
Alkanediamide-Linked Bisbenzamidines Are Promising Antiparasitic Agents

 ,
, 

Abstract
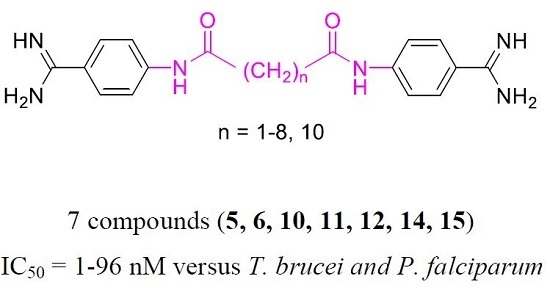
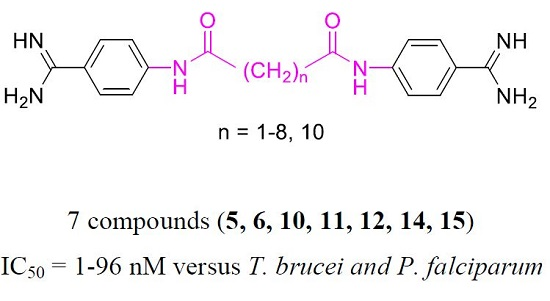
:
1. Introduction
2. Results and Discussion
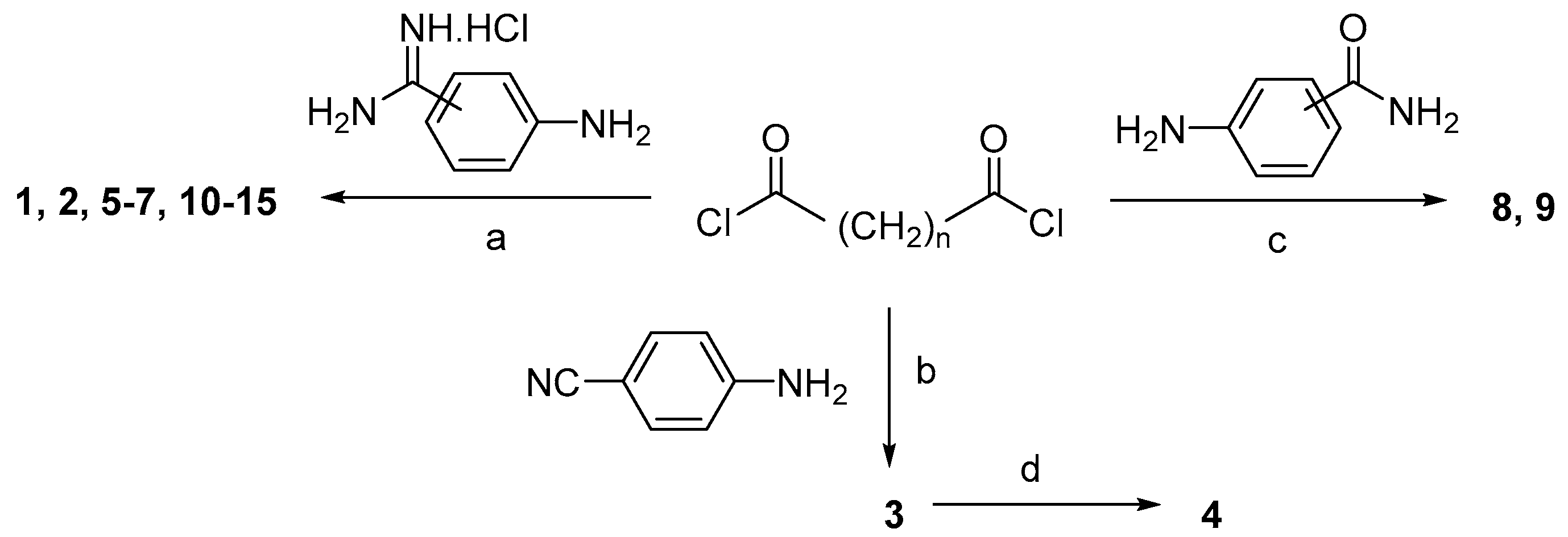
2.1. Chemistry
2.2. Pharmacology
2.3. Molecular Modeling Studies
3. Experimental Section
3.1. Chemistry
General Synthesis of Bisbenzamidines 1, 2, 7, 10–15
3.2. Biology
3.2.1. In Vitro Antiparasitic Activity
3.2.2. In Vivo Anti-Trypanosomal Activity in Mice
3.2.3. DNA Binding Affinity Measurements
3.3. Molecular Modeling Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vanden Eynde, J.J.; Mayence, A.; Huang, T.L.; Collins, M.S.; Rebholz, S.; Walzer, P.D.; Cushion, M.T. Novel bisbenzamidines as potential drug candidates for the treatment of Pneumocystis carinii pneumonia. Bioorg. Med. Chem. Lett. 2004, 14, 4545–4548. [Google Scholar] [CrossRef] [PubMed]
- Cushion, M.T.; Walzer, P.D.; Ashbaugh, A.; Rebholz, S.; Brubaker, R.; Vanden Eynde, J.J.; Mayence, A.; Huang, T.L. In vitro selection and in vivo efficacy of piperazine- and alkanediamide-linked bisbenzamidines against Pneumocystis pneumonia in mice. Antimicrob. Agents Chemother. 2006, 50, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Vanden Eynde, J.J.; Mayence, A.; Collins, M.S.; Cushion, M.T.; Rattendi, D.; Londono, I.; Mazumder, L.; Bacchi, C.J.; Yarlett, N. Synthesis and SAR of alkanediamide-linked bisbenzamidines with antitrypanosomal and anti-Pneumocystis activity. Bioorg. Med. Chem. Lett. 2009, 19, 5884–5886. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, C.J.; Nathan, H.C.; Livingston, T.; Valladares, G.; Saric, M.; Sayer, P.D.; Njogu, A.R.; Clarkson, A.B., Jr. Differential susceptibility to DL-α-difluoromethylornithine in clinical isolates of Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 1990, 34, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Mayence, A.; Vanden