
Another Look at the Contribution of Oral Microbiota to the Pathogenesis of Rheumatoid Arthritis: A Narrative Review

 , ,
, , 
Abstract
:1. Introduction

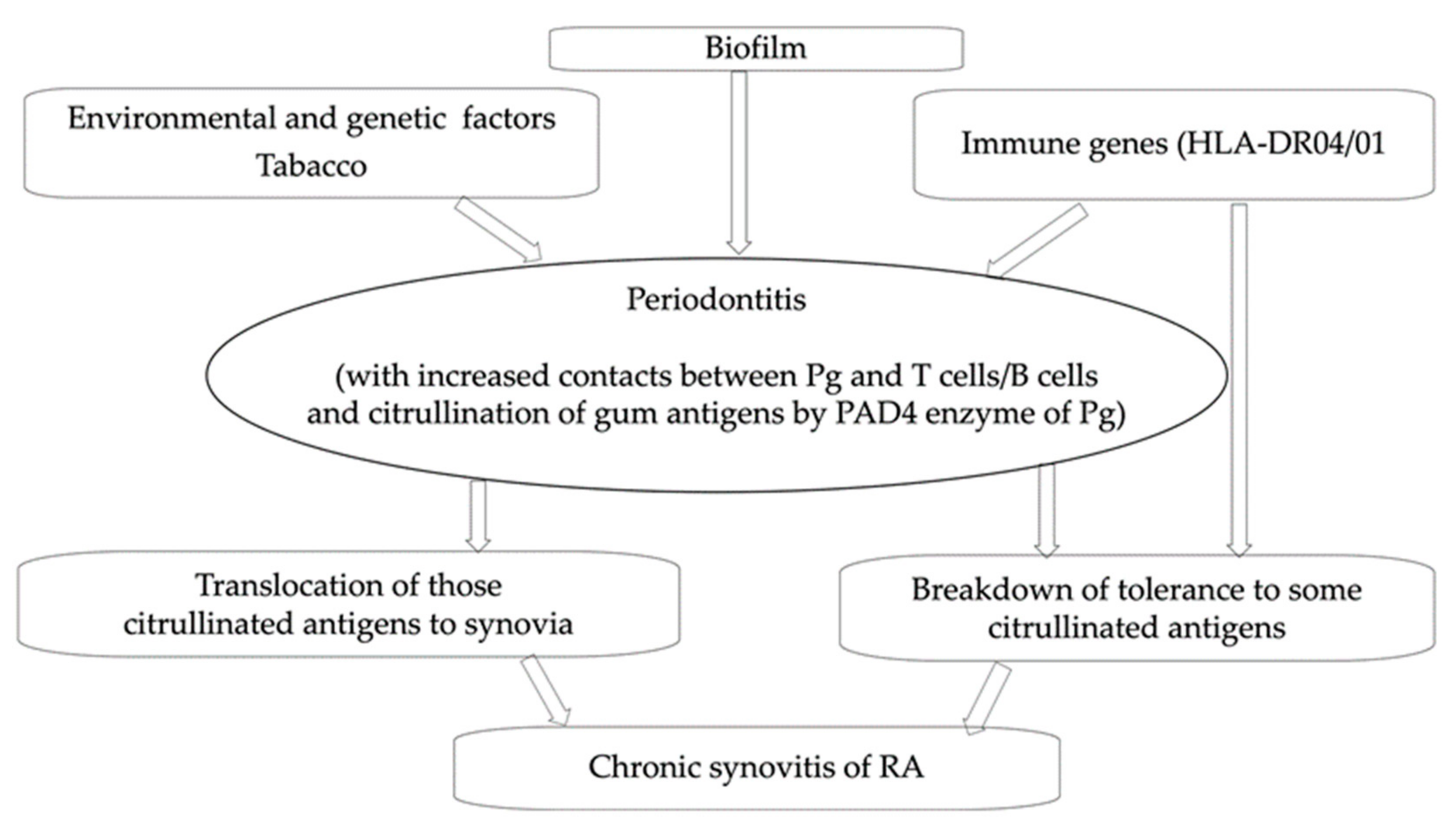{kind=link}
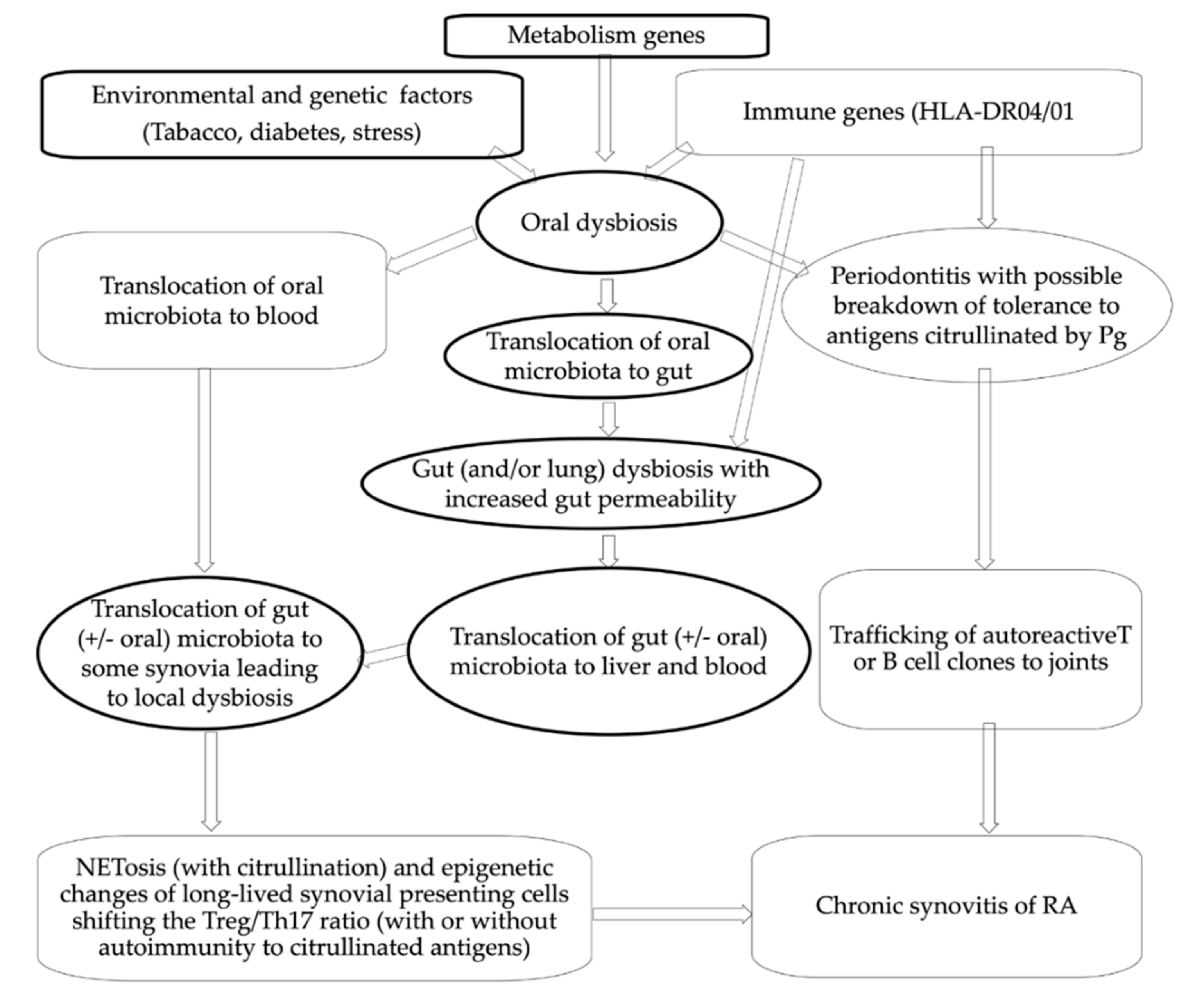{kind=link}
| Clinical and In Vitro Studies | |||||
|---|---|---|---|---|---|
| Author, Year | Objective | Patients (Mean Age) | Study Design | Study Group | Main Results |
| Rotsein, I. and Katz, J., 2021 [10] | To assess the prevalence of periapical abscesses in patients with RA, and to evaluate the effects of commonly used antirheumatic medications on such prevalence | NR | A cross-sectional study | Patients with RA were included and data of diagnosis of RA antirheumatic medications, and the presence of periapical abscess were recorded. | Statistically higher prevalence of periapical abscesses in patients with RA than in the general patient population (OR: 2.60) (p < 0.0001). Treatment with TNF-α inhibitors may lower the prevalence of periapical abscesses in such patients |
| Xiao, F. et al., 2021 [11] | To explore the correlation between RA and PD | 631 (40.93) | A cross-sectional comparative study | 307 patients with RA (RA group) and 324 healthy individuals (control group) were selected, the incidence of PD in both groups was analyzed, and ELISA was used to detect the IL-1β and TNF-α levels in the (GCF) of both groups | The prevalence of PD in the RA group was significantly higher than that in the control group and increased with age and disease duration in patients with RA. The presence of RA can increase risk of PD occurrence and is positively correlated with levels of IL-1β and TNF-α in the GCF. |
| González, D.A. et al., 2021 [12] | To identify clinical and/or serological variables in patients with RA that are associated with their periodontal severity | 110 (48.5) | A cross-sectional study | Subjects with RA and chronic periodontitis were included. RA clinical parameters and rheumatoid factor, presence of bone erosions, and rheumatic nodules) and corticosteroid therapy were considered. Periodontitis was evaluated according to the American Academy of Periodontology (1999). | Periodontitis severity is associated with higher RA disease activity (DAS-284.1; OR: 51.4; 95%, CI 9.4–281.5), longer disease duration, and rheumatoid nodules (OR: 6.; 95% CI: 1.3–31.6). |
| Moentadj, R. et al., 2021 [13] | To study the oral microbiota in a prospective cohort of patients with RA, FDR, and HC, then to genomically and functionally characterize streptococcal species from each group to understand their potential contribution to RA development. | 222 (45.7–53.5) | A cross-sectional comparative study | 116 patients with RA, 63 FDR, and 43 HC were included; salivary samples were taken by tongue swabbing for analysis of microbial dysbiosis by using 16S rRNA gene sequencing technology | Dysbiosis-associated periodontal inflammation and barrier dysfunction may permit arthritogenic insoluble pro-inflammatory pathogen-associated molecules, such as streptococcal cell walls, to reach synovial tissue. Streptococcus species can also be enriched in the oral cavity of some RA and their FDRs, and are the source of peptidoglycan-polysaccharide polymers that can induce arthritis in mice |
| Lehenaff, R. et al., 2021 [14] | To analyze the subgingival microbiome of both shallow (health-associated) and deep (disease-associated) subgingival sites in patients with RA, and then compare them with non-RA household controls. | 18 (53.2–55.1) | A case-control study | RA (n = 8) ok and non-RA (n = 10) subjects were recruited and subgingival plaque samples from both shallow (periodontal health-associated, probing depth ≤3 mm) and deep subgingival sites (periodontal disease-associated, probing depth ≥4 mm) were collected. RA subjects also had rheumatological evaluation. Plaque community profiles were analyzed using 16 S rRNA sequencing. | Lack of consistency of gingival microbiomes in RA may relate to numerous explanations: differences in RA subtypes, timing of sampling (RA onset or flares, versus RA in remission), treatments given, and how subgingival plaques are sampled: samples were collected from both shallow (periodontal health-associated, probing depth ≤3 mm) and deep subgingival sites (periodontal disease-associated, probing depth ≥4 mm). In RA, the microbiomes of deep and shallow sites in patients with RA were more similar to each other. |
| Arévalo-Caro, C. et al., 2021 [15] | To determine the relationship between titres of anti-Porphyromonas gingivalis (P. gingivalis) antibody andthe RA, HLA-DRB1 susceptibility region associated with SE using the Gregersen’s and de Vries’s classification methods. | 100 | A case-control study | Results of IgG1 and IgG2 anti-P. gingivalis antibodies, ACPA, diagnosis for RA, and PD, and a genetic study of the HLA-DRB1 region were obtained from 50 patients with RA and 50 control individuals. | Although no association was found between SE and anti-P. gingivalis antibodies; according to the de Vries’s classification, an association existed between HLA-DRB1 neutral alleles, and high titres of IgG anti-P.gingivalis antibodies for RA, focusing on novel associations between P.gingivalis and RA. NETosis or intracellular infections by gut pathobionts may also contribute to citrullination. This could explain why anti-Pg antibodies were not clearly associated with SE or ACPAs in patients with RA. |
