
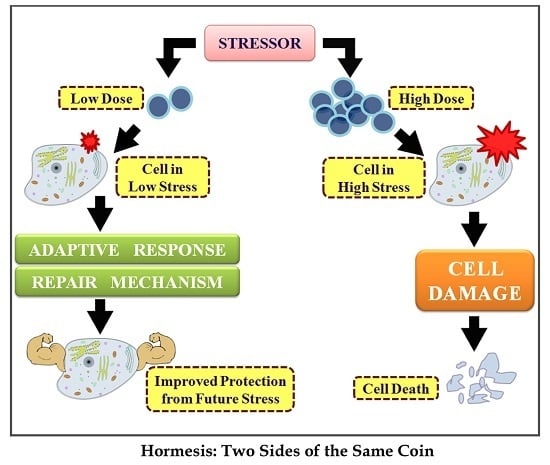
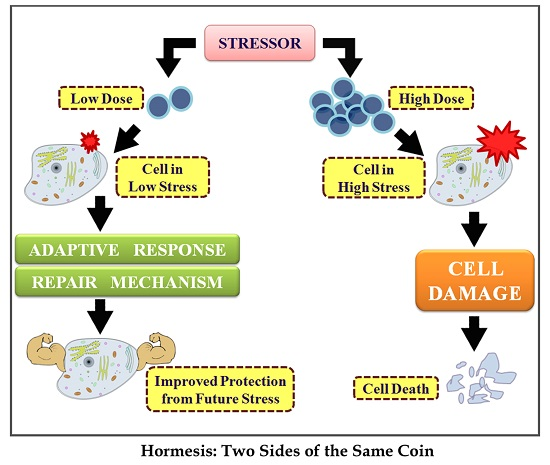
Hormesis: Decoding Two Sides of the Same Coin



Abstract
:
