
X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy

Abstract
:1. Introduction
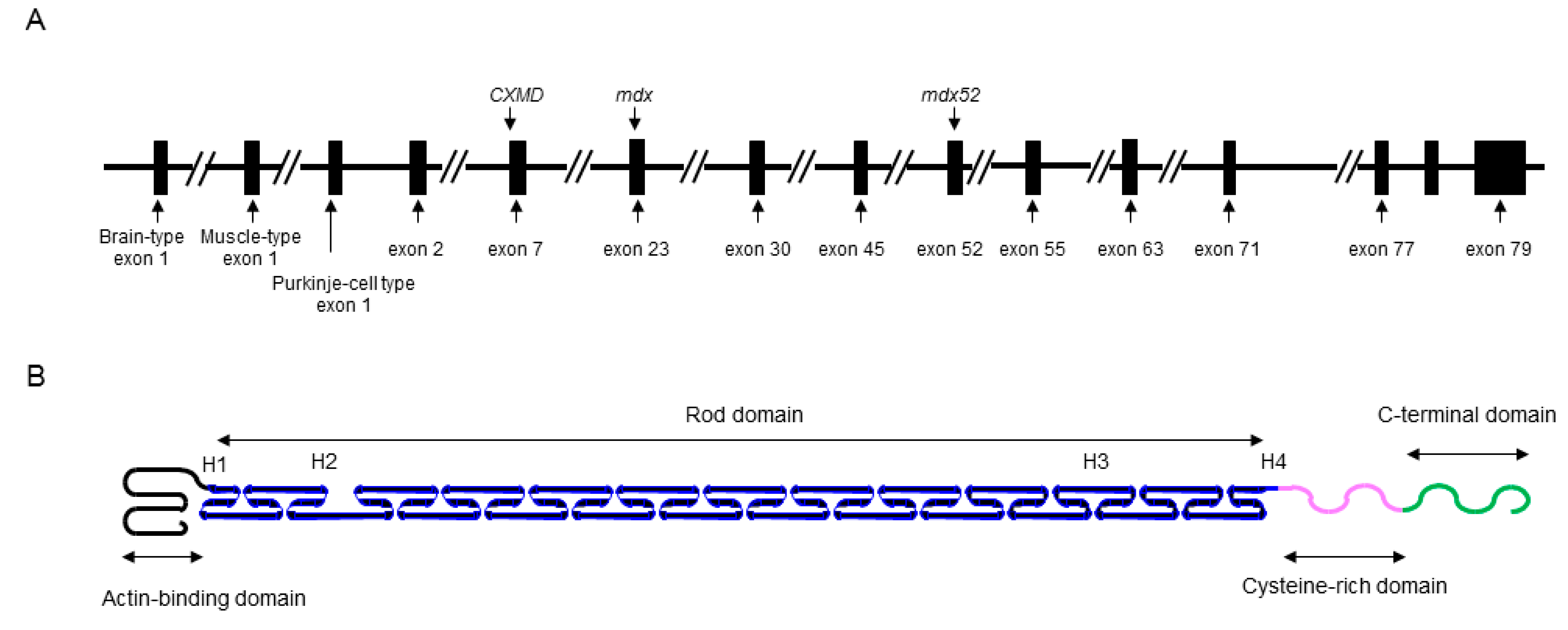
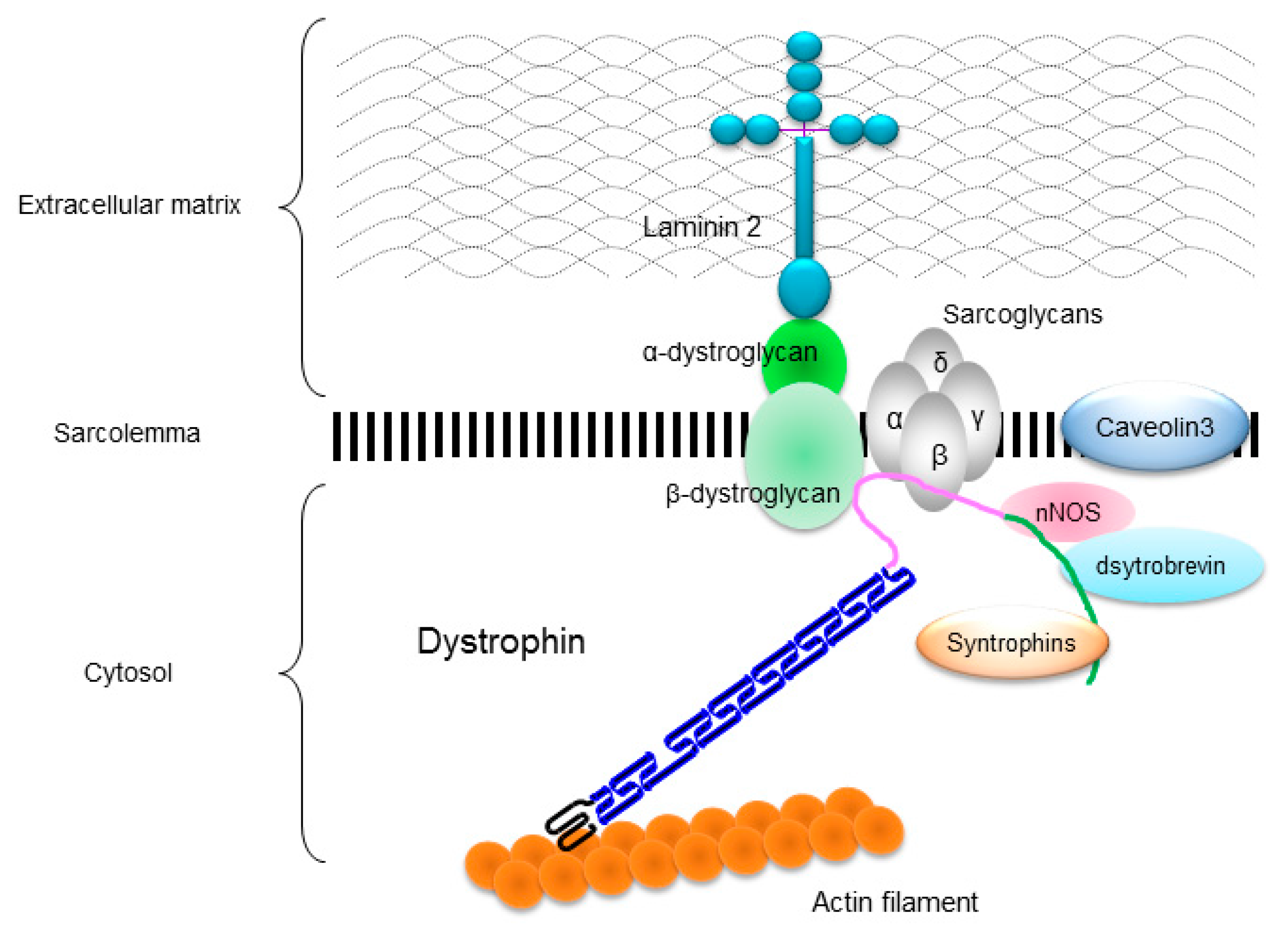
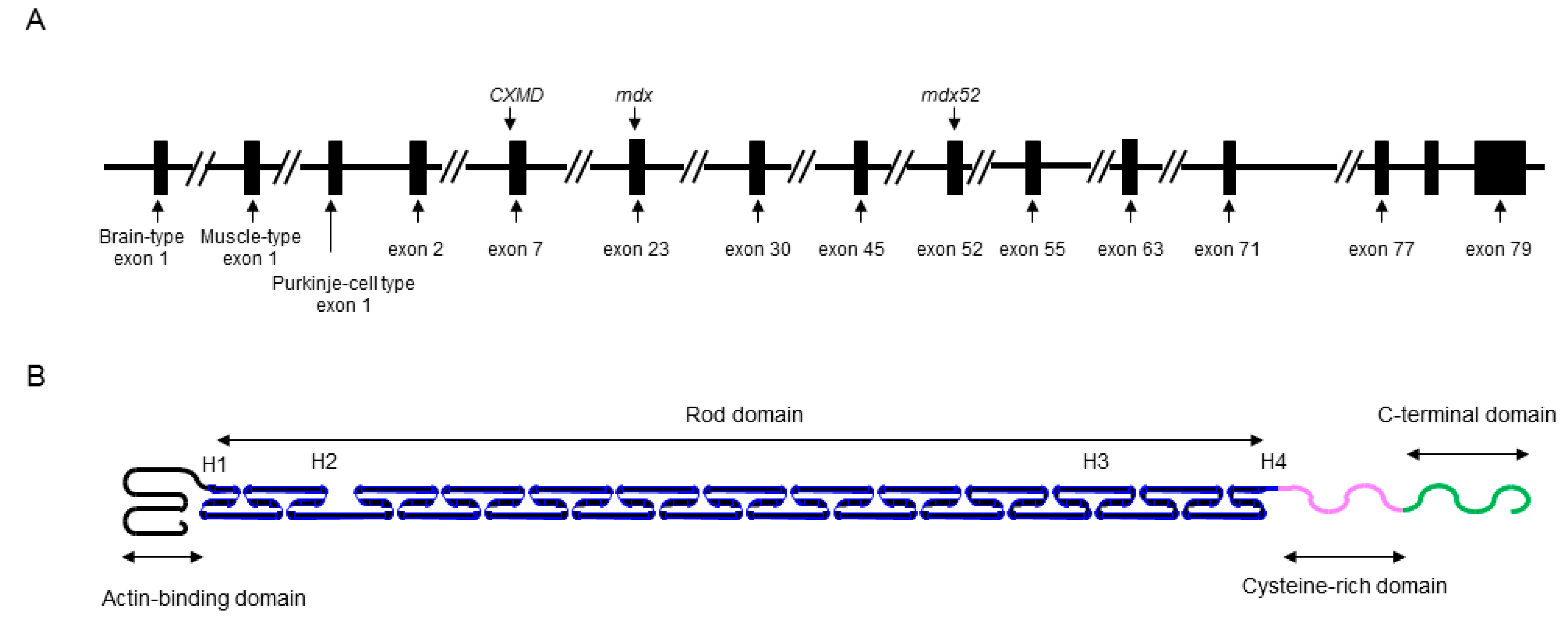
2. Identification of XLDCM
3. DMD Mutations in Patients with XLDCM
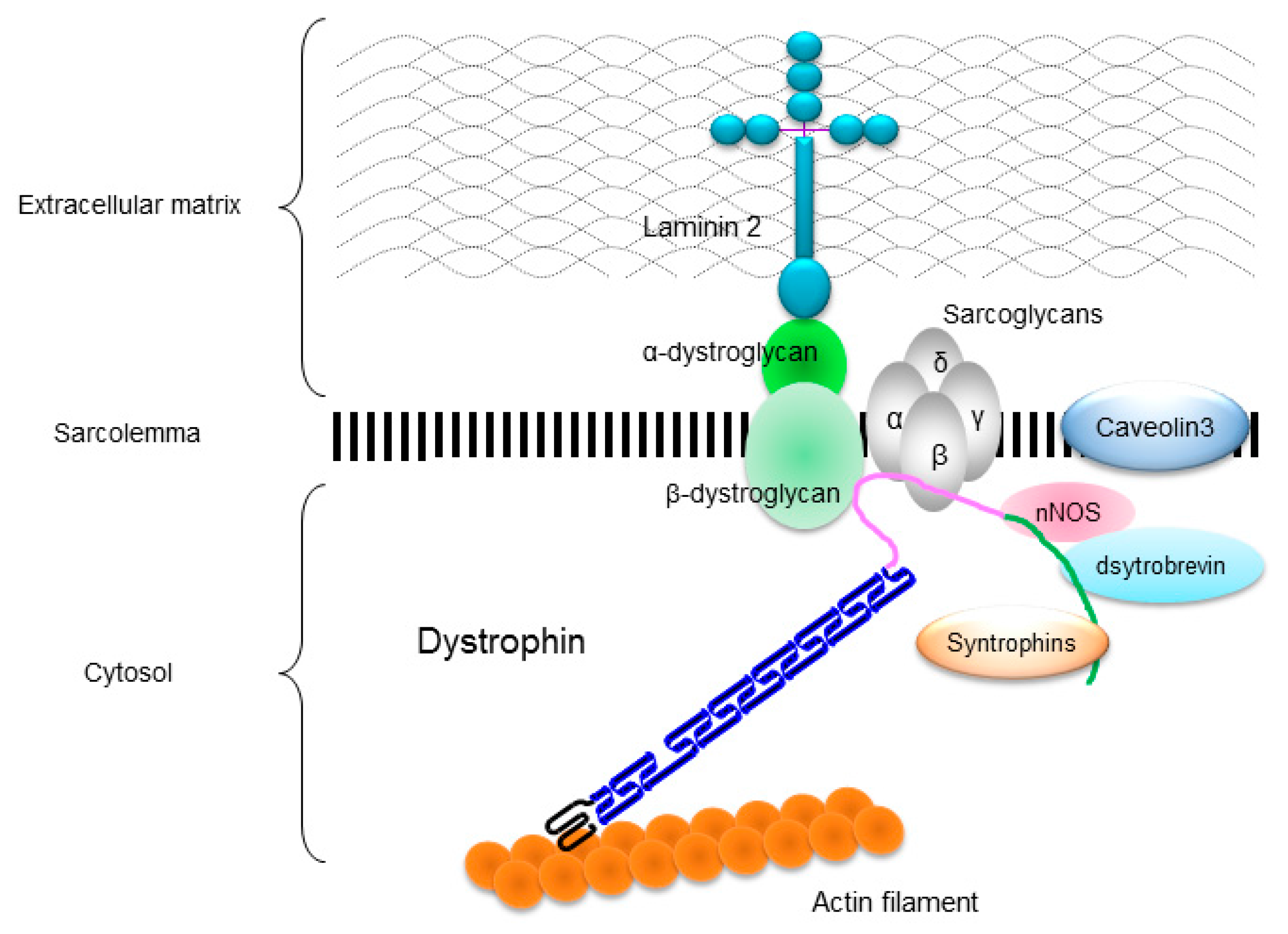
4. Pathogenic Mechanisms in XLDCM
4.1. Hypothesis 1: The Transcriptional Regulation of the DMD Gene Differs between the Skeletal and Cardiac Muscles

{kind=link}
{kind=link}
{kind=link}
| . | Mutation type | Family history | Serum CK level | Reference [No.] |
|---|---|---|---|---|
| Muscle promoter-exon 1 | Deletion | + | High | Muntoni, et al. 1993 [19] |
| Muscle exon 1 | Insertion of L1sequence | + | High | Yoshida, et al. 1993 [11], 1998 [20] |
| Intron 1 | Point mutation at splice donor site | + | Normal~High | Milasin, et al. 1996 [21] |
| Exon 2 | Missense mutation | − | High | Feng, et al. 2002 [22] |
| Exon 2-7 | Deletion | High | Gold, et al. 1992 [23), Nigro, et al. 1995 [24] | |
| Exon 2-7 | Duplication | + | High | Bies, et al. 1997 [25] |
| Exon 3-7 | Deletion | Unknown | Nigro, et al. 1995 [24] | |
| Intron 5 | IVS5+1 | − | Unknown | Feng, et al. 2002 [22] |
| Exon (5) 6-13(14/15) | Deletion (actual range was unknown) | − | Unknown | Oldfors, et al. 1994 [26] |