| Manoil, D. et al., 2021 [16] | To examine potential correlations between detached subgingival bacteria collected in GCF and RA parameters. | 149 (34.95–48.04) | A case-control study | Patients with RA (n = 52) and patients with BD, (n = 40) as another systemic inflammatory disease, were studied along with a systemically healthy control group (HC; n = 57). Full mouth periodontal parameters were recorded. RA activity was assessed using the 28-joint DAS-28. RFs-IgM and -IgA were measured using ELISA. GCF samples were investigated by using fluorescent in situ hybridization for 10 different bacterial taxa. | Altogether, although RA and ACPAs could also be driven by other triggers such as gut bacteria, the PPAD of Pg might contribute to ACPAs in a subset of patients with RA. Pg displayed significant correlations with not only plaque scores and bleeding on probing, but also with RF-IgA. Patients with RA displaying RF-IgA levels >75 IU/mL exhibited fivefold more abundant Pg levels than those with levels below this threshold did. This association with RF-IgA levels appeared even more pronounced, by sixfold more Pg levels (p = 0.025), in patients with a DAS-28 score >3.2, indicative of being moderately/very active. |
| Peng, H.Y. et al., 2020 [17] | To determine the role of Pg in RA and to identify novel therapeutic targets for auto-inflammatory diseases. | 238 | A case-control study | Serum samples were obtained from patients with RA (n = 155), PD subjects (n = 48), and HCs (n = 35). The profile of antibody response to gingipain RgpA-specific domains, a cysteine protease produced by Pg, was determined in all included patients’ sera, and the potential protective effects of RgpA domains in an experimental arthritis model were also tested. | The pathobiont Pg is the bacteria detected most often in patients with both PD and RA, especially in smokers; smoking also strongly worsens PD and RA. Several lines of evidence suggest that Pg might contribute to some PD and RA pathogenesis: Pg produces cysteine proteases, such as gingipain RgpA, endowed with the potential to induce significant bone loss in animal model systems and in patients in either alveolar or subchondral bone. |
| Frid, P. et al. 2020 [18] | To characterize the salivary oral microbiome associated with JIA, and correlate it with the disease activity, including gingival inflammation | 93 (12.6) | A case-control study | Patients with JIA (n = 59) and healthy controls (HC; n = 34) were recruited, and microbiome profiling of saliva samples was performed by sequencing the V1 V3 region of the 16S rRNA gene. | No differences were found in alpha and beta diversity of oral bacteria among children with JIA and HC. Several taxa associated with chronic inflammation were found to be associated with JIA and disease activity. |
| Lopez-Oliva, I. et al., 2018 [19] | To characterize the periodontal microbiome in periodontally healthy individuals with and without RA, using next-generation sequencing | 41 (36–60) | A case-control study | Patients with RA (n = 22) and non-RA controls (n = 19) were recruted. All participants were periodontally healthy. Subgingival plaque samples were collected and analyzed using 16S rDNA sequencing. | In periodontally healthy individuals with RA, the oral microbiome is enriched for pro inflammatory organisms, and those capable of producing substantial amounts of citrulline (pro-antigenic). |
| Dong, X.H. et al., 2018 [20] | To present evidence showing that P. gingivalis OMVs promote the mucosal transmission of HIV-1 | NA | In vitro | The host cells were co-incubated with HIVNL4.3 alone, HIV/P. gingivalis 33,277 vesicle complexes, or HIV/KDP 128 vesicle complexes for 30 min. Cell-free complexes were removed by washing the cells with PBS, and the cells were then fixed, permeabilized, immunostained, and analyzed using confocal microscopy. | RANK overexpression and an increase in the ratio of RANK-L to osteoprotegerin was observed in both PD and RA, with a high level of RANK-L expression on gingival B cells, most notably those capable of recognizing Pg. Infection with the virus worsens both periodontitis and RA, which act by promoting the growth of organisms such as Pg. Reciprocally, Pg could foster infection of oral keratinocytes by viruses such as EBV and CMV, as Pg OMVs promote the mucosal transmission of viruses |
| Stobernack, T. et al., 2018 [21] | To identify possible functions of PPAD in the periodontal environment. | NA | In vitro | Human paraffin-embedded gingival tissues were collected from patients with Pg-colonized periodontitis. Deparaffinization of 5 m sections was performed by several xylene, ethanol, and water washes. Endogenous peroxidase activity was inhibited by the addition of hydrogen peroxide in methanol, followed by blocking of nonspecific antibody binding with 1% bovine serum albumin and 1% normal goat serum in PBS. | PPAD induces the citrullination of various autoantigens. The rationale for this Pg-induced citrullination may be linked to the ability of PPAD to citrullinate the histone H3, thereby facilitating bacterial escape from NETs, and immune evasion of Pg. The PAD enzyme, detectable in the gingiva of patients with PD, neutralizes human innate immune defenses at three other distinct levels: bacterial phagocytosis, capture in NETs, and killing by the lysozyme-derived cationic antimicrobial peptide LP9 |