1. Introduction
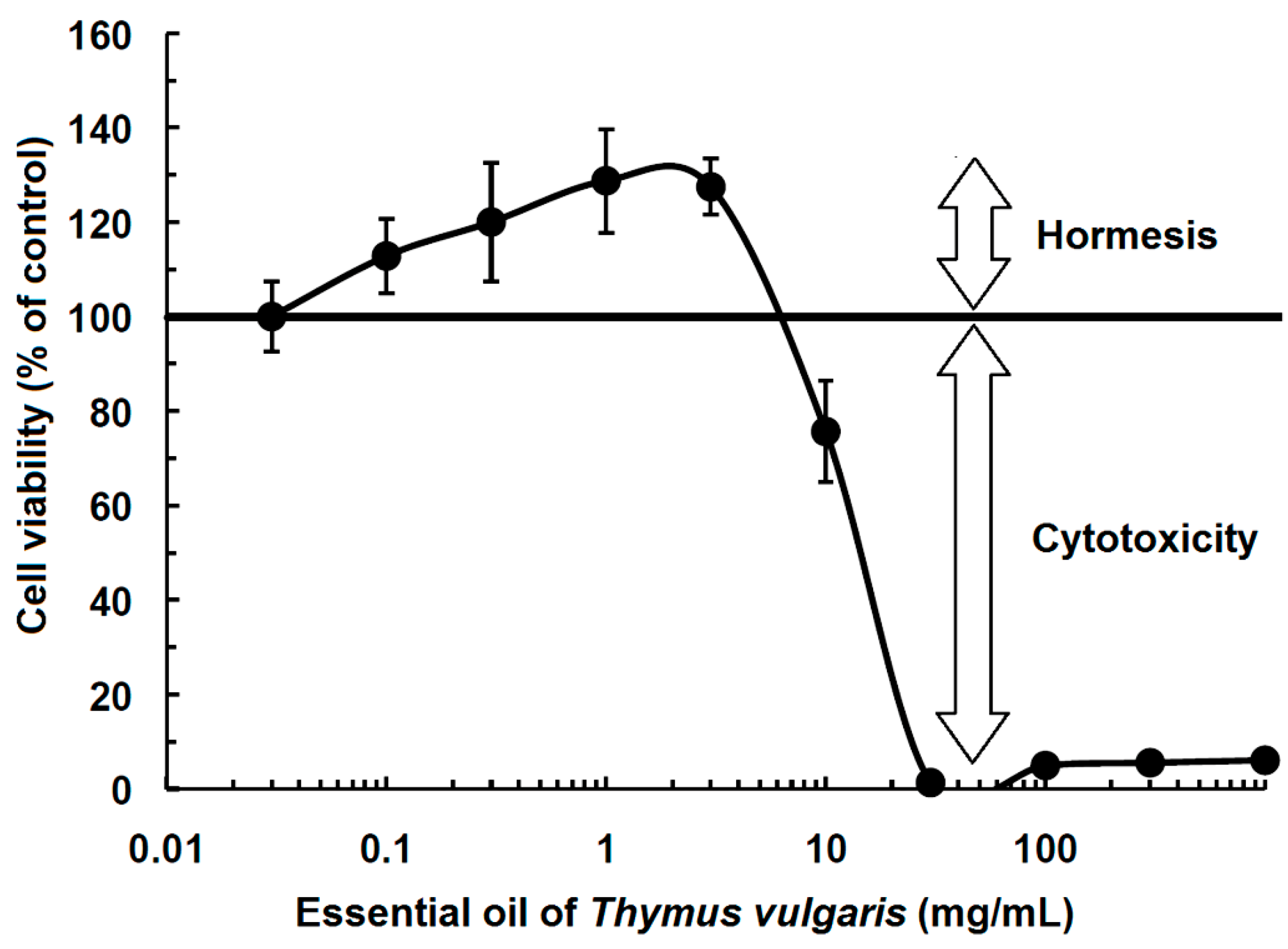
2. History
3. What Triggers Hormesis?
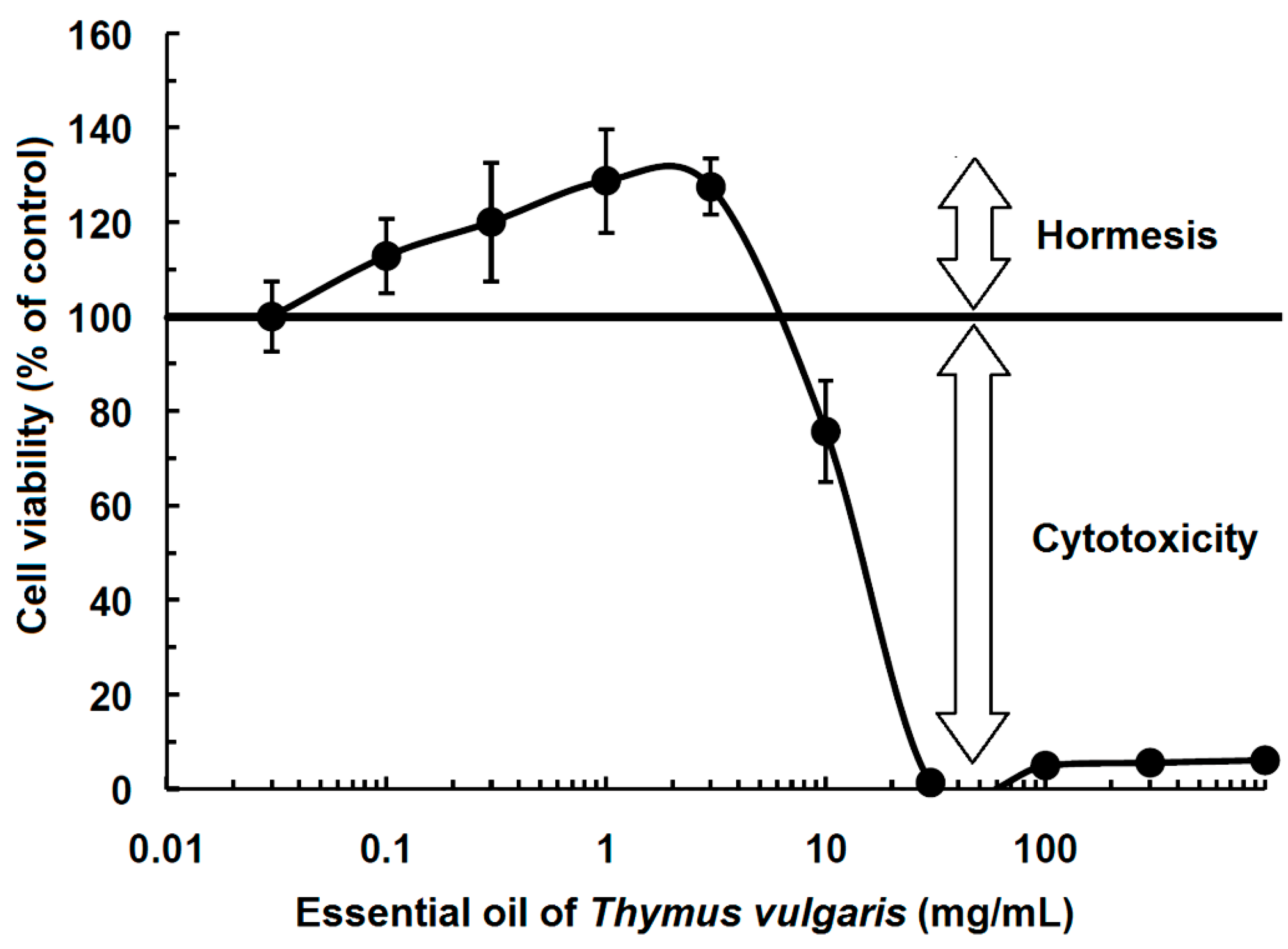
3.1. Phytochemicals
3.2. Temperature
3.3. Caloric Restriction
3.4. Exercise
4. Hormesis in Stressor-Mediated Pathways
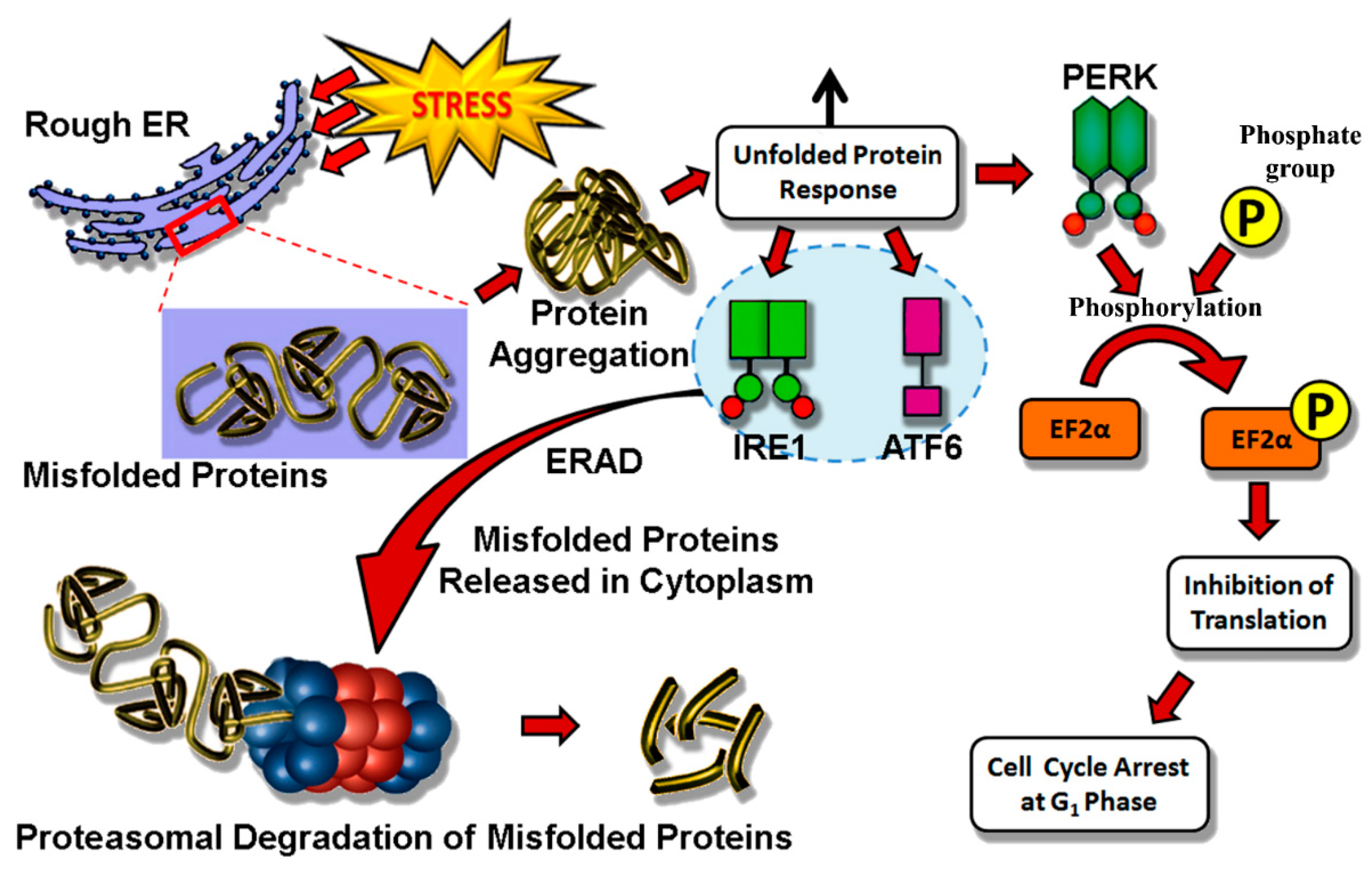
4.1. Endoplasmic Reticulum Stress
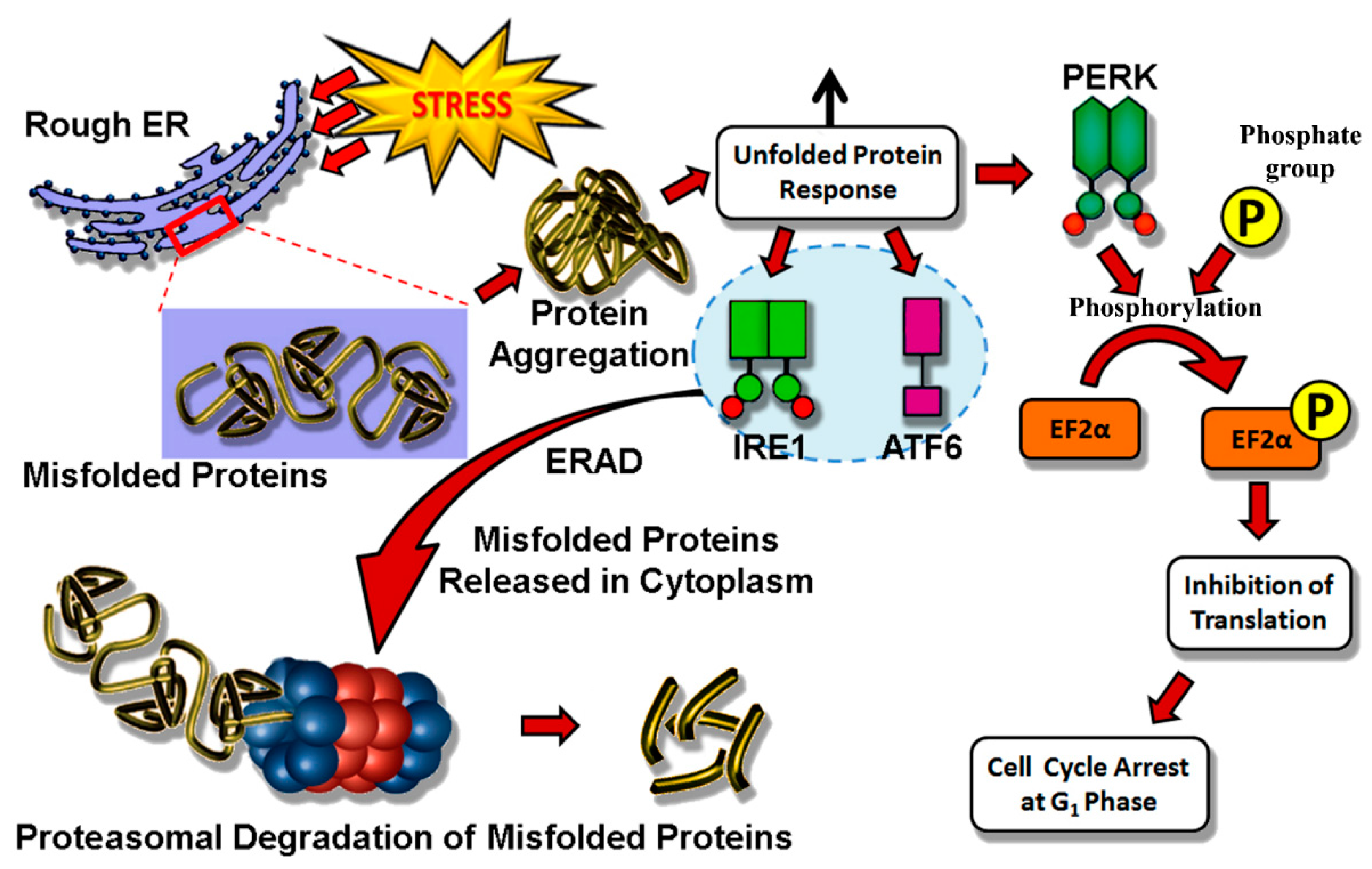
4.2. Mitochondria and ROS
4.3. Insulin or Insulin-Like Growth Factor 1 Signaling
5. Hormesis and Cancer: A Potential Rescue Path
5.1. Nrf2-Keap1 Signaling Pathway
5.2. NFκB Signaling Pathway
5.3. Sirtuin-FOXO Signaling Pathway
6. Hormetic Compounds in Cancer

{kind=link}
{kind=link}
{kind=link}
| Compound | Low Dose | High Dose | Plausible Mode of Mechanism | Cancer |
|---|---|---|---|---|
| ATN-161 | ↓angiogenesis | ↑cytotoxic | Integrin inhibitor [90] | Colon |
| Chalcone | non-toxic | cytotoxic | Nrf2 activation; inhibits NFκB; TRAIL-mediated hypoxia-induced apoptosis [91] | Ovarian, hepatic |
| Cilengitide | ↑angiogenesis | ↓angiogenesis | Inhibits αvβ3 and αvβ5 integrins; endothelial cell migration [92] | Subcutaneous tumor graft |
| Curcumin | ↑HO-1, neuro-protection | ↑DNA damage, apoptosis | Reduces matrix metalloproteinases’ expression through downregulation of NFκB and AP-1 [93,94] | Breast |
| Dithiolethione | neuro-protection | cytotoxic | Increases Nrf2/ARE pathway-mediated transcriptional activity of phase II enzymes [66] | - |
| Endostatin | ↓angiogenesis | cytotoxic | Inhibits endothelial cell proliferation, migration [95] | Pancreatic |
| Epigallocatechin | neuro-protection | pro-oxidant, ↑apoptosis | Phosphorylates Bad at Ser-112,136 through ERK, AKT pathways; Bcl-2:Bax increases [96] | Neuroblastoma |
| Genistein | ↑proliferation | ↓proliferation | Increases cleaved PARP expression; inhibits NFκB; inhibits Akt [97] | Prostate |
| Isothiocyanates | ↑proliferation | ↓proliferation | Alters cell growth and migration pattern [98] | Colon |
| Kaempferol | estrogen agonist | growth inhibitor | Depletes estrogen-induced malignancy; suppresses COX-2; induces caspase-3, apoptosis inducing factor (AIF), MnSOD [99] | Breast |
| Metformin | anti-diabetic | anti-cancer | Suppresses mTOR/S6K1; inhibits tyrosine kinase receptors HER1/2 [100] | Epidermoid, breast, prostate |
| Quercetin | anti-oxidant | pro-oxidant | Suppresses NFκB activity, G1 cell cycle arrest, ↑p21, p53; inhibits ubiquitination [15] | Pancreatic, colon, hepatic |
| Resveratrol | ↑proliferation cardio protection | ↓proliferation anti-cancer | Activates Nrf2; upregulates FOXO [101,102] | endometrial |
| Rosiglitazone | ↓angiogenesis | ↑cytotoxicity | Inhibits endothelial proliferation and vascular endothelial growth factor (VEGF) activation; upregulates matrix metalloproteinase (MMP) inhibitors [103] | Bladder, breast, thyroid |
| Secoiridoid | ↓pro-aging effect | ↑cytotoxicity | Activates ER stress, unfolded protein-mediated response, SIRT1 and Nrf2 [21] | Breast |
| Sulforaphane | ↑proliferation ↑angiogenesis | ↓proliferation ↓angiogenesis | Activates Nrf2/ARE pathway; regulates NFκB and AP-1 to induce apoptosis; activates autophagy [98] | Bladder |
| Thrombo-spondin-1 | ↑cell migration | ↓cell migration | Inhibits endothelial cell migration. [104] | Oral |
7. Hormesis Mimetics: A World of Endless Opportunities
7.1. Heat Mimetics
7.2. CR Mimetics
7.3. Radiation Mimetics
7.4. Hibernation Mimetics
| Compound | Type of Mimetic | Plausible Mode of Mechanism | Disease |
|---|---|---|---|
| Carnitine | caloric restriction | Upregulates HO-1, sirtuin, thioredoxin, ↓pro-oxidant activity, mediates fatty acid metabolism [66] | Neurodegenerative damage |
