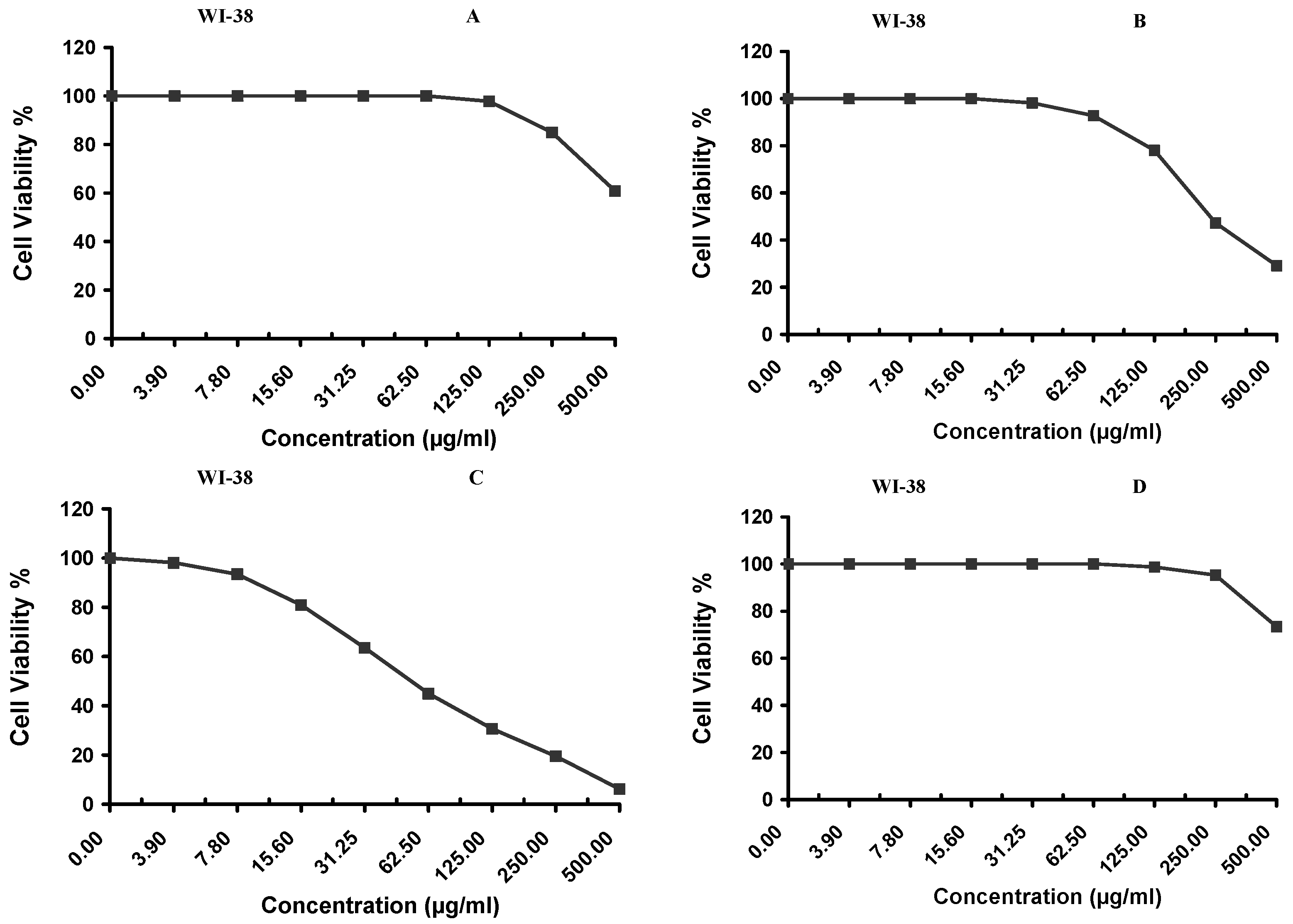
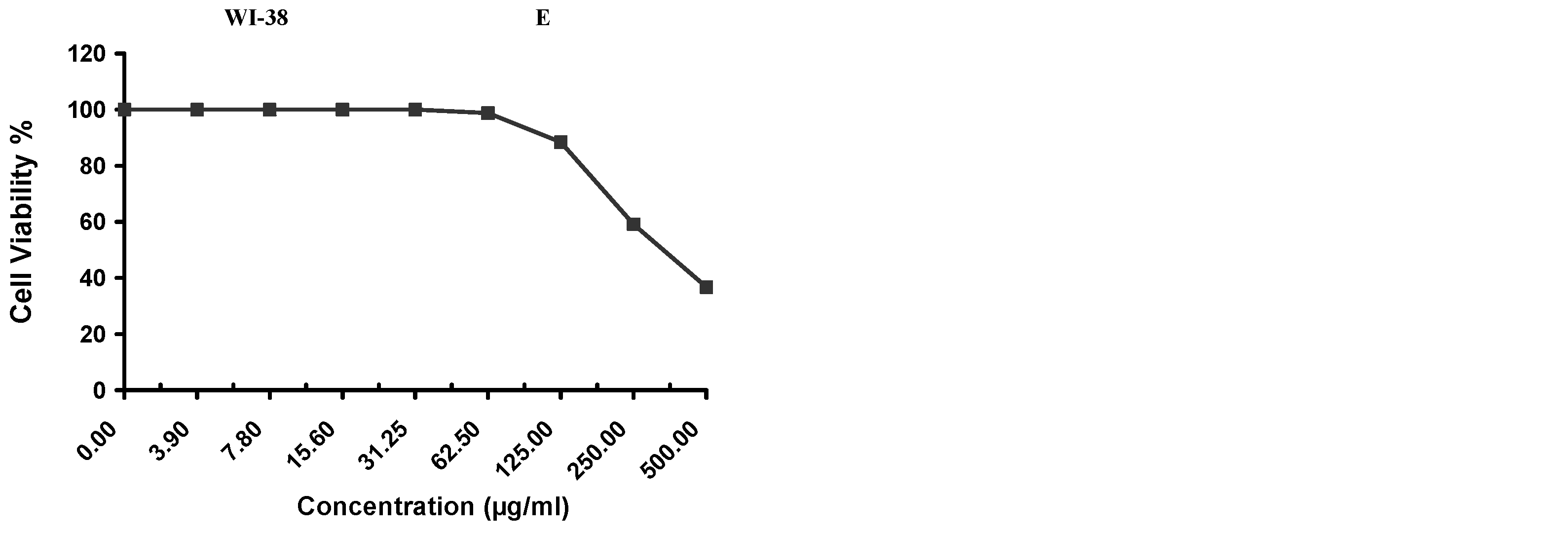
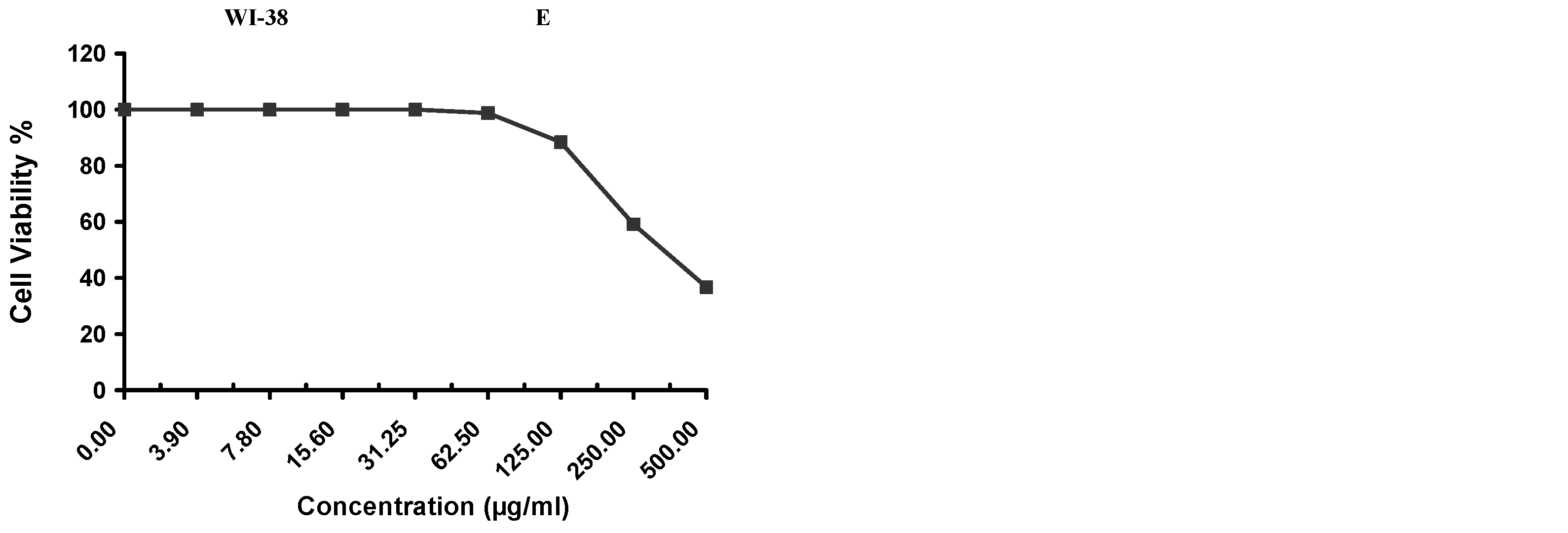
Cytotoxicity of the samples (NPs and pure Cf) were evaluated against four cancerous cell lines, MCF-7 (human breast cancer), A-549 (lung cancer), HeLa (cervical cancer) and HepG-2 (liver cancer), to assess their potential anticancer activity as well as against human normal lung fibroblast (WI-38) to assess their biocompatibility. The assessment was performed through the protocol of MTT assay as identified elsewhere [
43]. All cell lines were allowed to incubate for 72 h. The numbers of viable cells and the percent cell viability were also determined, applied similarly for all types of cell lines, and calculated according to Equation (5). The relation between surviving cells and drug concentration (dose–response curve) was plotted to get the survival curve of each tumor cell line after treatment with the specified sample