| Exon 9 | Missense mutation | + | High | Ortiz-Lopez, et al. 1997 [27] |
| Intron 11 | Insertion of Alu like sequence | + | High | Ferlini, et al. 1998 [28] |
| Exon 27 | Frameshift mutation | + | High | Tsuda, et al. 2014 [29] |
| Exon 27-30 | Deletion | High | Franz, et al. 2000 [30] | |
| Exon 29 | Missense mutation | + | High | Franz, et al. 2000 [31] |
| Exon 45-51 | Deletion | + | High | Arbustini, et al. 2000 [32] |
| Exon 45-55 | Deletion | +/− | Normal~High | Beroud, et al. 2007 [33]; Nakamura, et al. 2008 [16] |
| Exon 45-48 | Deletion | − | High | Arbustini, et al. 2000 [32]; Shimizu, et al. 2005 [34] |
| Exon 48 | Deletion | − | Normal | Arbustini, et al. 2000 [32] |
| Exon 48-51 | Deletion | − | High | Arbustini, et al. 2000 [32] |
| Exon 48-49 | Deletion | +/− | High | Piccolo, et al. 1994 [13], Muntoni, et al. 1997 [35] |
| Exon 48-52 | Deletion | + | Normal | Shimizu, et al. [34] |
| Exon 48-53 | Deletion | − | Normal | Arbustini, et al. 2000 [31] |
| Exon 49-51 | Deletion | − | Normal | Gold, et al. 1992 [23], Muntoni, et al. 1997 [35] |
| Exon 67 | Missense mutation | − | Unknown | Feng, et al. 2002 [22] |
4.2. Hypothesis 2: Dystrophin Stability and Protein-Binding Mechanisms Differ between the Skeletal and Cardiac Muscles
4.3. Hypothesis 3: Exercise Overload Affects Cardiac Damage
5. Clinical and Laboratory Features in XLDCM