| Resveratrol | caloric restriction | Sirtuin activator, ↓UV/H2O2-induced apoptosis, ↑SIRT mediated FOXO activation [109,110] | Longevity, oxidative damage, toxicity resistance |
| Secoiridoid | caloric restriction | ↑Nrf2, SIRT1 signaling, mediates ER stress response, regulates mTOR pathway [21] | Longevity, age-associated diseases |
| PPARδ agonists | caloric restriction | ↓Glucose consumption in skeletal muscles [111] | Insulin sensitivity |
| Ethanol | heat | ↑Hsp70 in brain; ↓β amyloid-induced neurotoxicity and apoptosis [112] | Alzheimer’s; dementia |
| Geranyl-geranylactone (GGA) | heat | Induces bone osteoblasts, upregulates thioredoxin, forms apoptosome on binding to Apaf-1, inhibits c-Jun N-terminal kinase [113,35] | Osteoporosis, increases immunity, apoptosis in normal cells |
| Delta 2 opioid receptors (DADLE) | hibernation | ↓Neuronal damage. [114] | Neurodegeneration |
| Oltipraz | radiation | ↑Nrf2-ARE binding, ↑transcriptional induction of carcinogen detoxification gene cascade [115] | Oxidative stress, cancer |
| Ferritins | radiation | ARE activation, ↓ROS-mediated damage [35] | Oxidative stress |
| Thiols and metals | radiation | ↑Antioxidant gene expression, ↑glutathione peroxidase activity [116,117] | Radiation induced oxidative damage, cancer |
| Oligonucleotides | radiation | ↓Mutagenesis, photo-carcinogenesis, ↑DNA repair, mitochondria hyperpolarization [108,118] | UV-induced mutation, cancer |
| Conserved peptide sequences, CpG oligo | ↑TH2-mediated inflammation, ↓cytokine dysfunction, ↑adaptive immunity [35] | Increases immunity |
8. Conclusions
Acknowledgements
Conflicts of interest
References
- Calabrese, E.J.; Baldwin, L.A. The frequency of U-shaped dose responses in the toxicological literature. Toxicol. Sci. Off. J. Soc. Toxicol. 2001, 62, 330–338. [Google Scholar] [CrossRef]
- Mattson, M.P. Awareness of hormesis will enhance future research in basic and applied neuroscience. Crit. Rev. Toxicol. 2008, 38, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Trovato, A.; Cavallaro, M.; Mancuso, C.; Di Rienzo, L.; Condorelli, D.; de Lorenzo, A.; Calabrese, E.J. The hormetic role of dietary antioxidants in free radical-related diseases. Curr. Pharm. Des. 2010, 16, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Nitric oxide: biphasic dose responses. Crit. Rev. Toxicol. 2001, 31, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-S.; Zheng, R.-L. Biphasic regulation of angiogenesis by reactive oxygen species. Pharm. 2006, 61, 223–229. [Google Scholar]
- Day, R.M.; Suzuki, Y.J. Cell proliferation, reactive oxygen and cellular glutathione. Dose-Response 2006, 3, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormesis: From mainstream to therapy. J. Cell Commun. Signal. 2014, 8, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Partridge, L. Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell MeTable 2008, 7, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Kouda, K.; Iki, M. Beneficial effects of mild stress (hormetic effects): Dietary restriction and health. J. Physiol. Anthropol. 2010, 29, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Galluzzi, L.; Kroemer, G. Hormesis, Cell death and aging. Aging 2011, 3, 821–828. [Google Scholar] [PubMed]
- Schumacher, B. Transcription-blocking dna damage in aging: a mechanism for hormesis. BioEssays News Rev. Mol. Cell. Dev. Biol. 2009, 31, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Freiberger, J.; Coulombe, K.; Suliman, H.; Carraway, M.; Piantadosi, C. Superoxide dismutase responds to hyperoxia in rat hippocampus. J. Undersea Hyperb. Med. Soc. Inc. 2004, 31, 227–232. [Google Scholar]
- Mach, W.J.; Thimmesch, A.R.; Pierce, J.T.; Pierce, J.D. Consequences of hyperoxia and the toxicity of oxygen in the lung. Nurs. Res. Pract. 2011, 2011, 260482. [Google Scholar] [CrossRef] [PubMed]