Eynde, J.J. Some non-conventional biomolecular targets for diamidines. A short survey. Bioorg. Med. Chem. 2014, 22, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.F.; Wang, M.Z.; Generaux, C.N.; Boykin, D.W.; Wilson, W.D.; De Koning, H.P.; Olson, C.A.; Pohlig, G.; Burri, C.; Brun, R.; et al. Diamidines for human African trypanosomiasis. Curr. Opin. Investig. Drugs 2010, 11, 876–883. [Google Scholar] [PubMed]
- Soeiro, M.N.; Werbovetz, K.; Boykin, D.W.; Wilson, W.D.; Wang, M.Z.; Hemphill, A. Novel amidines and analogues as promising agents against intracellular parasites: A systematic review. Parasitology 2013, 140, 929–951. [Google Scholar] [CrossRef] [PubMed]
- Thuita, J.K.; Wolf, K.K.; Murilla, G.A.; Bridges, A.S.; Boykin, D.W.; Mutuku, J.N.; Liu, Q.; Jones, S.K.; Gem, C.O.; Ching, S.; et al. Chemotherapy of second stage human African trypanosomiasis: Comparison between the parenteral diamidine DB829 and its oral prodrug DB868 in vervet monkeys. PLoS Negl. Trop. Dis. 2015, 9, e0003409. [Google Scholar] [CrossRef] [PubMed]
- Kode, N.R.; Vanden Eynde, J.J.; Mayence, A.; Wang, G.; Huang, T.L. Design and synthesis of N1,N5-bis[4-(5-alkyl-1,2,4-oxadiazol-3-yl)phenyl]glutaramides as potential antifungal prodrugs. Molecules 2013, 18, 11250–11263. [Google Scholar] [CrossRef] [PubMed]
- Eynde, J.J.V.; Mayence, A.; Johnson, M.T.; Huang, T.L.; Collins, M.S.; Rebholz, S.; Walzer, P.D.; Cushion, M.T.; Donkor, I.O. Antitumor and anti-Pneumocystis carinii activities of novel bisbenzamidines. Med. Chem. Res. 2005, 14, 143–157. [Google Scholar] [CrossRef]
- Wilson, W.D.; Tanious, F.A.; Mathis, A.; Tevis, D.; Hall, J.E.; Boykin, D.W. Antiparasitic compounds that target DNA. Biochimie 2008, 90, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Chemaxon. Available online: https://www.chemaxon.com (accessed on 18 April 2016).
- Schrodinger suite. Available online: http://www.schrodinger.com (accessed on 18 April 2016).