. The 50% inhibitory concentration (IC
50), the concentration required to cause toxic effects in 50% of intact cells, was also reported.
where OD
t is the mean optical density of wells treated with the tested sample and OD
c is the mean optical density of untreated cells.
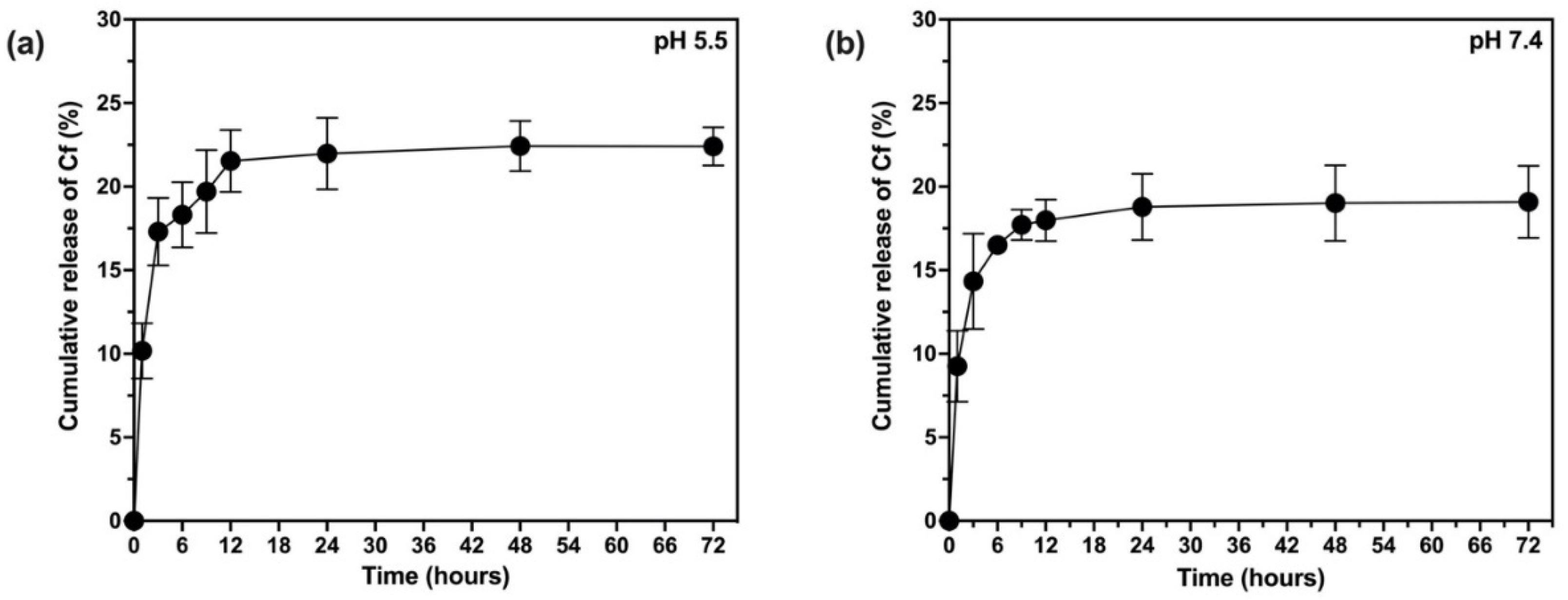
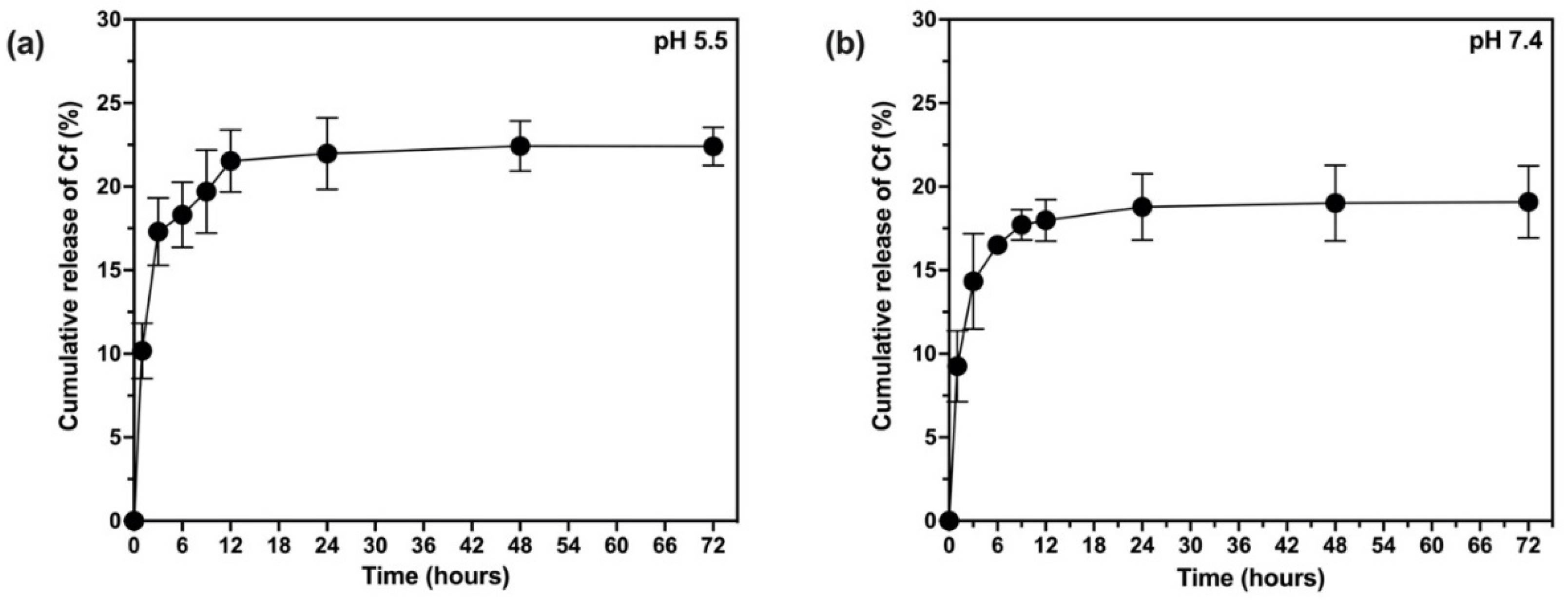
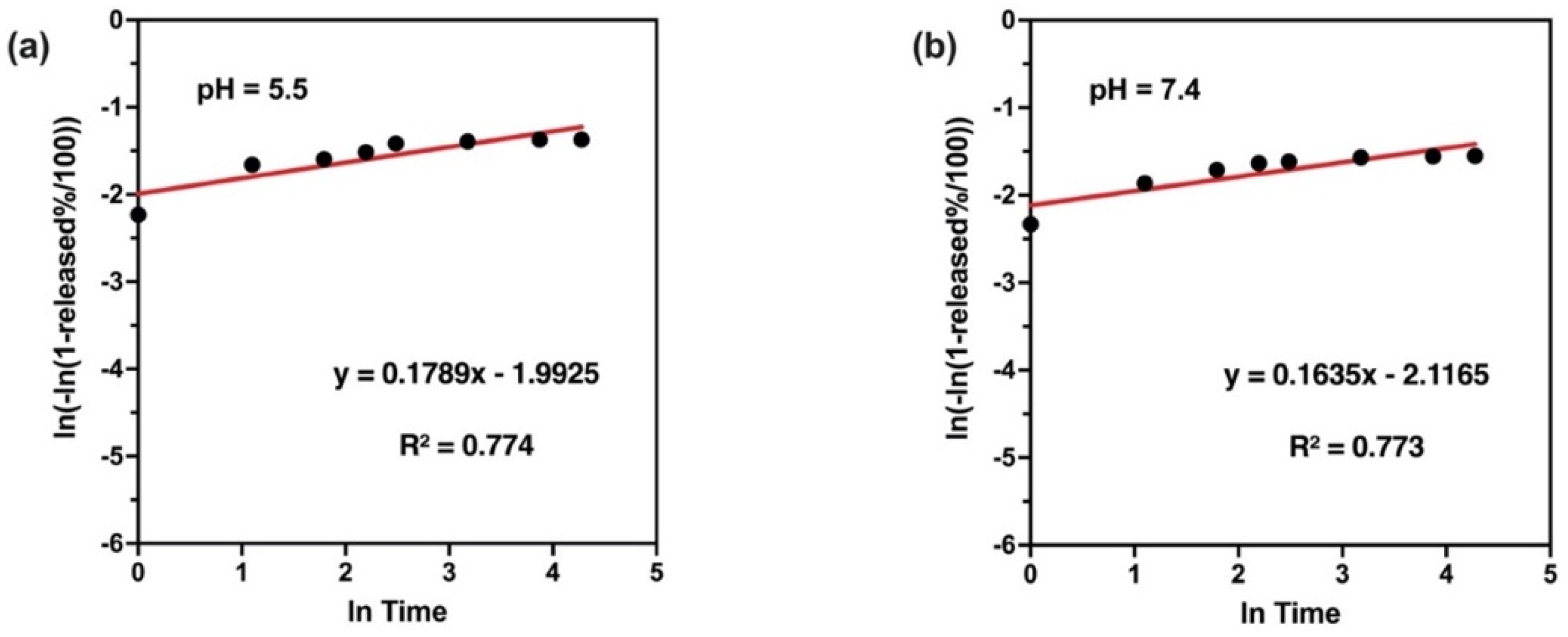
2.10. In Vitro Release and Release Kinetics Studies
The release rate of Cf from the NPs was investigated based on the quantification of the extract released from the NPs using UV-Vis spectroscopy in two different pH media (5.5 and 7.4) [
39,
42]. To achieve the sink condition, the release medium of pH 7.4 was prepared of 60% phosphate buffer saline (PBS) and 40% methanol (where the extract dissolves) whereas the release medium of pH 5.5 was prepared from 60% acetate buffer and 40% methanol. Typically, a