| Zhang, X. et al., 2015 [2] | To characterize the oral and gut microbiomes in patients with RA compared to heathly controls. | 191 (18–65) | A case-control study | A metagenomic shotgun sequencing and a metagenome-wide association study of fecal, dental, and salivary samples from treatment-naive individuals with RA (n = 94), and HC (n = 97) was conducted. | Compared to heathly controls, dysbiosis was detected in the gut and oral microbiomes of patients with RA, but it was partially resolved after RA treatment. Alterations in the gut, dental, or saliva microbiome distinguished individuals with RA from HC. |
| Scher, J.U. et al., 2013 [22] | To determine if particular intestinal bacteria are associated with RA | 114 (42.4–50) | A cross-sectional comparative study | Subjects with NORA (n = 44), chronic, treated RA (n = 26), PsA (n = 16), and HCs (n = 28) were included, and 16S sequencing (of the 16S gene (regions V1–V2, 454 platform) on 114 stool samples from patients with RA and controls, and shotgun sequencing on a subset of 44 such samples were performed. | Many cases of RA may later be induced by pathobionts from gut microbiota, such as Prevotella Copri, indicating a potential role of this bacterium in the pathogenesis of RA. |
| Review articles | |||||
| Alghamdi, M.A., 2021 [23] | To highlight the effect of both gut and oral microbiota dysbiosis on the development of RA, as well as to discuss how the alteration in microbiota composition leads to the overgrowth of some bacterial species entangled in RA pathogenicity. | NA | Review | NA | Pg-induced citrullination may contribute to the occurrence of antibodies to citrullinated peptides (ACPA), the most specific signature of RA, especially in patients with the SE HLA-DRB1-04/01 |
| Nik-Azis, N.M. et al., 2021 [24] | To discuss how RA and specifically ACPA-positive RA link to PD, and to appraise the epidemiologicalevidenceonthe relationship between ACPA-positive RA and PD | NA | Review | Articles were searched following the PRISMA guidelines across the Medline, Web of Science, Scopus, and Cochrane Library databases | The severity manifestations of periodontal disease did not differ much in patients with RA with and without ACPA |
| Chu, X.J. et al., 2021 [25] | To describe a possible relationship between RA and the microbiome of the oral cavity and gut. | NA | A systematic review | A bibliographic search was performed in three databases (EMBASE, Cochrane Library, and PubMed) from inception to 7 June 2020 to identify case control studies that compared the oral and gut microbiomes in adult patients with RA to those of controls | Of 26 articles reviewed, ≥3 articles reported decreased Streptococcus and Haemophilus contrasting with increased Prevotella in the oral cavity of patients with RA compared with heathy controls. In addition, some Prevotella species, including P. histicola and P. oulorum, showed increased trends in oral cavites of patients with RA, compared with HCs. |
| Gonzalez-Febles, J. et al., 2021 [26] | To update existing information on the epidemiological association between RA and PD and the biological mechanisms linking these two diseases. To determine whether treatment of PD could influence the initiation and progression of RA. | NA | Review | PubMed, Medline, and Cochrane Library | There was a clear association between PD and RA, with ORs ranging from 1.82 to 20.57 and patients with RA having a high prevalence of PD and tooth loss. The presence of both periodontal inflammation and high numbers of periodontopathic bacteria (Pg and Aa) have been associated with the onset of RA and increased RA disease activity. Nonsurgical periodontal therapy seems to play a role in the control of RA disease activity. |
| Zorba, M. et al. 2020 [27] | To review current literature on the possible role of the oral microbiome in the pathogenesis of autoimmune diseases. | NA | Review | PubMed, Medline, Research Gate, and Google Scholar | Oral dysbiosis has also been reported in other adult disorders sometimes overlapping with RA (Sjögren’s syndrome, systemic lupus erythematosus, RA, BD, Crohn’s disease, and psoriasis). |
| Berthelot, J.M and Le Goff, B., 2010 [28] | To assess the prevalence of PD in patients with RA. | NA | Review | PubMed, Medline, and EMBASE | Modest but increased prevalence of PD among patients with RA compared to the general population, unrelated to secondary Sjögren’s syndrome. Indeed, the prevalence of the SE HLA-DRB1-04 is increased in both RA and PD, and exacerbated T-cell responsiveness with high tissue levels of IL-17 and exaggerated B-cell/plasma cells responses are found in both the synovium and gingival tissue affected with PD. |
2. Translocation of Oral Microbiota toward Synovium through Blood May Occur Either Directly or Following Gut Colonization