- Donkor, I.O.; Huang, T.L.; Tao, B.; Rattendi, D.; Lane, S.; Vargas, M.; Goldberg, B.; Bacchi, C. Trypanocidal activity of conformationally restricted pentamidine congeners. J. Med. Chem. 2003, 46, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Mayence, A.; Vanden Eynde, J.J.; Krogstad, F.M.; Krogstad, D.J.; Cushion, M.T.; Huang, T.L. Parallel solution-phase synthesis of conformationally restricted congeners of pentamidine and evaluation of their antiplasmodial activities. J. Med. Chem. 2004, 47, 2700–2705. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, C.C.; McClernon, D.R.; Elwell, L.P.; Tidwell, R.R. Selective inhibition of topoisomerases from Pneumocystis carinii compared with that of topoisomerases from mammalian cells. Antimicrob. Agents Chemother. 1994, 38, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.; Hurley, L.H. Molecular struggle for transcriptional control. Nat. Med. 1995, 1, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Purfield, A.E.; Tidwell, R.R.; Meshnick, S.R. The diamidine DB75 targets the nucleus of Plasmodium falciparum. Malar. J. 2009, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Stead, A.M.; Bray, P.G.; Edwards, I.G.; DeKoning, H.P.; Elford, B.C.; Stocks, P.A.; Ward, S.A. Diamidine compounds: Selective uptake and targeting in Plasmodium falciparum. Mol Pharmacol 2001, 59, 1298–1306. [Google Scholar] [PubMed]
- Rodriguez, F.; Rozas, I.; Kaiser, M.; Brun, R.; Nguyen, B.; Wilson, W.D.; Garcia, R.N.; Dardonville, C. New bis(2-aminoimidazoline) and bisguanidine DNA minor groove binders with potent in vivo antitrypanosomal and antiplasmodial activity. J. Med. Chem. 2008, 51, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Basselin, M.; Badet-Denisot, M.A.; Lawrence, F.; Robert-Gero, M. Effects of pentamidine on polyamine level and biosynthesis in wild-type, pentamidine-treated, and pentamidine-resistant leishmania. Exp. Parasitol. 1997, 85, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Dardonville, C.; Barrett, M.P.; Brun, R.; Kaiser, M.; Tanious, F.; Wilson, W.D. DNA binding affinity of bisguanidine and bis(2-aminoimidazoline) derivatives with in vivo antitrypanosomal activity. J. Med. Chem. 2006, 49, 3748–3752. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.J.; Jenkins, T.C.; Neidle, S. Crystal structure of a pentamidine-oligonucleotide complex: Implications for DNA-binding properties. Biochemistry 1992, 31, 7104–7109. [Google Scholar] [CrossRef] [PubMed]
- Mayence, A.; Vanden Eynde, J.J.; Kaiser, M.; Brun, R.; Yarlett, N.; Huang, T.L. Bis(oxyphenylene)benzimidazoles: A novel class of anti-Plasmodium falciparum agents. Bioorg. Med. Chem. 2011, 19, 7493–7500. [Google Scholar] [CrossRef] [PubMed]
