
The Genomic Physics of COVID-19 Pathogenesis and Spread



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Gene Networks as a Driver of Interpersonal Variability and Transmissibility
2. A Physical Model for Contextualizing Genomic Networks
2.1. The Integration of Allometric Scaling Law and Evolutionary Game Theory
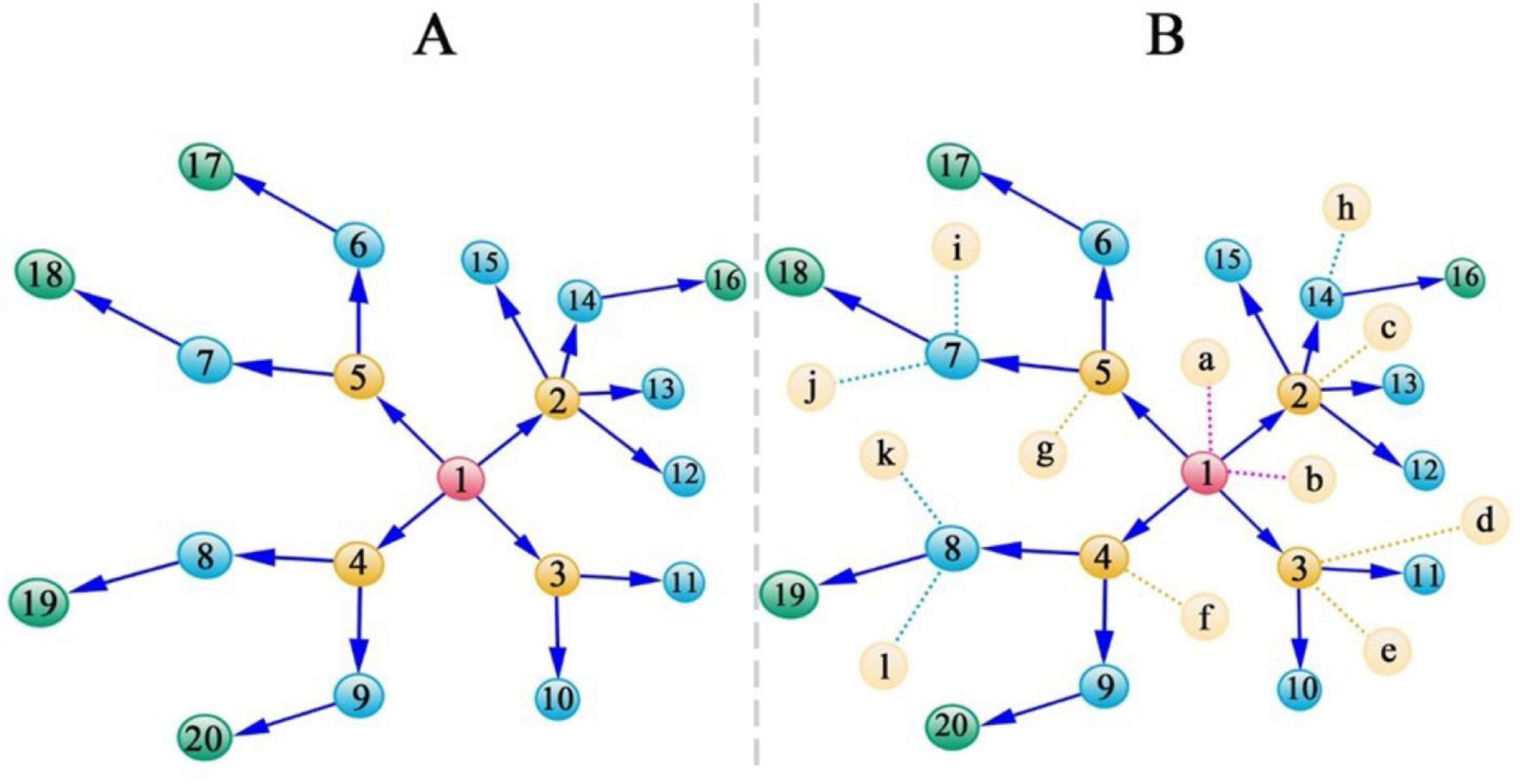
2.2. Modularity Theory and Dunbar’s Law
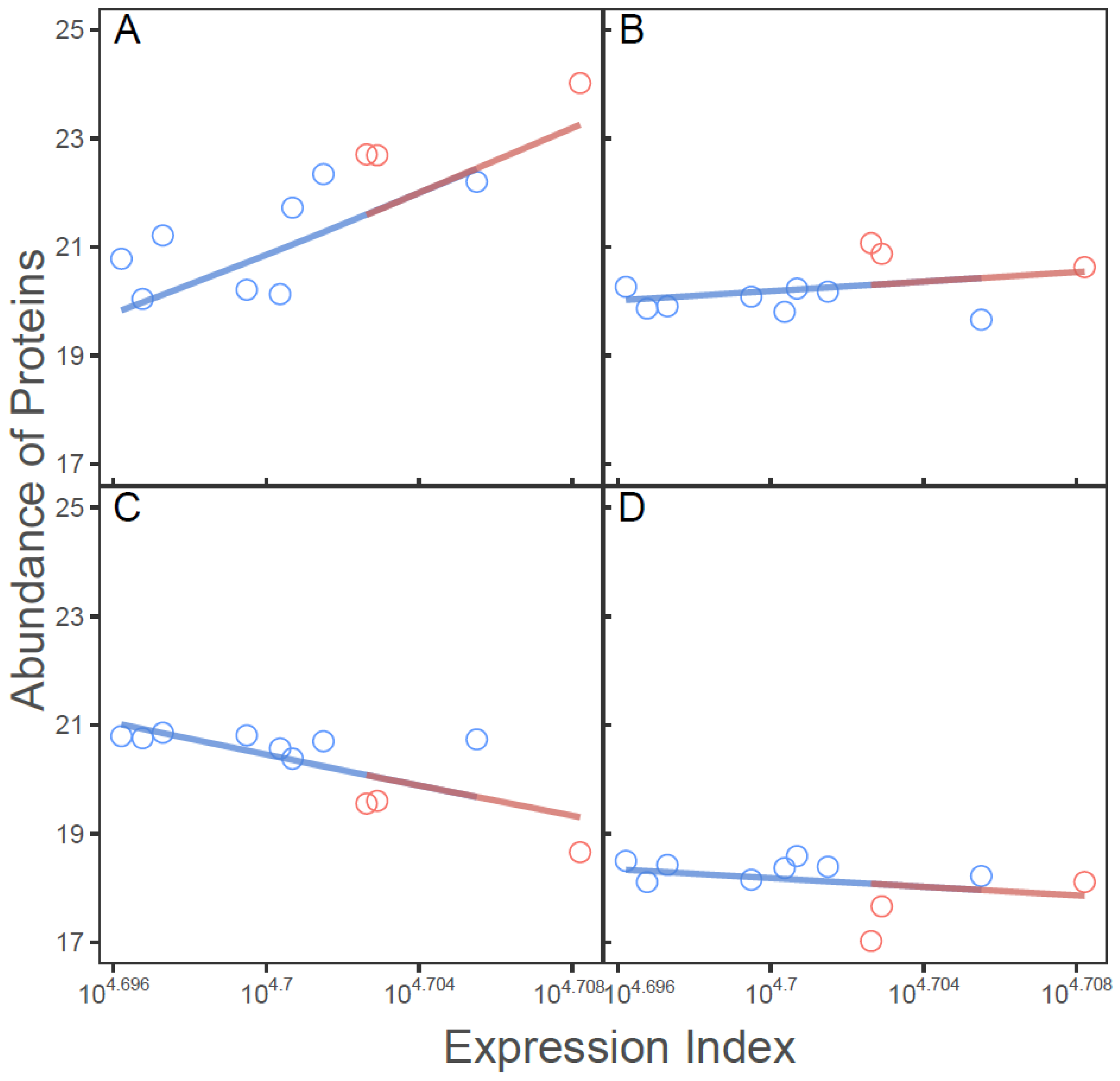
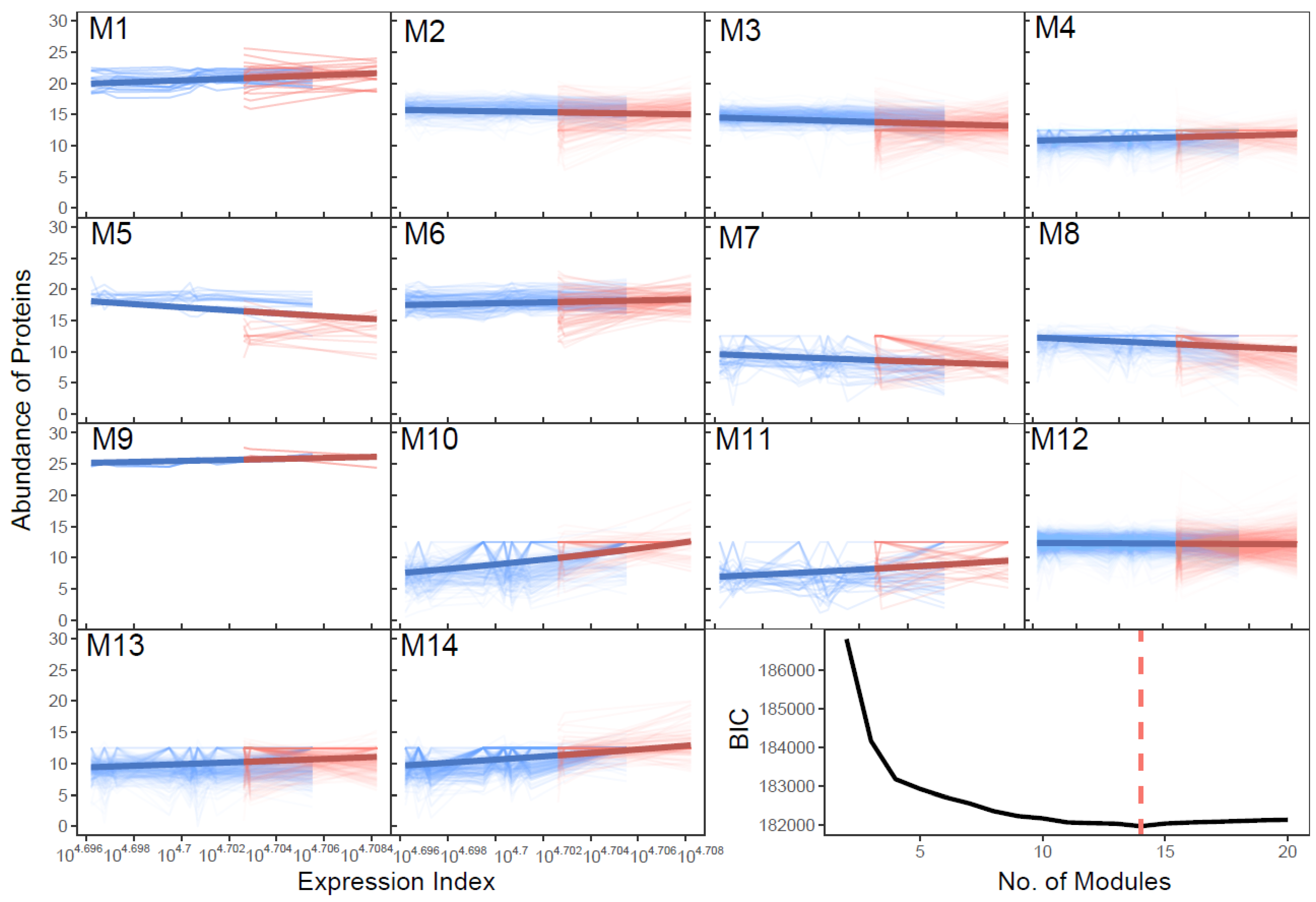
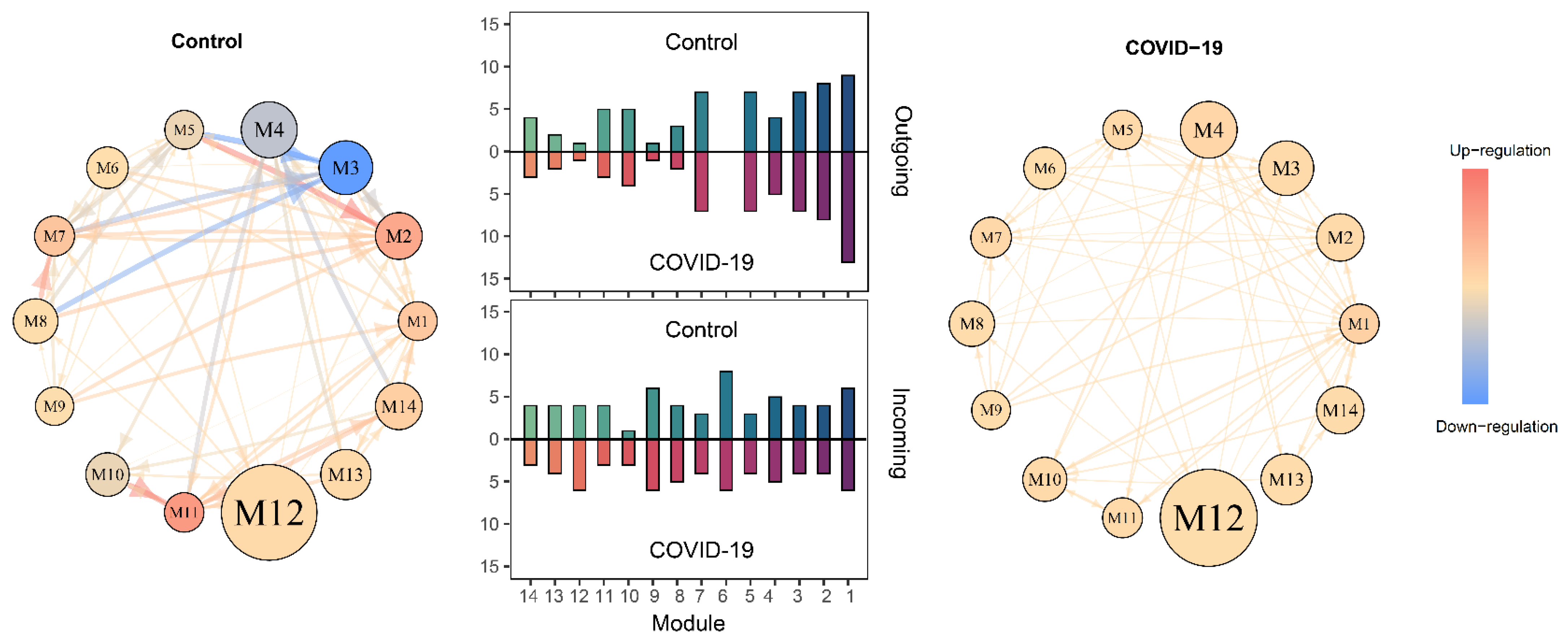
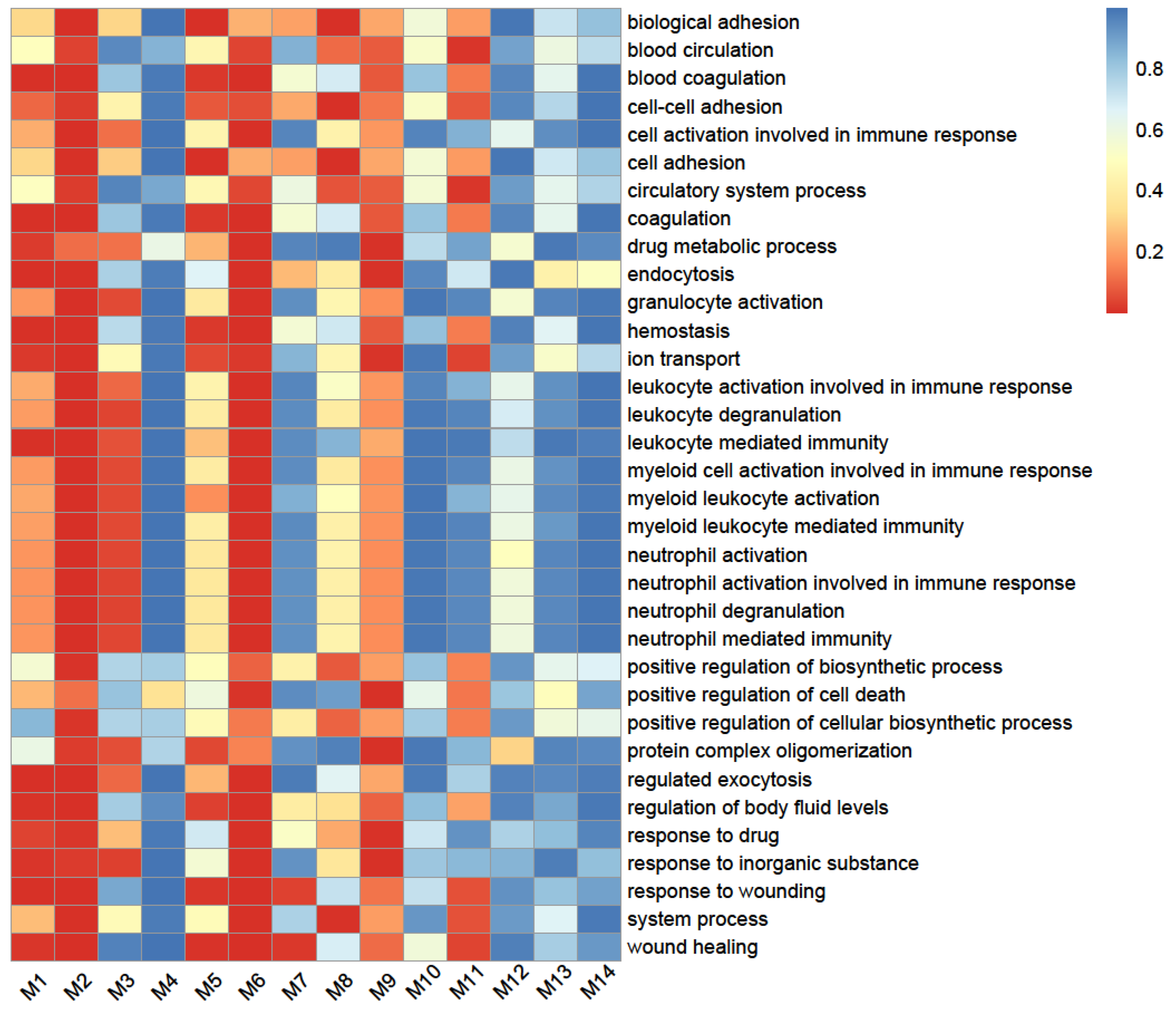
2.3. SARS-CoV-2-Induced Network Change