dialysis bag (12,000–14,000 kDa) contained certain weight of lyophilized NPs and 5 mL of the release medium, then formed a total of 20 mL to be incubated in an orbital shaker at 37 °C and 100 rpm over a period of three days. Samples were withdrawn at predetermined time intervals throughout the 72 h and replaced by an equal volume of fresh medium. The samples were subjected to UV-Vis analysis (at 273 nm) to estimate the total cumulative amount of the extract (mg) in a unit volume of the release medium (mL). Finally, the cumulative amount of the released Cf extract was expressed as the cumulative release percent as in Equation (6). To study the kinetic profile of Cf release from the PLGA NPs, data were treated according to zero-order, first-order, Higuchi, Korsmeyer–Peppas, and Hixson–Crowell, Weibull, and Noyes–Whitney Equations [
38].
where
is the cumulative amount of the Cf extract released at each time interval while
is the initial amount of the extract added to the NP formula.
2.11. Statistical Analysis
One-way ANOVA was carried out, followed by Tukey’s test and statistical significance was expressed at p < 0.05. The values of the results were reported as the mean ± standard deviation (SD) of the triplicate determinations.
5. Conclusions
The present study evidently demonstrated a successful encapsulation of coffee extract into the biopolymeric nano-system, PLGA/PVA via the SE-SE method (owing to the hydrophobic nature of the extract). Primarily, the influence of three independent factors (PVA concentration, homogenization speed and duration) on the particle diameter, PDI, ZP, EE% and LC% was evaluated and statistically analyzed by BBD to identify the optimum formulation conditions. The results of this phase were very close to the expected values (with an average particle size of 273.750 nm, a PDI of 