2.1. Direct Translocation from Gingiva to Synovia
2.2. Translocation of Some Oral Bacteria in Blood May Occur Following a First Step of Migration of Those Bacteria to Gut
2.3. Translocation of Oral Bacteria in Blood Has Been Observed, including in Patients with RA
2.4. Microbiotal DNA, including DNA from Oral Bacteria, Has Been Found in Synovia of Patients with RA and Healthy Controls
2.5. Acquired Dysbiosis in Oral Mucosa Might Translate in Lasting Dysbiosis in Some RA Synovia, Following Translocation of Various Pathobionts
3. Methodologies Other Than Transversal Studies of Periodontal Microbiota in Cohorts of Patients with RA versus Controls Are Required
3.1. Studies of Individuals at Risk of RA Even before the Onset of Arthritis, and Very Early RA
3.2. Longitudinal Studies of Microbiota in Individual Patients with RA (before, during, and Following Flares) with Simultaneous Assessments of Synovial, Blood, Oral, and Gut Microbiota
3.3. Separate Analysis according to HLA Background and Other Parameters That May Affect Oral and Synovial Dysbiosis, Such as Smoking, Metabolic and Hormonal Status (Age and Sex), and Ongoing Treatments
3.3.1. Genetics
3.3.2. Stress, Hormonal Status, and Tobacco
3.3.3. Ongoing Treatments
3.4. Concurrent Assessments of Immune Responses (Including Regulatory T Cell and Th17 Responses) through Transcriptomic Analyses in Gingival Sulcus, Blood, and Synovium
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Möller, B.; Kollert, F.; Sculean, A.; Villiger, P.M. Infectious triggers in periodontitis and the gut in rheumatoid arthritis (RA): A complex story about association and causality. Front. Immunol. 2020, 11, 1108. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Zaura, E.; Brandt, B.W.; Buijs, M.J.; Brun, J.G.; Crielaard, W.; Bolstad, A.I. Subgingival microbiome of rheumatoid arthritis patients in relation to their disease status and periodontal health. PLoS ONE 2018, 13, e0202278. [Google Scholar] [CrossRef] [Green Version]
- Teles, F.; Wang, Y.; Hajishengallis, G.; Hazturk, H.; Marchesan, J.T. Impact of systemic factors in shaping the periodontal microbiome. Periodontology 2000 2021, 85, 126–160. [Google Scholar] [CrossRef]
- Buschhart, A.L.; Bolten, L.; Volzke, J.; Ekat, K.; Kneitz, S.; Mikkat, S.; Kreikemeyer, B.; Müller-Hilke, B. Periodontal pathogens alter the synovial proteome. Periodontal pathogens do not exacerbate macroscopic arthritis but alter the synovial proteome in mice. PLoS ONE 2020, 15, e0242868. [Google Scholar] [CrossRef]
- Dehner, C.; Fine, R.; Kriegel, M.A. The microbiome in systemic autoimmune disease: Mechanistic insights from recent studies. Curr. Opin. Rheumatol. 2019, 31, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.W.; Wang, X. Mobile microbiome: Oral bacteria in extra-oral infections and inflammation. J. Dent. Res. 2013, 92, 485–491. [Google Scholar]
- Davison, E.; Johnston, W.; Piela, K.; Rosier, B.T.; Paterson, M.; Mira, A.; Culshaw, S. The Subgingival plaque microbiome, systemic antibodies against bacteria and citrullinated proteins following periodontal therapy. Pathogens 2021, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Moura, M.F.; Cota, L.O.M.; Silva, T.A.; Cortelli, S.C.; Ferreira, G.A.; López, M.M.; Cortelli, J.R.; Costa, F.O. Clinical and microbiological effects of non-surgical periodontal treatment in individuals with rheumatoid arthritis: A controlled clinical trial. Odontology 2021, 109, 484–493. [Google Scholar] [CrossRef]
- Rotsein, I.; Katz, J. Prevalence of periapical abscesses in patients with rheumatoid arthritis. A cross sectional study Am. J. Dent. 2021, 34, 211–214. [Google Scholar]
- Xiao, F.; Li, C.; Lin, Y.; Peng, Z.; Xu, X.; Wen, Y.; Huang, N.; Zhang, P. Increased risk of periodontitis occurrence in patients with rheumatoid arthritis and its association with the levels of IL-1β and TNF-α in gingival crevicular fluid. Ann. Palliat. Med. 2021, 10, 9078–9087. [Google Scholar] [CrossRef]
- González, D.A.; Bianchi, M.L.; Salgado, P.A.; Armada, M.; Seni, S.; Isnardi, C.A.; Citera, G.; Ferrary, T.; Orman, B. Disease activity and subcutaneous nodules are associated to severe periodontitis in patients with rheumatoid arthritis. Rheumatol. Int. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Moentadj, R.; Wang, Y.; Bowerman, K.; Rehaume, L.; Nel, H.; Cuiv, P.O.; Stephens, J.; Baharom, A.; Maradana, M.; Lakis, V.; et al. Streptococcus species enriched in the oral cavity of patients with RA are a source of peptidoglycan-polysaccharide polymers that can induce arthritis in mice. Ann. Rheum Dis. 2021, 80, 573–581. [Google Scholar] [CrossRef]
- Lehenaff, R.; Tamashiro, R.; Nascimento, M.M.; Lee, K.; Jenkins, R.; Whitlock, J.; Li, E.C.; Sidhu, G.; Anderson, S.; Progulske-Fox, A.; et al. Subgingival microbiome of deep and shallow periodontal sites in patients with rheumatoid arthritis: A pilot study. BMC Oral. Health 2021, 21, 248. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Caro, C.; Romero-Sánchez, C.; Garavito-Rodríguez, E. Relation between anti- Porphyromonas gingivalis antibody titers and HLA-DRB1 neutral alleles in individuals with rheumatoid arthritis. Acta Odontol. Scand. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Manoil, D.; Bostanci, N.; Mumcu, G.; Inanc, N.; Can, M.; Direskeneli, H.; Belibasakis, G.N. Novel and known periodontal pathogens residing in gingival crevicular fluid are associated with rheumatoid arthritis. J. Periodontol. 2021, 92, 359–370. [Google Scholar] [CrossRef]