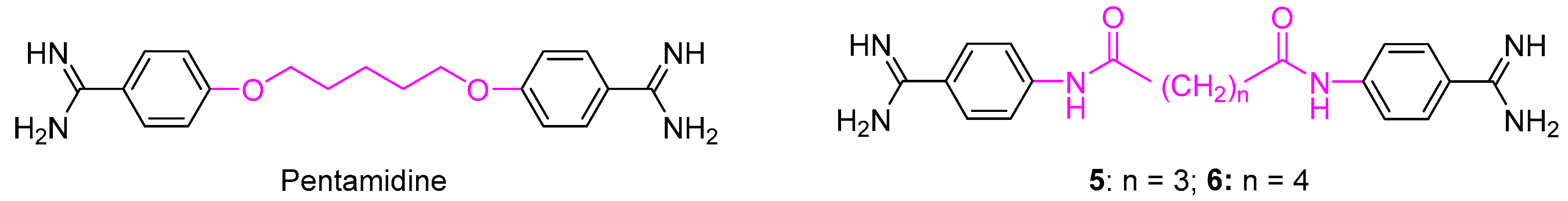

{kind=link}
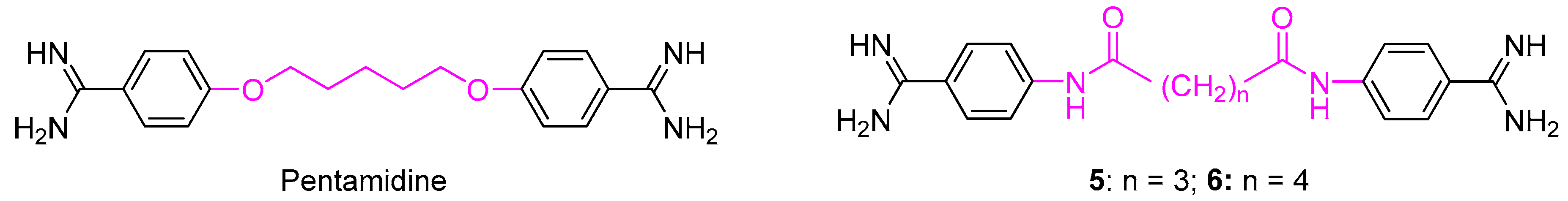{kind=link}
{kind=link}
| Compd. | n |  | IC50 (µM) a | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T.b. brucei b | T.b. rhod c | T. cruzi | L. donovani | P. falcipNF54 d | P. falcipK1 e | L6 Cells | ||||
| 1 | 1 | p-Am | p-Am | ND | ND | >100 | >100 | 0.008 | 0.003 | >100 |
| 2 | 2 | p-Am | p-Am | 9.0 | 2.19 | >100 | >100 | ND | 0.29 | 28.0 |
| 3 | 3 | p-CN | p-CN | 6.50 | 7.90 | >100 | >100 | ND | 44.8 | >100 |
| 4 | 3 | p-C(=NOH)NH2 | p-C(=NOH)NH2 | 7.30 | 10.0 | >100 | >100 | ND | 3.16 | >100 |
| 5 | 3 | p-Am | p-Am | 0.009 | 0.096 | >100 | >100 | 0.002 | 0.002 | 69.0 |
| 6 | 4 | p-Am | p-Am | 0.003 | 0.002 | >100 | >100 | 0.004 | 0.018 | >100 |
| 7 | 4 | m-Am | m-Am | 0.041 | 0.021 | >100 | >100 | ND | 0.38 | >100 |
| 8 | 4 | p-CONH2 | p-CONH2 | 2.80 | 1.10 | >100 | >100 | ND | >100 | >100 |
| 9 | 4 | m-CONH2 | m-CONH2 | NT | 1.97 | >100 | >100 | ND | >100 | 46.4 |
| 10 | 4 | p-Am | m-Am | 0.012 | 0.007 | >100 | >100 | 0.015 | 0.004 | >100 |
| 11 | 5 | p-Am | p-Am | 0.002 | 0.002 | >100 | >100 | 0.002 | 0.002 | 43.0 |
| 12 | 6 | p-Am | p-Am | 0.003 | 0.001 | >100 | >100 | 0.002 | 0.006 | 38.4 |
| 13 | 7 | p-Am | p-Am | 0.400 | 0.240 | 99.5 | 67.4 | 0.002 | 0.008 | 49.2 |
| 14 | 8 | p-Am | p-Am | 0.002 | 0.004 | 76.4 | 68.5 | 0.014 | 0.012 | 78.6 |
| 15 | 10 | p-Am | p-Am | 0.008 | 0.007 | 80.9 | 10.7 | 0.032 | 0.015 | 80.2 |
| Ref | 0.002 f | 0.002 f | 1.13 g | 0.44 h | 0.006 i | 0.18 i | 0.010 j | |||
| Compd. | MolWt | LogP | a H-don Count | b H-acc Count | c Ring # | LogD | d Rot Bonds | e PSA | Lipinki Rule of 5 | Lead Likeness |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 338.37 | 0.38 | 6 | 6 | 2 | −4.44 | 6 | 157.94 | 3 of 4 | 4 of 6 |
| 2 | 352.40 | 0.31 | 6 | 6 | 2 | −4.5 | 7 | 157.94 | 3 of 4 | 4 of 6 |
| 3 | 332.36 | 2.63 | 2 | 4 | 2 | 2.63 | 6 | 105.78 | 4 of 4 | 6 of 6 |
| 4 | 398.42 | 0.75 | 6 | 8 | 2 | 0.74 | 8 | 175.42 | 3 of 4 | 5 of 6 |
| 5 | 366.42 | 0.76 | 6 | 6 | 2 | −4.06 | 8 | 157.94 | 3 of 4 | 4 of 6 |
| 6 | 380.45 | 1.20 | 6 | 6 | 2 | −3.61 | 9 | 157.94 | 3 of 4 | 5 of 6 |
| 7 | 380.45 | 1.20 | 6 | 6 | 2 | −3.6 | 9 | 157.94 | 3 of 4 | 5 of 6 |
| 8 | 382.42 | 1.06 | 4 | 6 | 2 | 1.06 | 9 | 144.38 | 4 of 4 | 6 of 6 |
| 9 | 382.42 | 1.06 | 4 | 4 | 2 | 1.06 | 9 | 144.38 | 4 of 4 | 6 of 6 |
| 10 | 380.45 | 1.20 | 6 | 6 | 2 | −3.61 | 9 | 157.94 | 3 of 4 | 5 of 6 |
| 11 | 394.48 | 1.64 | 6 | 6 | 2 | −3.17 | 10 | 157.94 | 3 of 4 | 5 of 6 |