3. Statistical Genetic Physics of COVID-19 Spread
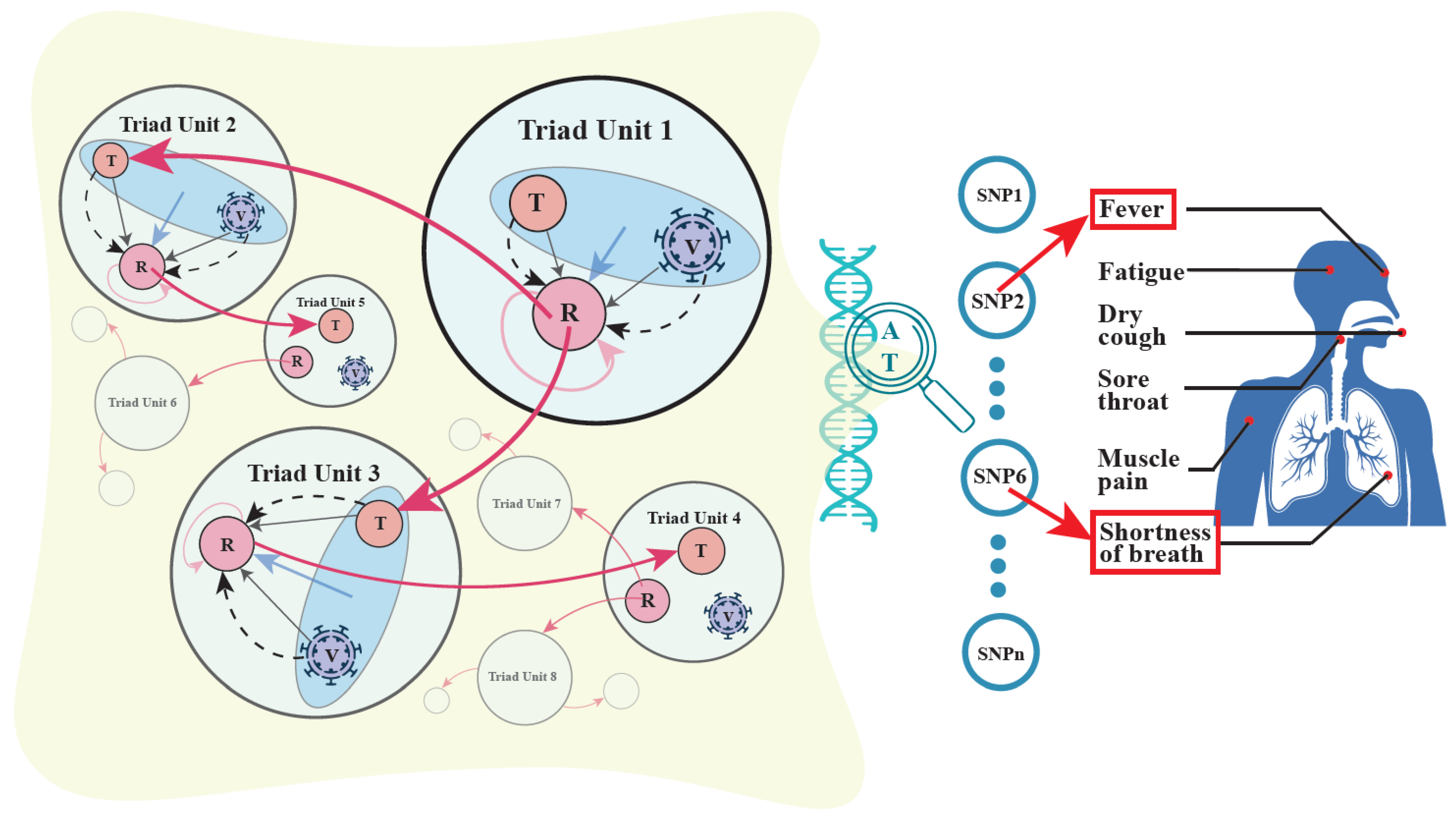
3.1. Horizontal Epistasis: An Emerging Concept
3.2. Genetic Hypergraphs
- Direct main effects of the gene of the recipient on its own phenotype;
- Indirect main effects of the gene of the transmitter on the phenotype of the recipient;
- Indirect genetic effect of the virus gene on the phenotype of the recipient;
- Horizontal two-way epistatic effects between the transmitter gene and recipient gene on the phenotype of the recipient;
- Horizontal two-way epistatic effects between the virus gene and transmitter gene on the phenotype of the recipient;
- Horizontal two-way epistatic effects between the virus gene and recipient gene on the phenotype of the recipient;
- Horizontal three-way epistatic effects among the virus gene, transmitter gene, and recipient gene on the phenotype of the recipient.