- Peng, H.Y.; Chen, S.Y.; Siao, S.H.; Chang, J.T.; Xue, T.Y.; Lee, Y.H.; Jan, M.S.; Tsay, G.J.; Zouali, M. Targeting a cysteine protease from a pathobiont alleviates experimental arthritis. Arthritis Res. Ther. 2020, 22, 114. [Google Scholar] [CrossRef] [PubMed]
- Frid, P.; Baraniya, D.; Halbig, J.; Rypdal, V.; Songstad, N.T.; Rosèn, A.; Berstad, J.R.; Flatø, B.; Alakwaa, F.; Gil, E.G.; et al. Salivary oral microbiome of children with juvenile idiopathic arthritis: A norwegian cross-sectional study. Front. Cell Infect. Microbiol. 2020, 10, 602239. [Google Scholar] [CrossRef]
- Lopez-Oliva, I.; Paropkari, A.D.; Saraswat, S.; Serban, S.; Yonel, Z.; Sharma, P.; De Pablo, P.; Raza, K.; Filer, A.; Chapple, I.; et al. Dysbiotic subgingival microbial communities in periodontally healthy patients with rheumatoid arthritis. Arthritis Rheumatol. 2018, 70, 1008–1013. [Google Scholar] [CrossRef]
- Dong, X.H.; Ho, M.H.; Liu, B.; Hildreth, J.; Dash, C.; Goodwin, J.S.; Balasubramaniam, M.; Chen, C.H.; Xie, H. Role of Porphyromonas gingivalis outer membrane vesicles in oral mucosal transmission of HIV. Sci. Rep. 2018, 8, 8812. [Google Scholar] [CrossRef] [Green Version]
- Stobernack, T.; Du Teil Espina, M.; Mulder, L.M.; Palma Medina, L.M.; Piebenga, D.R.; Gabarrini, G.; Zhao, X.; Janssen, K.M.J.; Hulzebos, J.; Brouwer, E.; et al. A Secreted bacterial peptidylarginine deiminase can neutralize human innate immune defense. mBio 2018, 9, e01704-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Alghamdi, M.A.; Redwan, E.M. Interplay of microbiota and citrullination in the immunopathogenesis of rheumatoid arthritis. Probiotics Antimicrob. Proteins. 2021, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nik-Azis, N.M.; Mohd, N.; Baharin, B.; Said, M.S.M.; Fadzilah, F.M.; Haflah, N.H.M. Periodontal disease in seropositive rheumatoid arthritis: Scoping review of the epidemiological evidence. Germs 2021, 11, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.J.; Cao, N.W.; Zhou, H.Y.; Meng, X.; Guo, B.; Zhang, H.Y.; Li, B.Z. The oral and gut microbiome in rheumatoid arthritis patients: A systematic review. Rheumatology 2021, 60, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Febles, J.; Sanz, M. Periodontitis and rheumatoid arthritis: What have we learned about their connection and their treatment? Periodontology 2000 2021, 87, 181–203. [Google Scholar] [CrossRef] [PubMed]
- Suárez, L.J.; Garzón, H.; Arboleda, S.; Rodríguez, A. Oral dysbiosis and autoimmunity: From local periodontal responses to an imbalanced systemic immunity. A review. Front. Immunol. 2020, 11, 591255. [Google Scholar] [CrossRef]
- Berthelot, J.M.; Le Goff, B. Rheumatoid arthritis and periodontal disease. Joint Bone Spine 2010, 77, 537–541. [Google Scholar] [CrossRef]
- Barzaghi, F.; Passerini, L.; Bacchetta, R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: A paradigm of immunodeficiency with autoimmunity. Front. Immunol. 2012, 3, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttierez, M.J.; Gilson, J.; Zacharias, J.; Ishmael, F.; Bingham, C.A. Childhood polyarthritis as early manifestation of autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy syndrome. Front. Immunol. 2017, 8, 377. [Google Scholar] [CrossRef]
- Generali, E.; Bose, T.; Selmi, C.; Voncken, J.W.; Damoiseaux, J.G.M.C. Nature versus nurture in the spectrum of rheumatic diseases: Classification of spondyloarthritis as autoimmune or autoinflammatory. Autoimmun. Rev. 2018, 17, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Asquith, M.; Sternes, P.R.; Costello, M.E.; Karstens, L.; Diamond, S.; Martin, T.M.; Li, Z.; Marshall, M.S.; Spector, T.D.; Le Cao, K.A.; et al. HLA alleles associated with risk of ankylosing spondylitis and rheumatoid arthritis influence the gut microbiome. Arthritis Rheumatol. 2019, 71, 1642–1650. [Google Scholar] [CrossRef]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid arthritis and the mucosal origins hypothesis: Protection turns to destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Barra, L.; Scinocca, M.; Saunders, S.; Bhayana, R.; Rohekar, S.; Racapé, M.; Coles, R.; Cairns, E.; Bell, D.A. Anti-Citrullinated protein antibodies in unaffected first-degree relatives of rheumatoid arthritis patients. Arthritis Rheum. 2013, 65, 1439–1447. [Google Scholar] [CrossRef]
- Generali, E.; Isailovic, N.; De Santis, M.; Ceribelli, A.; Alborghetti, F.; Colloredo, G.; Porrati, L.; Matthias, T.; Zucchi, A.; Guidelli, G.M.; et al. Prevalence and predictive value over 16 years of serum rheumatoid factor and anti-cyclic citrullinated peptide antibodies in the general population. Arthritis Rheumatol. 2017, 69 (Suppl. 10). Available online: https://acrabstracts.org/abstract/prevalence-and-predictive-value-over-16-years-of-serum-rheumatoid-factor-and-anti-cyclic-citrullinated-peptide-antibodies-in-the-general-population/ (accessed on 28 December 2021).
