
Seatbelts in CAR therapy: How Safe Are CARS?

Abstract
:1. Introduction
2. CAR T-Cells in the Autologous Setting
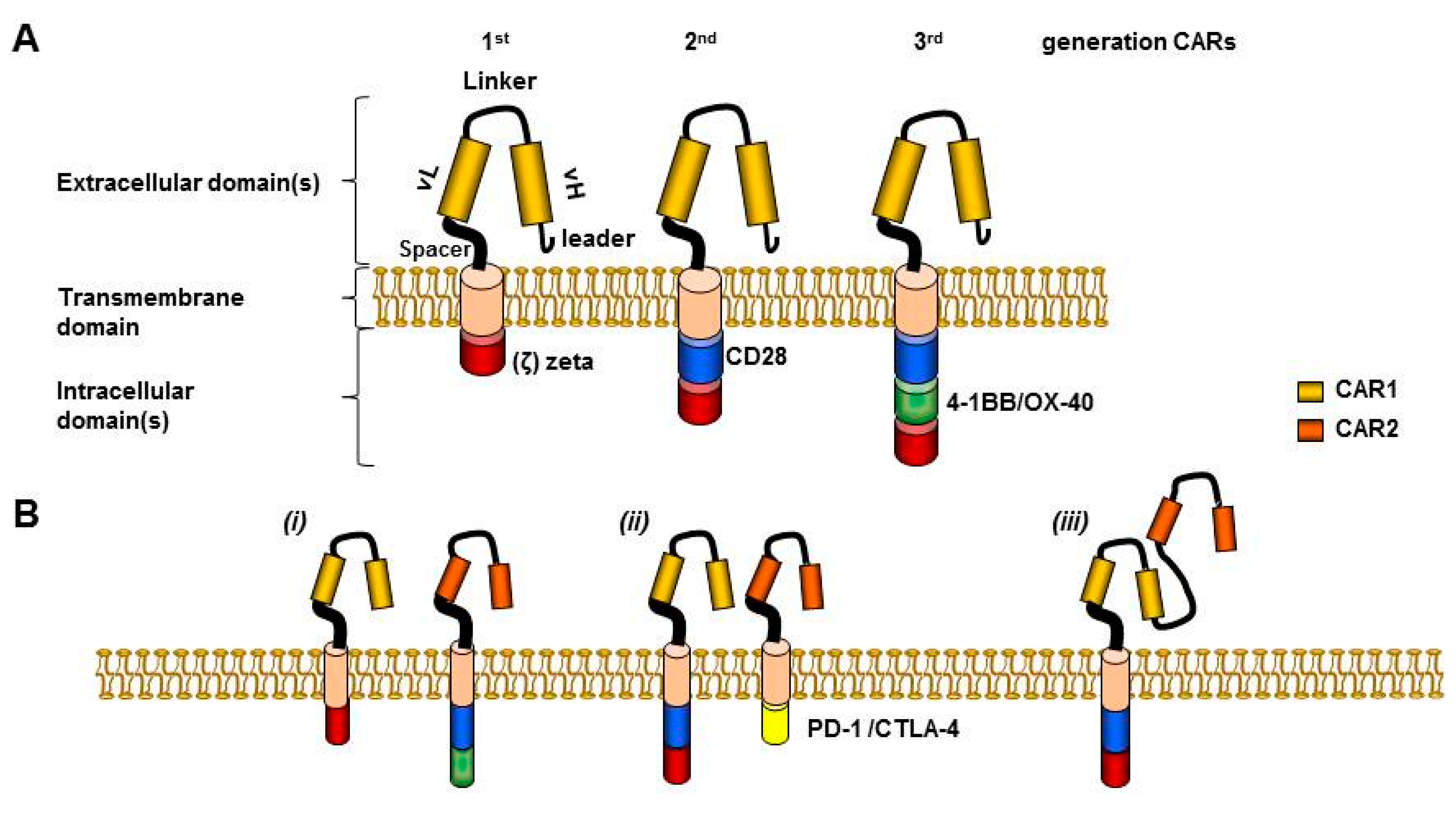
3. Side Effects from CAR T-Cells Infused in the Autologous Setting
4. Recommendations from the Recombinant DNA Advisory Committee of the National Institute of Health
5. Therapeutic Approach to Cytokine Release Syndrome
6. Strategies to Ensure Safety
6.1. Suicide Gene Applications

{kind=link}
| Mechanism of Action (Source) | Activating Agent | Mechanism of Action | Percentage of Transduced Cell Elimination in Patients | Advantages | Drawback(s) | Refs. |
|---|---|---|---|---|---|---|
| Metabolic (viral) e.g., HSV-TK | Ganciclovir | -Ganciclovir triphosphate mediated Interference of DNA synthesis; -Apoptosis through CD95 aggregation | NR, in vivo depletion of alloreactive cells | -Gradual onset -Eliminates alloreactive cells when used in allo setting | -Preclude therapeutic use of ganciclovir -Immunogenic | [61,71,72] |
| Dimerization inducing (human) e.g., iCasp9 | Non-therapeutic small molecule dimerizer | -iCasp9 dimerization and activation of downstream caspases resulting in apoptosis | Incomplete, but >=90% with in vivo depletion of alloreactive cells | -Rapid onset -Eliminates alloreactive cells, and non-immunogenic when used in allo setting -Uses non-therapeutic agent | -Kills ≥90% of cells -Uses non commercially available dimerizer | [64,73] |
| Therapeutic mAb mediated (human) e.g., CD20 | mAb | -Antibody/complement dependent cellular cytotoxicity | Not done | -Rapid onset -Non-immunogenic when used in allo setting | On-target toxicity from each specific mAb used needs to be considered | [66,67,68,69,70,74] |
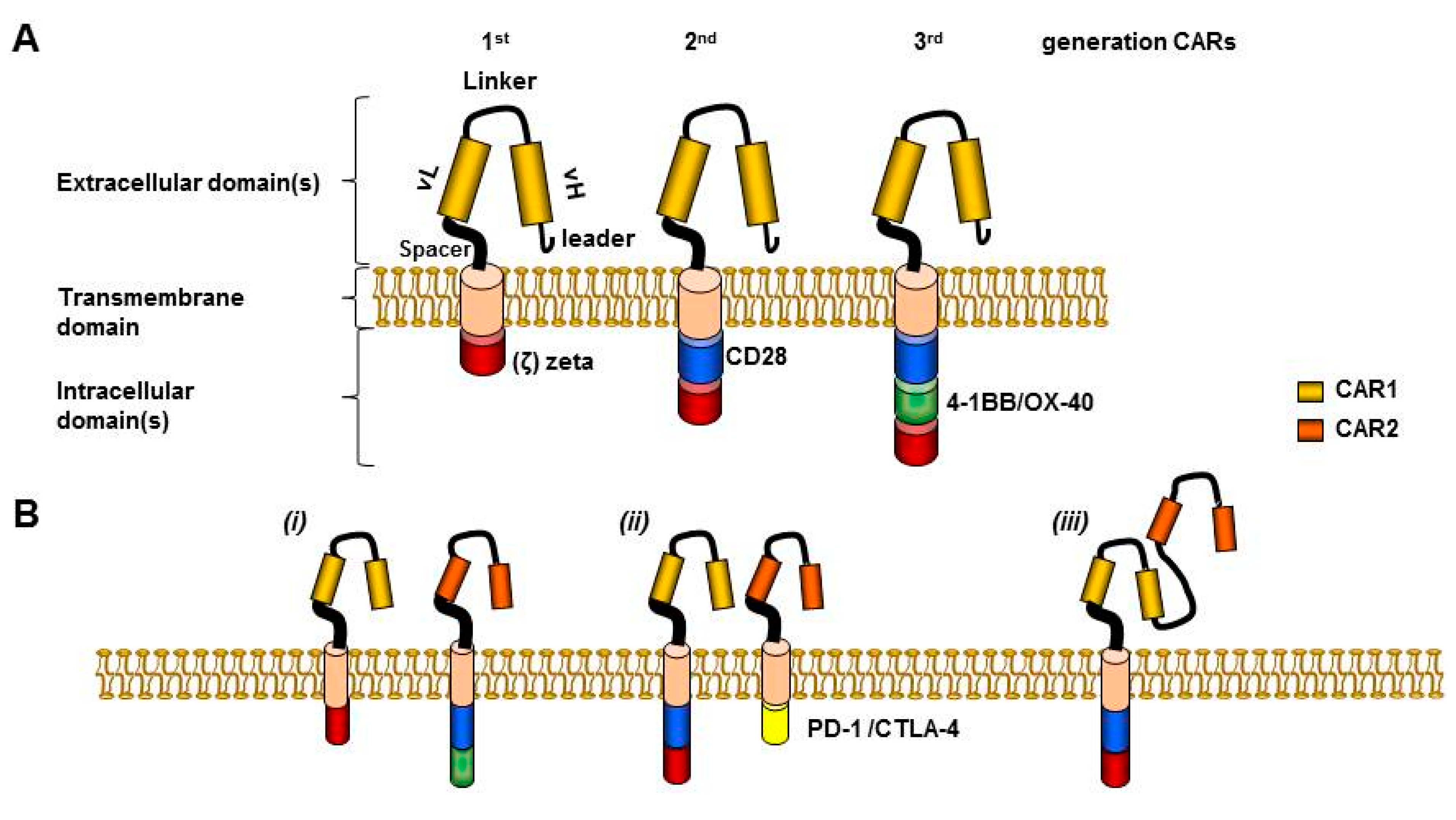
6.2. Dual Targeting Strategies to Ensure Safety
7. CAR T-Cells in the Allogeneic Setting
8. Conclusions
Acknowledgement
Author Contributions
Conflict of Interests
References
- Copelan, E.A. Hematopoietic stem-cell transplantation. New Engl. J. Med. 2006, 354, 1813–1826. [Google Scholar] [PubMed]
- Rambaldi, A.; Biagi, E.; Bonini, C.; Biondi, A.; Introna, M. Cell-based strategies to manage leukemia relapse: Efficacy and feasibility of immunotherapy approaches. Leukemia 2015, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ringden, O.; Labopin, M.; Gorin, N.C.; Schmitz, N.; Schaefer, U.W.; Prentice, H.G.; Bergmann, L.; Jouet, J.P.; Mandelli, F.; Blaise, D.; et al. Is there a graft-versus-leukaemia effect in the absence of graft-versus-host disease in patients undergoing bone marrow transplantation for acute leukaemia? Br. J. Haematol. 2000, 111, 1130–1137. [Google Scholar]