3.3. Mobile Hypergraphs Encapsulated in a Genome-Wide Atlas
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef]
- Heymann, D.L.; Shindo, N. COVID-19: What is next for public health? Lancet 2020, 395, 542–545. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Leng, L.; Cao, R.; Ma, J.; Mou, D.; Zhu, Y.; Li, W.; Lv, L.; Gao, D.; Zhang, S.; Gong, F.; et al. Pathological features of COVID-19-associated lung injury: A preliminary proteomics report based on clinical samples. Signal Transduct. Target. Ther. 2020, 5, 240. [Google Scholar] [CrossRef]
- Sen, R. High-throughput approaches of diagnosis and therapies for COVID-19: Antibody panels, proteomics and metabolomics. Future Drug Discov. 2020, 3, FDD55. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 2020, 182, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Bessy, C.; Plantier, L.; Lefevre, A.; Piver, E.; Bernard, L.; Marlet, J.; Stefic, K.; Benz-de Bretagne, I.; Cannet, P.; et al. The specific metabolome profiling of patients infected by SARS-CoV-2 supports the key role of tryptophan-nicotinamide pathway and cytosine metabolism. Sci. Rep. 2020, 10, 16824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Govindavari, J.P.; Davis, B.D.; Chen, S.S.; Kim, J.T.; Song, J.; Lopategui, J.; Plummer, J.T.; Vail, E. Analysis of genomic characteristics and transmission routes of patients with confirmed SARS-CoV-2 in southern California during the early stage of the US COVID-19 pandemic. JAMA Netw. Open 2020, 3, e2024191. [Google Scholar] [CrossRef]
- Hu, J.; Thomas, C.; Brunak, S. Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 2016, 17, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.R.; Weiss, S.T.; Glass, K.; Sharma, A. Network medicine in the age of biomedical big data. Front. Genet. 2019, 10, 294. [Google Scholar] [CrossRef]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. 2020, 94, e00510–e00520. [Google Scholar] [CrossRef]
- Benetti, E.; Tita, R.; Spiga, O.; Ciolfi, A.; Birolo, G.; Bruselles, A.; Doddato, G.; Giliberti, A.; Marconi, C.; Musacchia, F.; et al. ACE2 variants underlie interindividual variability and susceptibility to COVID-19 in Italian population. Eur. J. Hum. Genet. 2020, 28, 1602–1614. [Google Scholar] [CrossRef]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Yu, P.; Zhu, J.; Zhang, Z.; Han, Y. A familial cluster of infection associated with the 2019 novel coronavirus indicating potential person-to-person transmission during the incubation period. J. Infect. Dis. 2020, 221, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yao, L.; Wei, T.; Tian, F.; Jin, D.Y.; Chen, L.; Wang, M. Presumed asymptomatic carrier transmission of COVID-19. JAMA 2020, 323, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Science. Available online: https://www.sciencemag.org/news/2020/03/how-sick-will-coronavirus-make-you-answer-may-be-your-genes (accessed on 9 October 2020).
- Discover. Available online: https://www.discovermagazine.com/health/who-gets-sickest-from-covid-19 (accessed on 9 October 2020).
- The Scientist. Available online: https://www.the-scientist.com/news-opinion/dna-could-hold-clues-to-varying-severity-of-covid-19-67435 (accessed on 9 October 2020).
- Bloomberg. Available online: https://www.bloomberg.com/news/articles/2020-04-16/your-risk-of-getting-sick-from-covid-19-may-lie-in-your-genes (accessed on 9 October 2020).
- News Medical. Available online: https://www.news-medical.net/news/20200923/Common-genetic-variants-may-influence-susceptibility-to-COVID-19.aspx (accessed on 9 October 2020).
- Newman, M.E. The structure and function of complex networks. SIAM Rev. 2003, 45, 167–256. [Google Scholar] [CrossRef]
- Homas, L.D.; Vyshenska, D.; Shulzhenko, N.; Yambartsev, A.; Morgun, A. Differentially correlated genes in co-expression networks control phenotype transitions. F1000Research 2016, 5, 2740. [Google Scholar]
- Han, S.W.; Chen, G.; Cheon, M.S.; Zhong, H. Estimation of directed acyclic graphs through two-stage adaptive lasso for gene network inference. J. Am. Stat. Assoc. 2016, 111, 1004–1019. [Google Scholar] [CrossRef]
- Vijesh, N.; Chakrabarti, S.; Sreekumar, J. Modeling of gene regulatory networks: A review. J. Biomed. Sci. Eng. 2013, 6, 223–231. [Google Scholar] [CrossRef]
- Wang, Y.X.; Huang, H. Review on statistical methods for gene network reconstruction using expression data. J. Theor. Biol. 2014, 362, 53–61. [Google Scholar] [CrossRef]