- Speciale, A.; Chirafisi, J.; Saija, A.; Cimino, F. Nutritional antioxidants and adaptive cell responses: an update. Curr. Mol. Med. 2011, 11, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Sertel, S.; Eichhorn, T.; Plinkert, P.K.; Efferth, T. Cytotoxicity of Thymus vulgaris essential oil towards human oral cavity squamous cell carcinoma. Anticancer Res. 2011, 31, 81–87. [Google Scholar] [PubMed]
- Mattson, M.P.; Cheng, A. Neurohormetic phytochemicals: low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Hursting, S.D.; Smith, S.M.; Lashinger, L.M.; Harvey, A.E.; Perkins, S.N. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis 2010, 31, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Yamaza, H.; Komatsu, T.; Wakita, S.; Kijogi, C.; Park, S.; Hayashi, H.; Chiba, T.; Mori, R.; Furuyama, T.; Mori, N.; et al. FoxO1 is involved in the antineoplastic effect of calorie restriction. Aging Cell 2010, 9, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Aragonès, G.; Barrajón-Catalán, E.; Beltrán-Debón, R.; Borrás-Linares, I.; Camps, J.; Corominas-Faja, B.; Cufí, S.; Fernández-Arroyo, S.; et al. Xenohormetic and anti-aging activity of secoiridoid polyphenols present in extra virgin olive oil: A new family of gerosuppressant agents. Cell Cycle Georget. Tex. 2013, 12, 555–578. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell MeTable 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-Response Publ. Int. Hormesis Soc. 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, B.; Manié, S.; Napoletano, F. Getting the better of ER stress. J. Cell Commun. Signal. 2014, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Maynard, K.I. Hormesis pervasiveness and its potential implications for pharmaceutical research and development. Dose-Response Publ. Int. Hormesis Soc. 2011, 9, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Mayor, A. The Poison King: The Life and Legend of Mithradates, Rome’s Deadliest Enemy; Princeton University Press: Princeton, NJ, USA, 2010; p. 242. [Google Scholar]
- Henschler, D. The origin of hormesis: Historical background and driving forces. Hum. Exp. Toxicol. 2006, 25, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef] [PubMed]
- Costantini, D. Oxidative Stress and Hormesis in Evolutionary Ecology and Physiology: A Marriage Between Mechanistic and Evolutionary Approaches, 1st ed.; Springer Science & Business Media: Berlin, Germany, 2014. [Google Scholar]
- Luch, A. Molecular toxicology. In Molecular, Clinical and Environmental Toxicology; Springer Science & Business Media: Berlin, Germany, 2012; Volume 1. [Google Scholar]
- Laughlin, R.B.; Ng, J.; Guard, H.E. Hormesis: A response to low environmental concentrations of petroleum hydrocarbons. Science 1981, 211, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, A.R. Hormesis—the stimulation of growth by low levels of inhibitors. Sci. Total Environ. 1982, 22, 213–234. [Google Scholar] [CrossRef]
- Sonneborn, J.S. Mimetics of hormetic agents: Stress-resistance triggers. Dose-Response Publ. Int. Hormesis Soc. 2010, 8, 97–121. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Hormetic mechanisms. Crit. Rev. Toxicol. 2013, 43, 580–606. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Dhawan, G.; Kapoor, R.; Iavicoli, I.; Calabrese, V. Hormesis: A fundamental concept with widespread biological and biomedical applications. Gerontology 2015. [CrossRef] [PubMed]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing A 2015, 12, 20. [Google Scholar] [CrossRef]