- De Bueger, M.; Bakker, A.; Van Rood, J.J.; Van der Woude, F.; Goulmy, E. Tissue distribution of human minor histocompatibility antigens. Ubiquitous versus restricted tissue distribution indicates heterogeneity among human cytotoxic t lymphocyte-defined non-mhc antigens. J. Immunol. 1992, 149, 1788–1794. [Google Scholar]
- Warren, E.H.; Fujii, N.; Akatsuka, Y.; Chaney, C.N.; Mito, J.K.; Loeb, K.R.; Gooley, T.A.; Brown, M.L.; Koo, K.K.; Rosinski, K.V.; et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood 2010, 115, 3869–3878. [Google Scholar]
- Thomas, S.; Stauss, H.J.; Morris, E.C. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology 2010, 129, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and t-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. New Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. Cd19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar]
- Brentjens, R.J.; Riviere, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous cd19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of b-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19–28z car t cell therapy in b cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-cd19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing cd19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating b-cell cancer with T cells expressing anti-cd19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing erbb2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J.; Zaia, J.; Rosenberg, S.A.; June, C.H.; Dotti, G.; Kahn, J.; Cooper, L.J.N.; Corrigan-Curay, J.; Strome, S.E. Considerations for the clinical application of chimeric antigen receptor T cells: Observations from a recombinant DNA advisory committee symposium held June 15, 2010. Cancer Res. 2011, 71, 3175–3181. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic t lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase i study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with caix car-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. Cd28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011, 121, 1822–1826. [Google Scholar]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J., Jr. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of erbb2 and muc1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schonfeld, K.; Koch, J.; Dotti, G.; et al. Tancar: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar]