6. Diagnosis and Differential Diagnosis
7. Treatment
| Disease | Inheritance | Gene | Symptoms and laboratory examination |
|---|---|---|---|
| Emery-Dreifuss muscular dystrophy | AR or AD | LMNA | joint contracture, hyper-CKemia, arrhythmias, muscle weakness of childhood onset |
| LGMD 1B | AR | LMNA | joint contracture (mild), hyper-CKemia, arrhythmias, weakness of limb-girdle muscle |
| McLoad syndrome | X-linked | XK | chorea, myopathy, hyper-CKemia, ancanthocyte |
| Barth syndrome | X-linked | TAZ | growth retardation, lactic acidosis, leukocytopenia, increase in 3-methylglutacon levels |
| Danon disease | X-linked | LAMP-2 | limb muscle weakness, atrophy and myalgia, hypertrophic or dilated cardiomyopathy, arrhythmias, mental retardation |
| Laing dystal myopathy | AD | MYH7 | childhood onset, Involvement of face, ankle, thumb, digital extensor, neck flexor muscles |
| Carvajal syndrome | AR | DSP | palmoplantar keratosis, kinky hair |
| Mitochondrial dilated cardiomyopathy | Maternal inheritance | mtDNA | focal glomerulosclerosis, Kearns-Sayre syndrome |
| HFE gene-related hereditary hemochromatosis | AD | HFE | liver cirrhosis, debates mellitus, deposition of melanin, increase in serum Fe and ferritin, DCM due to siderosis |
8. Conclusions
Acknowledgement
Abbreviations
| XLDCM | X-linked dilated cardiomyopathy |