- Berthelot, J.M.; Puéchal, X. Impaired intracellular pathogen clearance and inflammatory joint disease: Is whipple’s disease a guiding light? Joint Bone Spine 2018, 85, 531–536. [Google Scholar] [CrossRef]
- Ahn, J.K.; Hwang, J.; Seo, G.H. Incidence and risk of developing rheumatic diseases in 19, 724 patients with palindromic rheumatism in South Korea: A nationwide population-based study. Joint Bone Spine 2021, 88, 105128. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, J.M.; De Bandt, M.; Morel, J.; Benatig, F.; Constantin, A.; Gaudin, P.; Le Loet, X.; Maillefert, J.F.; Meyer, O.; Pham, T.; et al. A tool to identify recent or present rheumatoid arthritis flare from both patient and physician perspectives: The ‘FLARE’ instrument. Ann. Rheum Dis. 2012, 71, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Costa Del Amo, P.; Lahoz-Beneytez, J.; Boelen, L.; Ahmed, R.; Miners, K.L.; Zhang, Y.; Roger, L.; Jones, R.E.; Marraco, S.; Speiser, D.E.; et al. Human TSCM cell dynamics in vivo are compatible with long-lived immunological memory and stemness. PLoS Biol. 2018, 16, e2005523. [Google Scholar] [CrossRef]
- Berthelot, J.M.; Sibilia, J. Trained immunity and autoimmune disease: Did Eve sin before Adam? Joint Bone Spine 2019, 86, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Gabarrini, G.; Grasso, S.; Van Winkelhoff, A.J.; Van Dijl, J.M. Gingimaps: Protein localization in the oral pathogen Porphyromonas gingivalis. Microbiol Mol. Biol Rev. 2020, 84, e00032-19. [Google Scholar] [CrossRef]
- Gabarrini, G.; Palma Medina, L.M.; Stobernack, T.; Prins, R.C.; Du Teil Espina, M.; Kuipers, J.; Chlebowicz, M.A.; Rossen, J.W.A.; Van Winkelhoff, A.J.; Van Dijl, J.M. There’s no place like OM: Vesicular sorting and secretion of the peptidylarginine deiminase of Porphyromonas gingivalis. Virulence 2018, 9, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Fine, D.H.; Schreiner, H.; Velusamy, S.K. Aggregatibacter, a low abundance pathobiont that influences biogeography, microbial dysbiosis, and host defense capabilities in periodontitis: The history of a bug, and localization of disease. Pathogens 2020, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, J.M.; Le Goff, B.; Maugars, Y. Bone marrow mesenchymal stem cells in rheumatoid arthritis, spondyloarthritis, and ankylosing spondylitis: Problems rather than solutions? Arthritis Res. Ther. 2019, 21, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, H.C.; Wang, Z.; Wang, G.F.; El-Gabalawy, H.; Goldbach-Mansky, R.; Li, Y.; Majeed, W.; Zhang, H.; Ngai, N.; Hudson, A.P.; et al. Chromosomal DNA from a variety of bacterial species is present in synovial tissue from patients with various forms of arthritis. Arthritis Rheum. 2001, 44, 1689–1697. [Google Scholar] [CrossRef]
- Cai, W.W.; Yu, Y.; Zong, S.Y.; Wei, F. Metabolic reprogramming as a key regulator in the pathogenesis of rheumatoid arthritis. Inflamm. Res. 2020, 69, 1087–1101. [Google Scholar] [CrossRef]
- Berthelot, J.M.; Le Goff, B.; Neel, A.; Maugars, Y.; Hamidou, M. NETosis: At the crossroads of rheumatoid arthritis, lupus, and vasculitis. Joint Bone Spine 2017, 84, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Hwang, J.; Xuan, J.; Jung, Y.H.; Cha, H.S.; Kim, K.H. Global metabolite profiling of synovial fluid for the specific diagnosis of rheumatoid arthritis from other inflammatory arthritis. PLoS ONE 2014, 9, e97501. [Google Scholar] [CrossRef] [Green Version]
- Khor, B.; Snow, M.; Herrman, E.; Ray, N.; Mansukhani, K.; Patel, K.A.; Said-Al-Naief, N.; Maier, T.; Machida, C.A. Interconnections between the oral and gut microbiomes: Reversal of microbial dysbiosis and the balance between systemic health and disease. Microorganisms 2021, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Herrema, H.; Nieuwdorp, M.; Groen, A.K. Microbiome and cardiovascular disease. Handb Exp. Pharmacol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Davanian, H.; Gaiser, R.A.; Silfverberg, M.; Hugerth, L.W.; Sobkowiak, M.J.; Lu, L.; Healy, K.; Sandberg, J.K.; Näsman, P.; Karlsson, J.; et al. Mucosal-associated invariant T cells and oral microbiome in persistent apical periodontitis. Int. J. Oral. Sci. 2019, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jia, Z.; Zhang, B.; Peng, L.; Zhao, F. Tracing the accumulation of in vivo human oral microbiota elucidates microbial community dynamics at the gateway to the GI tract. Gut 2020, 69, 1355–1356. [Google Scholar] [CrossRef]
- Park, S.Y.; Hwang, B.O.; Lim, M.; Ok, S.H.; Lee, S.K.; Chun, K.S.; Park, K.K.; Hu, Y.; Chung, W.Y.; Song, N.Y. Oral-Gut microbiome axis in gastrointestinal disease and cancer. Cancers 2021, 13, 2124. [Google Scholar] [CrossRef]
- Olsen, I.; Yamazaki, K. Can oral bacteria affect the microbiome of the gut? J. Oral. Microbiol. 2019, 11, 1586422. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.C.; Bergsten, E.; et al. Extensive transmission of microbes along the gastrointestinal tract. Elife 2019, 8, e42693. [Google Scholar] [CrossRef]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Hein, R.; Schmidt, T.M.; Kamada, N. The bacterial connection between the oral cavity and the gut diseases. J. Dent. Res. 2020, 99, 1021–1029. [Google Scholar] [CrossRef]
- Read, E.; Curtis, M.A.; Neves, J.F. The role of oral bacteria in inflammatory bowel disease. The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 731–742. [Google Scholar] [CrossRef]