- Rattan, S.I.S. Hormetic modulation of aging and longevity by mild heat stress. Dose-Response 2006, 3, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.B. Ionizing radiation and aging: rejuvenating an old idea. Aging 2009, 1, 887–902. [Google Scholar] [PubMed]
- Heber, D. Vegetables, fruits and phytoestrogens in the prevention of diseases. J. Postgrad. Med. 2004, 50, 145–149. [Google Scholar] [PubMed]
- Parker, J.A.; Arango, M.; Abderrahmane, S.; Lambert, E.; Tourette, C.; Catoire, H.; Néri, C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 2005, 37, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xu, C.; Shen, G.; Jain, M.R.; Khor, T.O.; Gopalkrishnan, A.; Lin, W.; Reddy, B.; Chan, J.Y.; Kong, A.-N.T. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 2006, 243, 170–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, K.W.; Lee, H.J. Protective effects of piceatannol against beta-amyloid-induced neuronal cell death. Ann. N. Y. Acad. Sci. 2007, 1095, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, D.; Li, G.; Liu, J.; Tian, J.; Fu, F.; Liu, K. Neuroprotective effects of safflor yellow b on brain ischemic injury. Exp. Brain Res. 2007, 177, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Li, W.; Kong, A.-N.T. Natural dietary anti-cancer chemopreventive compounds: Redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells. Acta Pharmacol. Sin. 2007, 28, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell. Mol. Life Sci. CMLS 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Son, T.G.; Camandola, S.; Mattson, M.P. Hormetic dietary phytochemicals. Neuromolecular Med. 2008, 10, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Park, H.G.; Han, S.I.; Oh, S.Y.; Kang, H.S. Cellular responses to mild heat stress. Cell. Mol. Life Sci. CMLS 2005, 62, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Le Bourg, E.; Valenti, P.; Lucchetta, P.; Payre, F. Effects of mild heat shocks at young age on aging and longevity in Drosophila melanogaster. Biogerontology 2001, 2, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Hormesis does not make sense except in the light of tor-driven aging. Aging 2011, 3, 1051–1062. [Google Scholar] [PubMed]
- Moskalev, A.A.; Shaposhnikov, M.V. Pharmacological inhibition of phosphoinositide 3 and tor kinases improves survival of Drosophila melanogaster. Rejuvenation Res. 2010, 13, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. The Stress of Life. Hans Selye, M.D. New York, McGraw-Hill Book Company, Inc. 1956. $5.95. J. Bone Jt. Surg. 1957, 39, 479–479. [Google Scholar]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.-C.; Martínez, A.; Santangelo, G.; Pallardó, F.V.; Sastre, J.; Viña, J. Oxidative stress in marathon runners: interest of antioxidant supplementation. Br. J. Nutr. 2006, 96, S31–S33. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Parise, G.; Melov, S.; Safdar, A.; Tarnopolsky, M.A. Analysis of global mrna expression in human skeletal muscle during recovery from endurance exercise. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1498–1500. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Pani, G.; Calabrese, V. Bilirubin: an endogenous scavenger of nitric oxide and reactive nitrogen species. Redox Rep. Commun. Free Radic. Res. 2006, 11, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, C.; Koverech, G.; Crupi, R.; di Paola, R.; Koverech, A.; Lodato, F.; Scuto, M.; Salinaro, A.T.; Cuzzocrea, S.; Calabrese, E.J.; et al. Osteoporosis and alzheimer pathology: Role of cellular stress response and hormetic redox signaling in aging and bone remodeling. Front. Pharmacol. 2014, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.S.; Levet, C.; Chatelain, G.; Dourlen, P.; Fouillet, A.; Dichtel-Danjoy, M.