| DCM | dilated cardiomyopathy |
| DMD | Duchenne muscular dystrophy |
| BMD | Becker muscular dystrophy |
| DGC | dystrophin glycoprotein complex |
| nNOS | neuronal nitric oxide synthase |
| CK | creatine kinase |
| PCR | polymerase chain reaction |
| IVS | intervening sequence |
| ECG | echocardiography |
| MLPA | multiple ligation probe amplification |
| ACE-I | angiotensin converting enzyme inhibitor |
| TGF | tumor growth factor |
| ARB | angiotensin receptor II blocker |
| AOs | antisense oligonucleotides |
| hiPSCs | human induced pluripotent stem cells |
Conflicts of Interests
References
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.M.; Thambyayah, M.; Gardner-Medwin, D. Prevalence and incidence of Becker muscular dystrophy. Lancet 1991, 337, 1022–1024. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Campbell, K.P.; Kahl, S.D. Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, E.; Hagiwara, Y.; Yoshida, M. Creatine kinase, cell membrane and Duchenne muscular dystrophy. Mol. Cell. Biochem. 1999, 190, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Ball, E.V.; Stenson, P.D.; Phillips, A.D.; Howells, K.; Heywood, S.; Mort, M.E.; Horan, M.P. The Human Gene Mutation Database, at the Institute of Medical Genetics in Cardiff. Available online: http://www.hgmd.org (accessed on 4 June 2015).