0.091, a ZP of −23.050 mV, a 76.892% as EE% and a 3.383% as LC%), confirming the success of the established design in fabricating the nano-system. By characterizing the NPs, the optimum formulation conditions were achieved at a PVA concentration of 0.5% (w/v), at a homogenization speed of 10,000 rpm for a duration of 7.5 min. A secondary phase was conducted to study the influence of increasing the initial drug loading on the aforementioned responses. Characterizing the NPs suggested a full and tight shielding of the drug by PLGA, and spherically shaped NPs with slight irregularity upon increasing the initial drug amount with higher TPC. In vitro evaluation of the NPs showed that sample PLGA-Cf-F2 (of particle size 318.60 ± 5.65 nm, PDI 0.074 ± 0.015, ZP −20.50 ± 0.52 mV, EE% 85.92 ± 4.01%, LC% 12.274% and a loading ratio of 0.5:1) recorded the most desirable results among other NPs and the pure Cf, in terms of the highest antioxidant activity (90,082 ± 0.199% inhibition), and the highest cytotoxicity against the four selected cancer cell lines (with an of 29.40 ± 1.10 µg/mL against MCF-7 cells, 23.40 ± 3.20 µg/mL against A-549 cells, 34.70 ± 4.10 µg/mL against HeLa cells and 18.20 ± 2.60 µg/mL against HepG-2 cells) with preferential toxicity over the normal fibroblasts (WI-38). The in vitro release rate study was performed on the succeeded NP sample at pH 5.5 and pH 7.4 for three days and showed a biphasic release rate pattern in both pH media. Finally, we can conclude that nano-encapsulation is a very promising tool for stabilizing the physicochemical properties of plant-sourced extracts as coffee extract, and for enhancing their respective biological activities. Accordingly, it can pave the way for endless applications in the pharmaceutical, food and cosmetic sectors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}