- Pellicciotta, M.; Rigoni, R.; Falcone, E.L.; Holland, S.M.; Villa, A.; Cassani, B. The microbiome and immunodeficiencies: Lessons from rare diseases. J. Autoimmun. 2019, 98, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Tajik, N.; Frech, M.; Schulz, O.; Schälter, F.; Lucas, S.; Azizov, V.; Dürholz, K.; Steffen, F.; Omata, Y.; Rings, A.; et al. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat. Commun. 2020, 11, 1995. [Google Scholar] [CrossRef] [Green Version]
- Kuraji, R.; Sekino, S.; Kapila, Y.; Numabe, Y. Periodontal disease-related nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: An emerging concept of oral-liver axis. Periodontology 2000 2021, 87, 204–240. [Google Scholar] [CrossRef]
- Imai, J.; Kitamoto, S.; Kamada, N. The pathogenic oral-gut-liver axis: New understandings and clinical implications. Expert Rev. Clin. Immunol. 2021, 17, 727–736. [Google Scholar] [CrossRef]
- Wu, H.J.J. Methotrexate works remotely, from the gut. Cell Host Microbe 2021, 29, 325–326. [Google Scholar] [CrossRef]
- Lee, P.C.; Hsieh, Y.C.; Huo, T.I.; Yang, U.C.; Lin, C.H.; Li, C.P.; Huang, Y.H.; Hou, M.C.; Lin, H.C.; Lee, K.C. Active vitamin D3 treatment attenuated bacterial translocation via improving intestinal barriers in cirrhotic rats. Mol. Nutr. Food Res. 2021, 65, e2000937. [Google Scholar] [CrossRef]
- Berthelot, J.M.; Darrieutort-Laffite, C.; Trang, C.; Maugars, Y.; Le Goff, B. Contribution of mycobiota to the pathogenesis of spondyloarthritis. Joint Bone Spine 2021, 88, 105245. [Google Scholar] [CrossRef] [PubMed]
- Villar-García, J.; Güerri-Fernández, R.; Moya, A.; González, A.; Hernández, J.J.; Lerma, E.; Guelar, A.; Sorli, L.; Horcajada, J.P.; Artacho, A.D.; et al. Impact of probiotic Saccharomyces boulardii on the gut microbiome composition in HIV-treated patients: A double-blind, randomized, placebo-controlled trial. PLoS ONE 2017, 12, e0173802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, Y.; Ouhara, K.; Munenaga, S.; Shoji, M.; Ozawa, T.; Hisatsune, J.; Kado, I.; Kajiya, M.; Matsuda, S.; Kawai, T.; et al. Effect of Porphyromonas gingivalis infection on gut dysbiosis and resultant arthritis exacerbation in mouse model. Arthritis Res. Ther. 2020, 22, 249. [Google Scholar] [CrossRef]
- Whittle, E.; Leonard, M.; Harrison, R.; Gant, T.; Tonge, D. Multi-Method characterisation of the human circulating microbiome. Front. Microbiol. 2019, 9, 3266. [Google Scholar] [CrossRef] [Green Version]
- Emery, D.C.; Cerajewska, T.L.; Seong, J.; Davies, M.; Paterson, A.; Allen-Birt, S.J.; West, N.X. Comparison of blood bacterial communities in periodontal health and periodontal disease. Front. Cell Infect. Microbiol. 2021, 10, 577485. [Google Scholar] [CrossRef]
- Panaiotov, S.; Hodzhev, Y.; Tsafarova, B.; Tolchkov, V.; Kalfin, R. Culturable and non-culturable blood microbiota of healthy individuals. Microorganisms 2021, 9, 1464. [Google Scholar] [CrossRef] [PubMed]
- Panaiotov, S.; Filevski, G.; Equestre, M.; Nikolova, E.; Kalfin, R. Cultural issolation and characteristics of the blood microbiome of healthy individuals. Adv. Microbiol. 2018, 8, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Hammad, D.B.M.; Hider, S.L.; Liyanapathirana, V.C.; Tonge, D.P. Molecular characterization of circulating microbiome signatures in rheumatoid arthritis. Front. Cell Infect. Microbiol. 2019, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Kempsell, K.E.; Cox, C.J.; Hurle, M.; Wong, A.; Wilkie, S.; Zanders, E.D.; Gaston, J.S.; Crowe, J.S. Reverse transcriptase-PCR analysis of bacterial rRNA for detection and characterization of bacterial species in arthritis synovial tissue. Infect. Immun. 2000, 68, 6012–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalupova, M.; Skalova, A.; Hajek, T.; Geigerova, L.; Kralova, D.; Liska, P.; Hecova, H.; Molacek, J.; Hrabak, J. Bacterial DNA detected on pathologically changed heart valves using 16S rRNA gene amplification. Folia Microbiol 2018, 63, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.M.; Velasco, C.; Rivas, A.; Andrews, M.; Garman, C.; Jacob, P.B.; Jeffries, M.A. Identification of cartilage microbial DNA signatures and associations with knee and hip osteoarthritis. Arthritis Rheumatol. 2020, 72, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Esberg, A.; Johansson, L.; Johansson, I.; Dahlqvist, S.R. Oral microbiota identifies patients in early onset rheumatoid arthritis. Microorganisms 2021, 9, 1657. [Google Scholar] [CrossRef] [PubMed]
- Moen, K.; Brun, J.G.; Valen, M.; Skartveit, L.; Eribe, E.K.; Olsen, I.; Jonsson, R. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin. Exp. Rheumatol. 2006, 24, 656–663. [Google Scholar]
- Martinez-Martinez, R.E.; Abud-Mendoza, C.; Patiño-Marin, N.; Rizo-Rodríguez, J.C.; Little, J.W.; Loyola-Rodrıguez, J.P. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J. Clin. Periodontol. 2009, 36, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Témoin, S.; Chakaki, A.; Askari, A.; El-Halaby, A.; Fitzgerald, S.; Marcus, R.E.; Han, Y.W.; Bissada, N.F. Identification of oral bacterial DNA in synovial fluid of arthritis patients with native and failed prosthetic joints. J. Clin. Rheumatol. 2012, 18, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Reichert, S.; Haffner, M.; Keyßer, G.; Schäfer, C.; Stein, J.M.; Schaller, H.G.; Wienke, A.; Strauss, H.; Heide, S.; Schulz, S. Detection of oral bacterial DNA in synovial fluid. J. Clin. Periodontol. 2013, 40, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, B.; Li, S.; Yang, L.; Zhu, D.; Wang, Y.; Shi, B.; Gai, Z.; Yang, J.; Heng, X.; et al. Detection and characterization of bacterial nucleic acids in culture-negative synovial tissue and fluid samples from rheumatoid arthritis or osteoarthritis patients. Sci. Rep. 2018, 8, 14305. [Google Scholar] [CrossRef]