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Nakamura, H.; Kimura, E.; Mori-Yoshimura, M.; Komaki, H.; Matsuda, Y.; Goto, K.; Hayashi, Y.K.; Nishino, I.; Takeda, S.; Kawai, M. Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy). Orphanet. J. Rare Dis. 2013, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Ikeda, S.; Owa, M.; Haruta, S.; Yanagisawa, N.; Tanaka, E.; Watanabe, M. A family of Becker’s progressive muscular dystrophy with severe cardiomyopathy. Eur. Neurol. 1987, 27, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Ikeda, S.; Nakamura, A.; Kagoshima, M.; Takeda, S.; Shoji, S.; Yanagisawa, N. Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve 1993, 16, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Melacini, P.; Fanin, M.; Danieli, G.A.; Fasoli, G.; Villanova, C.; Angelini, C.; Vitiello, L.; Miorelli, M.; Buja, G.F.; Mostacciuolo, M.L.; et al. Cardiac involvement in Becker muscular dystrophy. J. Am. Coll. Cardiol. 1993, 22, 1927–1934. [Google Scholar] [CrossRef]
- Piccolo, G.; Azan, G.; Tonin, P.; Arbustini, E.; Gavazzi, A.; Banfi, P.; Mora, M.; Morandi, L.; Tedeschi, S. Dilated cardiomyopathy requiring cardiac transplantation as initial manifestation of Xp21 Becker type muscular dystrophy. Neuromuscul. Disord. 1994, 4, 143–146. [Google Scholar] [CrossRef]
- Yu, Y.; Yamabe, H.; Fujita, H.; Inoue, T.; Yokota, Y.; Nishio, H.; Wada, H.; Matsuo, M.; Yokoyama, M. Cardiac involvement in a family with Becker muscular dystrophy. Intern. Med. 1995, 34, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Melacini, P.; Fanin, M.; Danieli, G.A.; Villanova, C.; Martinello, F.; Miorin, M.; Freda, M.P.; Miorelli, M.; Mostacciuolo, M.L.; Fasoli, G.; et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation 1996, 94, 3168–3175. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.1.; Yoshida, K.; Fukushima, K.; Ueda, H.; Urasawa, N.; Koyama, J.; Yazaki, Y.; Yazaki, M.; Sakai, T.; Haruta, S.; et al. Follow-up of three patients with a large in-frame deletion of exons 45−55 in the Duchenne muscular dystrophy (DMD) gene. J. Clin. Neurosci. 2008, 15, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Berko, B.A.; Swift, M. X-linked dilated cardiomyopathy. N. Engl. J. Med. 1987, 316, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Cau, M.; Ganau, A.; Congiu, R.; Arvedi, G.; Mateddu, A.; Giovanna, M.; Carlo Cianchetti, M.; Realdi, G.; Cao, A.; et al. Deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N. Engl. J. Med. 1993, 329, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Nakamura, A.; Yazaki, M.; Ikeda, S.; Takeda, S. Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum. Mol. Genet. 1998, 7, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Milasin, J.; Muntoni, F.; Severini, G.M.; Bartoloni, L.; Vatta, M.; Krajinovic, M.; Mateddu, A.; Angelini, C.; Camerini, F.; Falaschi, A.; et al. A point mutation in the 5' splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy. Hum. Mol. Genet. 1996, 5, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yan, J.; Buzin, C.H.; Towbin, J.A.; Sommer, S.S. Mutations in the dystrophin gene are associated with sporadic dilated cardiomyopathy. Mol. Genet. Metab. 2002, 77, 119–126. [Google Scholar] [CrossRef]
- Gold, R.; Kress, W.; Meurers, B.; Meng, G.; Reichmann, H.; Müller, C.R. Becker muscular dystrophy: Detection of unusual disease courses by combined approach to dystrophin analysis. Muscle Nerve. 1992, 15, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Muntoni, F. 42nd ENMC Sponsored International Workshop: X-linked cardiomyopathies. 21–23 June 1996, Naarden, The Netherlands. Neuromuscul. Disord. 1997, 7, 397–403. [Google Scholar]