- Wu, R.L.; Jiang, L.B. recovering dynamic networks in big static datasets. Phys. Rep. 2021; in press. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, L.B.; Fu, G.F.; Wang, M.; Wang, Y.; Shen, B.; Liu, Z.; Wang, Z.; Hou, W.; Berceli, S.A.; et al. An omnidirectional visualization model of personalized gene regulatory networks. NPJ Syst. Biol. Appl. 2019, 5, 38. [Google Scholar] [CrossRef]
- Love, A.C.; Hüttemann, A. Comparing part-whole explanations in biology and physics. In Explanation, Prediction, and Confirmation; Springer: Berlin/Heidelberg, Germany, 2011; pp. 183–202. [Google Scholar]
- Healey, R.; Uffink, J. Part and whole in physics: An introduction. Stud. Hist. Phil. Mod. Phys. 2013, 44, 20–21. [Google Scholar] [CrossRef]
- Smith, J.M.; Price, G.R. The logic of animal conflict. Nature 1973, 246, 15–18. [Google Scholar] [CrossRef]
- Griffin, C.; Jiang, L.; Wu, R. Analysis of quasi-dynamic ordinary differential equations and the quasi-dynamic replicator. Phys. A Stat. Mech. Its Appl. 2020, 555, 124422. [Google Scholar] [CrossRef]
- May, R.M. Will a large complex system be stable? Nature 1972, 238, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.; Rudolf, L.; Levin, S.A.; Dieckmann, U. Generalized models reveal stabilizing factors in food webs. Science 2009, 325, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Allesina, S.; Tang, S. Stability criteria for complex ecosystems. Nature 2012, 483, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Busiello, D.M.; Suweis, S.; Hidalgo, J.; Maritan, A. Explorability and the origin of network sparsity in living systems. Sci. Rep. 2017, 7, 12323. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Soto, C. On the role of sparseness in the evolution of modularity in gene regulatory networks. PLoS Comput. Biol. 2018, 14, e1006172. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Pavlicev, M.; Cheverud, J.M. The road to modularity. Nat. Rev. Genet. 2007, 8, 921–931. [Google Scholar] [CrossRef]
- Kim, B.R.; Zhang, L.; Berg, A.; Fan, J.; Wu, R.L. A computational approach to the functional clustering of periodic gene expression profiles. Genetics 2008, 180, 821–834. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Xu, M.; Wang, Z.; Tao, M.; Zhu, J.; Wang, L.; Li, R.; Berceli, S.A.; Wu, R.L. How to cluster gene expression dynamics in response to environmental signals. Brief. Bioinform. 2012, 13, 162–174. [Google Scholar] [CrossRef]
- Dunbar, R.I.M. Neocortex size as a constraint on group size in primates. J. Hum. Evol. 1992, 22, 469–493. [Google Scholar] [CrossRef]
- Gonçalves, B.; Perra, N.; Vespignani, A. Modeling Users’ Activity on Twitter Networks: Validation of Dunbar’s Number. PLoS ONE 2011, 6, e22656. [Google Scholar] [CrossRef]
- Miritello, G.; Moro, E.; Lara, R.; Martínez-López, R.; Belchamber, J.; Roberts, S.G.B.; Dunbar, R.I.M. Time as a limited resource: Communication strategy in mobile phone networks. Soc. Netw. 2013, 35, 89–95. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Ghinai, I.; McPherson, T.D.; Hunter, J.C.; Kirking, H.L.; Christiansen, D.; Joshi, K.; Rubin, R.; Morales-Estrada, S.; Black, S.R.; Pacilli, M. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet 2020, 395, 1137–1144. [Google Scholar] [CrossRef]
- Anacleto, O.; Cabaleiro, S.; Villanueva, B.; Saura, M.; Houston, R.D.; Woolliams, J.A.; Doeschl-Wilson, A.B. Genetic differences in host infectivity affect disease spread and survival in epidemics. Sci. Rep. 2019, 9, 4924. [Google Scholar] [CrossRef] [PubMed]
- van Sluijs, L.; Pijlman, G.P.; Kammenga, J.E. Why do Individuals Differ in Viral Susceptibility? A Story Told by Model Organisms. Viruses 2017, 9, 284. [Google Scholar] [CrossRef]
- Thompson, J.N. The evolution of species interactions. Science 1999, 284, 2116–2118. [Google Scholar] [CrossRef]
- Lawrence, D.; Fiegna, F.; Behrends, V.; Bundy, J.G.; Phillimore, A.B.; Bell, T.; Barraclough, T.G. Species interactions alter evolutionary responses to a novel environment. PLoS Biol. 2012, 10, e1001330. [Google Scholar] [CrossRef]
- Barraclough, T.G. How do species interactions affect evolutionary dynamics across whole communities? Ann. Rev. Ecol. Evol. Syst. 2015, 46, 25–48. [Google Scholar] [CrossRef]
- Santostefano, F.; Wilson, A.J.; Niemelä, P.T.; Dingemanse, N.J. Indirect genetic effects: A key component of the genetic architecture of behaviour. Sci. Rep. 2017, 7, 10235. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, J.; Sang, M.; Zhang, Y.; Ye, M.; Zhang, H.; Wu, B.; Zhu, Y.; Xu, P.; Tai, R.; et al. A drive to driven model of mapping intraspecific interaction networks. iScience 2019, 22, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.N.; McAdam, A.G. Indirect genetic effects clarify how traits can evolve even when fitness does not. Evol. Let. 2019, 3, 4–14. [Google Scholar] [CrossRef]