| 12 | 408.50 | 2.09 | 6 | 6 | 2 | −2.72 | 11 | 157.94 | 3 of 4 | 4 of 6 |
| 13 | 422.53 | 2.53 | 6 | 6 | 2 | −2.28 | 12 | 157.94 | 3 of 4 | 4 of 6 |
| 14 | 436.56 | 2.98 | 6 | 6 | 2 | −1.84 | 13 | 157.94 | 3 of 4 | 4 of 6 |
| 15 | 466.61 | 3.87 | 6 | 6 | 2 | −0.95 | 15 | 157.94 | 3 of 4 | 4 of 6 |
| 16 f | 340.42 | 2.32 | 4 | 6 | 2 | −2.50 | 10 | 118.20 | 4 of 4 | 6 of 6 |
| Clinical Isolate | Compd. | Dosage (mg/kg/Day) | Mean Survival (Days) | No. of Mice Cured/Total (%) |
|---|---|---|---|---|
| Lab 110 EATRO b | None | - | 5.0 | 0/3 |
| Pentamidine | 1.0, 2.5, 5, 10 | >30 | 5/5 (100) c | |
| 5 | 1.0 | 6.0 | 0/3 | |
| 2.5 | 11.3 | 0/3 | ||
| 5 | 10 | 2/3 (66) | ||
| 10 | >30 | 3/3 (100) | ||
| 15 | >30 | 3/3 (100) | ||
| 6 | 1.0 | 6.7 | 0/3 | |
| 2.5 | 10.0 | 0/3 | ||
| 5 | >30 | 3/3 (100) | ||
| 10 | >30 | 3/3 (100) | ||
| 15 | >30 | 3/3 (100) | ||
| 11 | 1.0, 2.5, 5, 10 | >30 | 3/3 (100) c | |
| KETRI 2002 b | None | - | 9.0 | 0/3 |
| Pentamidine | 1.0, 5, 10 | >30 | 5/5 (100) c | |
| 5 | 10, 15, 25 | >30 | 3/3 (100) c | |
| 6 | 10, 15, 25 | >30 | 3/3 (100) c | |
| 11 | 1.0, 2.5, 5, 10 | >30 | 3/3 (100) c | |
| KETRI 2538 b | None | - | 4.3 | 0/3 |
| Pentamidine | 1.0, 5, 10 | >30 | 5/5 (100) c | |
| 5 | 10, 15, 25 | >30 | 3/3 (100) c | |
| 6 | 5, 10, 15 | >30 | 3/3 (100) c | |
| KETRI 1992 b | None | - | 7.4 | 0/5 |
| Pentamidine | 1 | 12.6 | 0/3 | |
| 5 | 17.0 | 0/3 | ||
| 10 | 22.2 | 0/3 | ||
| 5 | 10 | 17.5 | 0/3 | |
| 15 | 14.5 | 0/3 | ||
| 25 | 25 | 0/3 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanden Eynde, J.J.; Mayence, A.; Mottamal, M.; Bacchi, C.J.; Yarlett, N.; Kaiser, M.; Brun, R.; Huang, T.L. Alkanediamide-Linked Bisbenzamidines Are Promising Antiparasitic Agents. Pharmaceuticals 2016, 9, 20. https://doi.org/10.3390/ph9020020
Vanden Eynde JJ, Mayence A, Mottamal M, Bacchi CJ, Yarlett N, Kaiser M, Brun R, Huang TL. Alkanediamide-Linked Bisbenzamidines Are Promising Antiparasitic Agents. Pharmaceuticals. 2016; 9(2):20. https://doi.org/10.3390/ph9020020
Chicago/Turabian StyleVanden Eynde, Jean Jacques, Annie Mayence, Madhusoodanan Mottamal, Cyrus J. Bacchi, Nigel Yarlett, Marcel Kaiser, Reto Brun, and Tien L. Huang. 2016. "Alkanediamide-Linked Bisbenzamidines Are Promising Antiparasitic Agents" Pharmaceuticals 9, no. 2: 20. https://doi.org/10.3390/ph9020020
APA StyleVanden Eynde, J. J., Mayence, A., Mottamal, M., Bacchi, C. J., Yarlett, N., Kaiser, M., Brun, R., & Huang, T. L. (2016). Alkanediamide-Linked Bisbenzamidines Are Promising Antiparasitic Agents. Pharmaceuticals, 9(2), 20. https://doi.org/10.3390/ph9020020