- Bies, R.D.; Maeda, M.; Roberds, S.L.; Holder, E.; Bohlmeyer, T.; Young, J.B.; Campbell, K.P. A 5' dystrophin duplication mutation causes membrane deficiency of alpha-dystroglycan in a family with X-linked cardiomyopathy. J. Mol. Cell Cardiol. 1997, 29, 3175–3188. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A.; Eriksson, B.O.; Kyllerman, M.; Martinsson, T.; Wahlström, J. Dilated cardiomyopathy and the dystrophin gene: An illustrated review. Br. Heart J. 1994, 72, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Lopez, R.; Li, H.; Su, J.; Goytia, V.; Towbin, J.A. Evidence for a dystrophin missense mutation as a cause of X-linked dilated cardiomyopathy. Circulation 1997, 95, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Galié, N.; Merlini, L.; Sewry, C.; Branzi, A.; Muntoni, F. A novel Alu-like element rearranged in the dystrophin gene causes a splicing mutation in a family with X-linked dilated cardiomyopathy. Am. J. Hum. Genet. 1998, 63, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Fitzgerald, K.; Scavena, M.; Gidding, S.; Cox, M.O.; Marks, H.; Flanigan, K.M.; Moore, S.A. Early-progressive dilated cardiomyopathy in a family with Becker muscular dystrophy related to a novel frameshift mutation in the dystrophin gene exon 27. J. Hum. Genet. 2014, 60, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Franz, W.M.; Müller, M.; Müller, O.J.; Herrmann, R.; Rothmann, T.; Cremer, M.; Cohn, R.D.; Voit, T.; Katus, H.A. Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X-linked dilated cardiomyopathy. Lancet 2000, 355, 1781–1785. [Google Scholar] [CrossRef]
- Franz, W.M.; Cremer, M.; Herrmann, R.; Grünig, E.; Fogel, W.; Scheffold, T.; Goebel, H.H.; Kircheisen, R.; Kübler, W.; Voit, T.; et al. X-linked dilated cardiomyopathy. Novel mutation of the dystrophin gene. Ann. N. Y. Acad. Sci. 1995, 752, 470–491. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Diegoli, M.; Morbini, P.; Dal Bello, B.; Banchieri, N.; Pilotto, A.; Magani, F.; Grasso, M.; Narula, J.; Gavazzi, A.; et al. Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 2000, 35, 1760–1768. [Google Scholar] [CrossRef]
- Béroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multi-exon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Ino, H.; Yasuda, T.; Fujino, N.; Uchiyama, K.; Mabuchi, T.; Konno, T.; Kaneda, T.; Fujita, T.; Masuta, E.; et al. Gene mutations in adult Japanese patients with dilated cardiomyopathy. Circ. J. 2005, 69, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Di Lenarda, A.; Porcu, M.; Sinagra, G.; Mateddu, A.; Marrosu, G.; Ferlini, A.; Cau, M.; Milasin, J.; Melis, M.A.; et al. Dystrophin gene abnormalities in two patients with idiopathic dilated cardiomyopathy. Heart 1997, 78, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Melis, M.A.; Ganau, A.; Dubowitz, V. Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X-linked dilated cardiomyopathy. Am. J. Hum. Genet. 1995, 56, 151–157. [Google Scholar] [PubMed]
- Muntoni, F.; Wilson, L.; Marrosu, G.; Marrosu, M.G.; Cianchetti, C.; Mestroni, L.; Ganau, A.; Dubowitz, V.; Sewry, C. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J. Clin. Invest. 1995, 96, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Ikeda, S.; Yazaki, M.; Yoshida, K.; Kobayashi, O.; Yanagisawa, N.; Takeda, S. Up-regulation of the brain and Purkinje-cell forms of dystrophin transcripts, in Becker muscular dystrophy. Am. J. Hum. Genet. 1997, 60, 1555–1558. [Google Scholar] [CrossRef]