-L.; Gambis, A.; Ryoo, H.D.; Steller, H.; Mollereau, B. ER stress protects from retinal degeneration. EMBO J. 2009, 28, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, B. Establishing links between endoplasmic reticulum-mediated hormesis and cancer. Mol. Cell. Biol. 2013, 33, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Fouillet, A.; Levet, C.; Virgone, A.; Robin, M.; Dourlen, P.; Rieusset, J.; Belaidi, E.; Ovize, M.; Touret, M.; Nataf, S.; et al. ER stress inhibits neuronal death by promoting autophagy. Autophagy 2012, 8, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Marada, S.; Stewart, D.P.; Bodeen, W.J.; Han, Y.-G.; Ogden, S.K. The unfolded protein response selectively targets active smoothened mutants. Mol. Cell. Biol. 2013, 33, 2375–2387. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, C.; Perrotta, R.; Graziano, A.; Calabrese, E.J.; Calabrese, V. Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: mitochondria as a “chi”. Immun. Ageing 2013, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.-J.; Shaki, F.; Ghazi-Khansari, M.; Pourahmad, J. Toxicity of arsenic (iii) on isolated liver mitochondria: a new mechanistic approach. Iran. J. Pharm. Res. IJPR 2013, 12, 121–138. [Google Scholar]
- Schmeisser, S.; Schmeisser, K.; Weimer, S.; Groth, M.; Priebe, S.; Fazius, E.; Kuhlow, D.; Pick, D.; Einax, J.W.; Guthke, R.; et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell 2013, 12, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R.A.C. C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowska, L.; Hu, D.; Huntsman, S.; Sung, A.; Chu, C.; Chen, J.; Joyner, A.H.; Schork, N.J.; Hsueh, W.-C.; Reiner, A.P.; et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell 2009, 8, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Bhakta-Guha, D.; Saeed, M.E.M.; Greten, H.J.; Efferth, T. Dis-organizing centrosomal clusters: Specific cancer therapy for a generic spread? Curr. Med. Chem. 2015, 22, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the human development index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H.; Verhagen, A.; Secrest, P.; Zhang, J.; Varki, N.M.; Crocker, P.R.; Bui, J.D.; Varki, A. Inverse hormesis of cancer growth mediated by narrow ranges of tumor-directed antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 5998–6003. [Google Scholar] [CrossRef] [PubMed]
- Suganthi, M.; Sangeetha, G.; Gayathri, G.; Ravi Sankar, B. Biphasic dose-dependent effect of lithium chloride on survival of human hormone-dependent breast cancer cells (MCF-7). Biol. Trace Elem. Res. 2012, 150, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Hine, C.M.; Mitchell, J.R. NRF2 and the phase II response in acute stress resistance induced by dietary restriction. J. Clin. Exp. Pathol. 2012, S4, 7329. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 2010, 50, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the keap1-nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Diwan, B.A.; Sun, Y.; Liu, J.; Qu, W.; He, Y.; Styblo, M.; Waalkes, M.P. Arsenic-induced malignant transformation of human keratinocytes: involvement of nrf2. Free Radic. Biol. Med. 2008, 45, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Nakajima, A.; Sakon-Komazawa, S.; Piao, J.-H.; Xue, X.; Okumura, K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006, 13, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Oellerich, M.F.; Potente, M. FOXOs and sirtuins in vascular growth, maintenance, and aging. Circ. Res. 2012, 110, 1238–1251. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Presegué, L.; Vaquero, A. The dual role of sirtuins in cancer. Genes Cancer 2011, 2, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.