- Hammad, D.B.M.; Liyanapathirana, V.; Tonge, D.P. Molecular characterisation of the synovial fluid microbiome in rheumatoid arthritis patients and healthy control subjects. PLoS ONE 2019, 14, e0225110. [Google Scholar]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Do, T.; Mankia, K.; Meade, J.; Hunt, L.; Clerehugh, V.; Speirs, A.; Tugnait, A.; Emery, P.; Devine, D. Dysbiosis in the oral microbiomes of anti-CCP positive individuals at risk of developing rheumatoid arthritis. Ann. Rheum. Dis. 2021, 80, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Mankia, K.; Cheng, Z.; Do, T.; Hunt, L.; Meade, J.; Kang, J.; Clerehugh, V.; Speirs, A.; Tugnait, A.; Hensor, E.M.A.; et al. Prevalence of periodontal disease and periodontopathic bacteria in anti-cyclic citrullinated protein antibody-positive at-risk adults without arthritis. JAMA Netw. Open. 2019, 2, e195394. [Google Scholar] [CrossRef]
- Orange, D.E.; Yao, V.; Sawicka, K.; Fak, J.; Frank, M.O.; Parveen, S.; Blachere, N.E.; Hale, C.; Zhang, F.; Raychaudhuri, S.; et al. RNA identification of PRIME cells predicting rheumatoid arthritis flares. N. Engl. J. Med. 2020, 383, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Kapila, Y.L. Oral health’s inextricable connection to systemic health: Special populations bring to bear multimodal relationships and factors connecting periodontal disease to systemic diseases and conditions. Periodontology 2000 2021, 87, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Karydis, A.; Sandal, I.; Luo, J.; Prislovsky, A.; Gamboa, A.; Rosloniec, E.F.; Brand, D.D. Influence of the shared epitope on the elicitation of experimental autoimmune arthritis biomarkers. PLoS ONE 2021, 16, e0250177. [Google Scholar] [CrossRef]
- Zorba, M.; Melidou, A.; Patsatsi, A.; Ioannou, E.; Kolokotronis, A. The possible role of oral microbiome in autoimmunity. Int. J. Women’s Dermatol. 2020, 6, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Almeida, V.S.M.; Azevedo, J.; Leal, H.F.; Queiroz, A.T.L.; Da Silva Filho, H.P.; Reis, J.N. Bacterial diversity and prevalence of antibiotic resistance genes in the oral microbiome. PLoS ONE 2020, 15, e0239664. [Google Scholar] [CrossRef] [PubMed]
- Artacho, A.; Isaac, S.; Nayak, R.; Flor-Duro, A.; Alexander, M.; Koo, I.; Manasson, J.; Smith, P.B.; Rosenthal, P.; Homsi, Y.; et al. The pretreatment gut microbiome is associated with lack of response to methotrexate in new-onset rheumatoid arthritis. Arthritis Rheumatol. 2021, 73, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Morrow, C.D. Perturbation of the human gastrointestinal tract microbial ecosystem by oral drugs to treat chronic disease results in a spectrum of individual specific patterns of extinction and persistence of dominant microbial strains. PLoS ONE 2020, 15, e0242021. [Google Scholar] [CrossRef]
- Bertolini, M.; Dongari-Bagtzoglou, A.D. The relationship of candida albicans with the oral bacterial microbiome in health and disease. Adv. Exp. Med. Biol. 2019, 1197, 69–78. [Google Scholar] [PubMed]
- Mahanonda, R.; Champaiboon, C.; Subbalekha, K.; Sa-Ard-Iam, N.; Yongyuth, A.; Isaraphithakkul, B.; Rerkyen, P.; Charatkulangkun, O.; Pichyangkul, S. Memory T cell subsets in healthy gingiva and periodontitis tissues. J. Periodontol. 2018, 89, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Juanola, O.; Piñero, P.; Gómez-Hurtado, I.; Caparrós, E.; García-Villalba, R.; Marín, A.; Zapater, P.; Tarín, F.; González-Navajas, J.M.; Tomás-Barberán, F.A.; et al. Regulatory T Cells restrict permeability to bacterial antigen translocation and preserve short-chain fatty acids in experimental cirrhosis. Hepatol. Commun. 2018, 2, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Montaño, R.; Bernard-Medina, A.G.; Oregon-Romero, E.; Martínez-Rodríguez, V.M.D.C.; Pita-López, M.L.; Gómez-Meda, B.C.; Guerrero-Velázquez, C. IL-23/IL-17 axis and soluble receptors isoforms sIL-23R and sIL-17RA in patients with rheumatoid arthritis-presenting periodontitis. J. Clin. Lab. Anal. 2021, 35, e23963. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berthelot, J.-M.; Bandiaky, O.N.; Le Goff, B.; Amador, G.; Chaux, A.-G.; Soueidan, A.; Denis, F. Another Look at the Contribution of Oral Microbiota to the Pathogenesis of Rheumatoid Arthritis: A Narrative Review. Microorganisms 2022, 10, 59. https://doi.org/10.3390/microorganisms10010059
Berthelot J-M, Bandiaky ON, Le Goff B, Amador G, Chaux A-G, Soueidan A, Denis F. Another Look at the Contribution of Oral Microbiota to the Pathogenesis of Rheumatoid Arthritis: A Narrative Review. Microorganisms. 2022; 10(1):59. https://doi.org/10.3390/microorganisms10010059
Chicago/Turabian StyleBerthelot, Jean-Marie, Octave Nadile Bandiaky, Benoit Le Goff, Gilles Amador, Anne-Gaelle Chaux, Assem Soueidan, and Frederic Denis. 2022. "Another Look at the Contribution of Oral Microbiota to the Pathogenesis of Rheumatoid Arthritis: A Narrative Review" Microorganisms 10, no. 1: 59. https://doi.org/10.3390/microorganisms10010059
APA StyleBerthelot, J.-M., Bandiaky, O. N., Le Goff, B., Amador, G., Chaux, A.-G., Soueidan, A., & Denis, F. (2022). Another Look at the Contribution of Oral Microbiota to the Pathogenesis of Rheumatoid Arthritis: A Narrative Review. Microorganisms, 10(1), 59. https://doi.org/10.3390/microorganisms10010059