- Neri, M.; Valli, E.; Alfano, G.; Bovolenta, M.; Spitali, P.; Rapezzi, C.; Muntoni, F.; Banfi, S.; Perini, G.; Gualandi, F.; Ferlini, A. The absence of dystrophin brain isoform expression in healthy human heart ventricles explains the pathogenesis of 5' X-linked dilated cardiomyopathy. BMC Med. Genet. 2012, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Sewry, C.; Melis, M.A.; Mateddu, A.; Muntoni, F. X-linked dilated cardiomyopathy and the dystrophin gene. Neuromuscul. Disord. 1999, 9, 339–346. [Google Scholar] [CrossRef]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 2009, 119, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.M.; Bandi, S.; Shah, D.D.; Armstrong, G.; Mallela, K.M. Missense mutation Lys18Asn in dystrophin that triggers X-Linked dilated cardiomyopathy decreases protein stability, increases protein unfolding, and perturbs protein structure, but does not affect protein function. PLoS ONE 2014, 9, e110439. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. The role of cytoskeletal proteins in cardiomyopathies. Curr. Opin. Cell. Biol. 1998, 10, 131–139. [Google Scholar] [CrossRef]
- Nakamura, A.; Yoshida, K.; Takeda, S.; Dohi, N.; Ikeda, S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002, 520, 18–24. [Google Scholar] [CrossRef]
- Cox, G.F.; Kunkel, L.M. Dystrophies and heart disease. Curr. Opin. Cardiol. 1997, 12, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Perloff, J.K.; Roberts, W.C.; de Leon, A.C., Jr.; O'Doherty, D. The distinct electrocardiogram of Duchenne’s progressive muscular dystrophy. An electrocardiographic-pathologic correlative study. Am. J. Med. 1967, 42, 179–188. [Google Scholar] [CrossRef]
- Sanyal, S.K.; Johnson, W.W.; Thapar, M.K.; Pitner, S.E. An ultrastructural basis for electrocardiographic alterations associated with Duchenne’s progressive muscular dystrophy. Circulation 1978, 57, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Perloff, J.K.; de Leon, A.C., Jr.; O'Doherty, D. The cardiomyopathy of progressive muscular dystrophy. Circulation 1966, 33, 625–648. [Google Scholar] [CrossRef] [PubMed]
- Frankel, K.A.; Rosser, R.J. The pathology of the heart in progressive muscular dystrophy: epimyocardial fibrosis. Hum. Pathol. 1976, 7, 375–386. [Google Scholar] [CrossRef]
- James, T.N. Observation on the cardiovascular involvement, including the cardiac conduction system, in progressive muscular dystrophy. Am. Heart J. 1962, 63, 48–56. [Google Scholar] [CrossRef]
- Finsterer, J.; Stöllberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.G.; Evans, E.B.; Vignos, P.J. Echocardiographic evaluation of left ventricular function in Duchenne’s muscular dystrophy. Am. J. Med. 1980, 69, 248–254. [Google Scholar] [CrossRef]
- D’Orsogna, L.; O’Shea, J.P.; Miller, G. Cardiomyopathy of Duchenne muscular dystrophy. Pediatr. Cardiology 1988, 9, 205–213. [Google Scholar] [CrossRef]
- Perloff, J.K. Cardiac rhythm and conduction in Duchenne’s muscular dystrophy: a prospective study of 20 patients. J. Am. Coll. Cardiol. 1984, 3, 1263–1268. [Google Scholar] [CrossRef]
- Janssen, B.; Hartmann, C.; Scholz, V.; Jauch, A.; Zschocke, J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics 2005, 6, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Beggs, A.H. Multiplex PCR for identifying DMD gene deletions. Curr. Protoc. Hum. Genet. 2006. [Google Scholar] [CrossRef]
- Hermans, M.C.; Pinto, Y.M.; Merkies, I.S.; de Die-Smulders, C.E.; Crijns, H.J.; Faber, C.G. Hereditary muscular dystrophies and the heart. Neuromuscul. Disord. 2010, 20, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.C. GeneTable of Neuromuscular Disorders. Available online: http://www.musclegenetable.fr/ (accessed on 4 June 2015).