-M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Fang, D. The roles of SIRT1 in cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hou, H.; Haller, E.M.; Nicosia, S.V.; Bai, W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005, 24, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Doñate, F.; Parry, G.C.; Shaked, Y.; Hensley, H.; Guan, X.; Beck, I.; Tel-Tsur, Z.; Plunkett, M.L.; Manuia, M.; Shaw, D.E.; et al. Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): Observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin. Cancer Res. 2008, 14, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, V.; Balasubramanian, R.; Sathe, P.; Murali Krishna, C.; Juvekar, A. In vitro anticancer activity of monosubstituted chalcone derivatives. Int. J. Tumor Ther. 2014, 3, 1–9. [Google Scholar]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Bachmeier, B.; Nerlich, A.G.; Iancu, C.M.; Cilli, M.; Schleicher, E.; Vené, R.; Dell’Eva, R.; Jochum, M.; Albini, A.; Pfeffer, U. The chemopreventive polyphenol curcumin prevents hematogenous breast cancer metastases in immunodeficient mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2007, 19, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Morvan, D. Metabolomics reveals metabolic targets and biphasic responses in breast cancer cells treated by curcumin alone and in association with docetaxel. PLoS ONE 2013, 8, e57971. [Google Scholar] [CrossRef] [PubMed]
- Celik, I.; Sürücü, O.; Dietz, C.; Heymach, J.V.; Force, J.; Höschele, I.; Becker, C.M.; Folkman, J.; Kisker, O. Therapeutic efficacy of endostatin exhibits a biphasic dose-response curve. Cancer Res. 2005, 65, 11044–11050. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Mandel, S.; Youdim, M.B.H. Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: a reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr. 2009, 4, 283–296. [Google Scholar] [CrossRef] [PubMed]
- El Touny, L.H.; Banerjee, P.P. Identification of a biphasic role for genistein in the regulation of prostate cancer growth and metastasis. Cancer Res. 2009, 69, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Wang, W.; Zhou, Z.; Sun, C. Benefits and risks of the hormetic effects of dietary isothiocyanates on cancer prevention. PLoS ONE 2014, 9, e114764. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.M.; Kim, Y.P.; Chung, K.H. Biphasic effects of kaempferol on the estrogenicity in human breast cancer cells. Arch. Pharm. Res. 2006, 29, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle Georget. Tex. 2010, 9, 1057–1064. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Resveratrol commonly displays hormesis: Occurrence and biomedical significance. Hum. Exp. Toxicol. 2010, 29, 980–1015. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Dudley, J.I.; Das, D.K. Dose-dependency of resveratrol in providing health benefits. Dose-Response 2010, 8, 478–500. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Singer, S.; Shen, L.Q.; Butterfield, C.E.; Freedman, D.A.; Chen, E.J.; Moses, M.A.; Kilroy, S.; Duensing, S.; Fletcher, C.; et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Invest. 2002, 110, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Motegi, K.; Harada, K.; Ohe, G.; Jones, S.J.; Ellis, I.R.; Crouch, D.H.; Schor, S.L.; Schor, A.M. Differential involvement of tgf-beta1 in mediating the motogenic effects of tsp-1 on endothelial cells, fibroblasts and oral tumour cells. Exp. Cell Res. 2008, 314, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Calabrese, E.J. Hormesis: A Revolution in Biology, Toxicology and Medicine, 1st ed.; Springer Science & Business Media: Berlin, Germany, 2009. [Google Scholar]