- Biscarini, F.; Bovenhuis, H.; van der Poel, J.; Rodenburg, T.B.; Jungerius, A.P.; van Arendonk, J.A. Across-line SNP association study for direct and associative effects on feather damage in laying hens. Behav. Genet. 2010, 40, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Mutic, J.J.; Wolf, J.B. Indirect genetic effects from ecological interactions in Arabidopsis thaliana. Mol. Ecol. 2007, 16, 2371–2381. [Google Scholar] [CrossRef]
- Cordell, H. Epistasis: What it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 2002, 11, 2463–2468. [Google Scholar] [CrossRef] [PubMed]
- Landolfo, S.; De Andrea, M.; Dell’Oste, V.; Gugliesi, F. Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape. World J. Virol. 2016, 5, 87–96. [Google Scholar] [CrossRef]
- Denzin, L.K.; Khan, A.A.; Virdis, F.; Wilks, J.; Kane, M.; Beilinson, H.A.; Dikiy, S.; Case, L.K.; Roopenian, D.; Witkowski, M. Neutralizing antibody responses to viral infections are linked to the non-classical MHC class II gene H2-Ob. Immunity 2017, 47, 310. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Holmes, E.C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Domingo-Calap, P. Viral evolution and immune responses. J. Clin. Microbiol. Biochem. Technol. 2019, 5, 013–018. [Google Scholar] [CrossRef]
- Xue, K.S.; Bloom, J.B. Linking influenza virus evolution within and between human hosts. Virus Evol. 2020, 6, veaa010. [Google Scholar] [CrossRef]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef]
- Pavlopoulos, G.A.; Secrier, M.; Moschopoulos, C.N.; Soldatos, T.G.; Kossida, S.; Aerts, J.; Schneider, R.; Bagos, P.G. Using graph theory to analyze biological networks. BioData Min. 2011, 4, 10. [Google Scholar] [CrossRef]
- Anholt, R.R.H. Evolution of epistatic networks and the genetic basis of innate behaviors. Trends Genet. 2020, 36, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Bretto, A. Hypergraph Theory: An Introduction; Springer: New York, NY, USA, 2013. [Google Scholar]
- Klamt, S.; Haus, U.-U.; Theis, F. Hypergraphs and cellular networks. PLoS Comput. Biol. 2009, 5, e1000385. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Ji, M.; Pei, F.; Zhao, Q.; Zhou, Y.; Hong, Y.; Han, S.; Wang, J.; Wang, Q.; et al. Transmission Routes Analysis of SARS-CoV-2: A Systematic Review and Case Report. Front. Cell Dev. Biol. 2020, 8, 618. [Google Scholar] [CrossRef]
- Bodó, Á.; Katona, G.Y.; Simon, P.L. SIS Epidemic Propagation on Hypergraphs. Bull. Math. Biol. 2016, 78, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Berg, A.; Chang, M.N.; Du, P.; Ahn, K.; Mauger, D.; Liao, D.; Wu, R. A statistical model for genetic mapping of viral infection by integrating epidemiological behavior. Stat. Appl. Genet. Mol. Biol. 2009, 8, 38. [Google Scholar] [CrossRef][Green Version]
- Sun, L.; Jiang, L.; Grant, C.N.; Wang, H.G.; Gragnoli, C.; Liu, Z.; Wu, R. Computational identification of gene networks as a biomarker of neuroblastoma risk. Cancers 2020, 12, 2086. [Google Scholar] [CrossRef]
- Stein, R.A. Super-spreaders in infectious diseases. Int. J. Infect. Dis. 2011, 15, e510–e513. [Google Scholar] [CrossRef]
- Talbert-Slagle, K.; Atkins, K.E.; Yan, K.K.; Khurana, E.; Gerstein, M.; Bradley, E.H.; Berg, D.; Galvani, A.P.; Townsend, J.P. Cellular superspreaders: An epidemiological perspective on HIV infection inside the body. PLoS Pathog. 2014, 10, e1004092. [Google Scholar] [CrossRef]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020, 30, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Fernández, L.; Sawalha, A.H. Genetic variability in the expression of the SARS-CoV-2 host cell entry factors across populations. Genes Immun. 2020, 21, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, A.; Zhao, J.; Griffin, C.; Wu, R. The Genomic Physics of COVID-19 Pathogenesis and Spread. Cells 2022, 11, 80. https://doi.org/10.3390/cells11010080
Dong A, Zhao J, Griffin C, Wu R. The Genomic Physics of COVID-19 Pathogenesis and Spread. Cells. 2022; 11(1):80. https://doi.org/10.3390/cells11010080
Chicago/Turabian StyleDong, Ang, Jinshuai Zhao, Christopher Griffin, and Rongling Wu. 2022. "The Genomic Physics of COVID-19 Pathogenesis and Spread" Cells 11, no. 1: 80. https://doi.org/10.3390/cells11010080
APA StyleDong, A., Zhao, J., Griffin, C., & Wu, R. (2022). The Genomic Physics of COVID-19 Pathogenesis and Spread. Cells, 11(1), 80. https://doi.org/10.3390/cells11010080