- Badorff, C.; Lee, G.H.; Lamphear, B.J.; Martone, M.E.; Campbell, K.P.; Rhoads, R.E.; Knowlton, K.U. Enteroviral protease 2A cleaves dystrophin: Evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999, 5, 320–326. [Google Scholar] [PubMed]
- Xiong, D.; Lee, G.H.; Badorff, C.; Dorner, A.; Lee, S.; Wolf, P.; Knowlton, K.U. Dystrophin deficiency markedly increases enterovirus-induced cardiomyopathy: a genetic predisposition to viral heart disease. Nat. Med. 2002, 8, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Bach, J.R.; Minami, R. Cardioprotection for Duchenne’s muscular dystrophy. Am. Heart J. 1999, 137, 895–902. [Google Scholar] [CrossRef]
- Weber, K.T. Anigiotensin II and connective tissue: Homeostasis and reciprocal regulation. Regul. Peptides 1999, 82, 1–17. [Google Scholar] [CrossRef]
- Kajimoto, H.; Ishigaki, K.; Okumura, K.; Tomimatsu, R.; Nakamzawa, M.; Saito, K.; Osawa, M.; Nakanishi, T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Cir. J. 2006, 70, 991–994. [Google Scholar] [CrossRef]
- Shirai, K.; Watanabe, K.; Ma, M.; Wahed, M.I.; Inoue, M.; Saito, Y.; Suresh, P.S.; Kashimura, T.; Tachikawa, H.; Kodama, M.; et al. Effects of angiotensin-II receptor blocker candesartan cilexetil in rats with dilated cardiomyopathy. Mol. Cell. Biochem. 2005, 269, 137–142. [Google Scholar] [CrossRef]
- De Mello, W.C.; Specht, P. Chronic blockade of angiotensin II AT1-receptors increased cell-to-cell communication, reduced fibrosis and improved impulse propagation in the failing heart. J. Renin Angiotensin Aldosterone Syst. 2006, 7, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Kass, D.A.; Thompson, W.R.; Wagner, K.R. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am. J. Cardiovasc. Drugs 2011, 11, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Cripe, L. Treatment of dystrophin cardiomyopathies. Nat. Rev. Cardiol. 2014, 11, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Markham, L.W.; Kinnett, K.; Wong, B.L.; Woodrow Benson, D.; Cripe, L.H. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul. Disord. 2008, 18, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Schram, G.; Fournier, A.; Leduc, H.; Dahdah, N.; Therien, J.; Vanasse, M.; Khairy, P. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2013, 61, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Romfh, A.; McNally, E.M. Cardiac assessment in Duchenne and Becker muscular dystrophies. Curr. Heart Fail. Rep. 2010, 7, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Ahn-Veelken, L.; Schlensak, C.; Berchtold-Herz, M.; Sarai, K.; Schaefer, M.; van de Loo, A.; Beyersdorf, F. Left ventricular reduction for idiopathic dilated cardiomyopathy as alternative to transplant-truth or dare? Thorac. Cardiovasc. Surg. 2001, 49, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.D.; Jefferies, J.L.; Sawnani, H.; Wong, B.L.; Gardner, A.; Del Corral, M.; Lorts, A.; Morales, D.L. Implantation of the HeartMate II and HeartWare left ventricular assist devices in patients with duchenne muscular dystrophy: Lessons learned from the first applications. ASAIO J. 2014, 60, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Iodice, F.; Testa, G.; Averardi, M.; Brancaccio, G.; Amodeo, A.; Cogo, P. Implantation of a left ventricular assist device as a destination therapy in Duchenne muscular dystrophy patients with end stage cardiac failure: management and lessons learned. Neuromuscul. Disord. 2015, 25, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Leprince, P.; Heloire, F.; Eymard, B.; Léger, P.; Duboc, D.; Pavie, A. Successful bridge to transplantation in a patient with Becker muscular dystrophy-associated cardiomyopathy. J. Heart Lung Transplant. 2002, 21, 822–824. [Google Scholar] [CrossRef]
- Wu, R.S.; Gupta, S.; Brown, R.N.; Yancy, C.W.; Wald, J.W.; Kaiser, P.; Kirklin, N.M.; Patel, P.C.; Markham, D.W.; Drazner, M.H.; et al. Clinical outcomes after cardiac transplantation in muscular dystrophy patients. J. Heart Lung Transplant. 2010, 29, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Blankinship, M.J.; Gregorevic, P.; Chamberlain, J.S. Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol. Ther. 2006, 13, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Takeda, S. Current challenges and future directions in recombinant AAV-mediated gene therapy of Duchenne muscular dystrophy. Pharmaceuticals 2013, 6, 813–836. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Duan, D. Progress in gene therapy of dystrophic heart disease. Gene Ther. 2012, 19, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Taghli-Lamallem, O.; Jagla, K.; Chamberlain, J.S.; Bodmer, R. Mechanical and non-mechanical functions of Dystrophin can prevent cardiac abnormalities in Drosophila. Exp. Gerontol. 2014, 49, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Takeda, S. Exon-skipping therapy for Duchenne muscular dystrophy. Neuropathology 2009, 29, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.A.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Arechavala-Gomezam, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Heemskerk, H.; de Winter, C.; van Kuik, P.; Heuvelmans, N.; Sabatelli, P.; Rimessi, P.; Braghettam, P.; van Ommenm, G.J.; de Kimpem, S.; Ferlini, A.; et al. Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther. 2010, 18, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.; Kalra, S.; Anderson, D.; George, V.; Ritso, M.; Laval, S.H.; Barresi, R.; Aartsma-Rus, A.; Lochmüller, H.; Denning, C. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013, 22, 2714–2724. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.; Turner, I.; Yasuda, S.; Martindale, J.; Davis, J.; Shillingford, M.; Kornegay, J.N.; Metzger, J.M. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Invest. 2010, 120, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals 2015, 8, 303-320. https://doi.org/10.3390/ph8020303
Nakamura A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals. 2015; 8(2):303-320. https://doi.org/10.3390/ph8020303
Chicago/Turabian StyleNakamura, Akinori. 2015. "X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy" Pharmaceuticals 8, no. 2: 303-320. https://doi.org/10.3390/ph8020303
APA StyleNakamura, A. (2015). X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals, 8(2), 303-320. https://doi.org/10.3390/ph8020303