- Ingram, D.K.; Zhu, M.; Mamczarz, J.; Zou, S.; Lane, M.A.; Roth, G.S.; deCabo, R. Calorie restriction mimetics: An emerging research field. Aging Cell 2006, 5, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Biasi, F.; Poli, G.; Chiarpotto, E. Calorie restriction and dietary restriction mimetics: A strategy for improving healthy aging and longevity. Curr. Pharm. Des. 2014, 20, 2950–2977. [Google Scholar] [CrossRef] [PubMed]
- Goukassian, D.A.; Helms, E.; van Steeg, H.; van Oostrom, C.; Bhawan, J.; Gilchrest, B.A. topical DNA oligonucleotide therapy reduces UV-induced mutations and photocarcinogenesis in hairless mice. Proc. Natl. Acad. Sci. USA 2004, 101, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Tertoolen, L.G.J.; de Vries-Smits, L.M.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M.T. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L.; Picard, F. Calorie restriction—the SIR2 connection. Cell 2005, 120, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Brunmair, B.; Staniek, K.; Dörig, J.; Szöcs, Z.; Stadlbauer, K.; Marian, V.; Gras, F.; Anderwald, C.; Nohl, H.; Waldhäusl, W.; et al. Activation of PPAR-δ in isolated rat skeletal muscle switches fuel preference from glucose to fatty acids. Diabetologia 2006, 49, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Belmadani, A.; Kumar, S.; Schipma, M.; Collins, M.A.; Neafsey, E.J. Inhibition of amyloid-β-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport 2004, 15, 2093–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, J.; Shidoji, Y.; Muto, Y.; Ohishi, N.; Yagi, K.; Ikegami, S.; Shinki, T.; Udagawa, N.; Suda, T.; et al. Effects of geranylgeranoic acid in bone: induction of osteoblast differentiation and inhibition of osteoclast formation. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2002, 17, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Wang, Y.; Su, T.-P. Delta opioid peptide (D-Ala 2, D-Leu 5) Enkephalin: Linking hibernation and neuroprotection. Front. Biosci. J. Virtual Libr. 2004, 9, 3392–3398. [Google Scholar] [CrossRef]
- Kwak, M.K.; Egner, P.A.; Dolan, P.M.; Ramos-Gomez, M.; Groopman, J.D.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat. Res. 2001, 480–481, 305–315. [Google Scholar] [CrossRef]
- Hamilton, K.L. Antioxidants and cardioprotection. Med. Sci. Sports Exerc. 2007, 39, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Kumar, A.; Balakrishnan, S.; Kushwaha, H.S.; Mishra, K.P. Radiation-induced micronucleus formation and dna damage in human lymphocytes and their prevention by antioxidant thiols. Mutat. Res. 2009, 676, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Iwado, E.; Eller, M.S.; Kondo, Y.; Fujiwara, K.; Li, G.-Z.; Hess, K.R.; Siwak, D.R.; Sawaya, R.; Mills, G.B.; et al. Telomere 3’ overhang-specific DNA oligonucleotides induce autophagy in malignant glioma cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 2918–2930. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M. Hormesis, adaptive epigenetic reorganization, and implications for human health and longevity. Dose-Response 2010, 8, 16–21. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhakta-Guha, D.; Efferth, T. Hormesis: Decoding Two Sides of the Same Coin. Pharmaceuticals 2015, 8, 865-883. https://doi.org/10.3390/ph8040865
Bhakta-Guha D, Efferth T. Hormesis: Decoding Two Sides of the Same Coin. Pharmaceuticals. 2015; 8(4):865-883. https://doi.org/10.3390/ph8040865
Chicago/Turabian StyleBhakta-Guha, Dipita, and Thomas Efferth. 2015. "Hormesis: Decoding Two Sides of the Same Coin" Pharmaceuticals 8, no. 4: 865-883. https://doi.org/10.3390/ph8040865
APA StyleBhakta-Guha, D., & Efferth, T. (2015). Hormesis: Decoding Two Sides of the Same Coin. Pharmaceuticals, 8(4), 865-883. https://doi.org/10.3390/ph8040865