- Hombach, A.; Sent, D.; Schneider, C.; Heuser, C.; Koch, D.; Pohl, C.; Seliger, B.; Abken, H. T-cell activation by recombinant receptors: Cd28 costimulation is required for interleukin 2 secretion and receptor-mediated t-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res. 2001, 61, 1976–1982. [Google Scholar] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4–1bb signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Snyder, K.M.; Suhoski, M.M.; Maus, M.V.; Kapoor, V.; June, C.H.; Mackall, C.L. 4–1bb is superior to cd28 costimulation for generating cd8+ cytotoxic lymphocytes for adoptive immunotherapy. J. Immunol. 2007, 179, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.C.; Latouche, J.B.; Krause, A.; Heston, W.D.; Bander, N.H.; Sadelain, M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human t-lymphocyte cytotoxicity and proliferation directed by a single chimeric tcrzeta /cd28 receptor. Nat. Biotech. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transpl. 2010, 16, 1245–1256. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both cd28 and 4–1bb domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Pure, E.; Milone, M.C.; et al. Multifactorial t-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase ix: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar]
- Hinrichs, C.S.; Borman, Z.A.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Huang, J.; Klebanoff, C.A.; Johnson, L.A.; Kerkar, S.P.; Yang, S.; et al. Human effector cd8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 2011, 117, 808–814. [Google Scholar]
- Zhang, Y.; Joe, G.; Hexner, E.; Zhu, J.; Emerson, S.G. Host-reactive cd8+ memory stem cells in graft-versus-host disease. Nat. Med. 2005, 11, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent t cell memory in primates. J. Clin. Invest. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. Il-7 and il-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar]
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting vegfr-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Invest. 2010, 120, 3953–3968. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Kloss, C.C.; Gunset, G.; Sadelain, M. Cd19 car-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PloS ONE 2013, 8, e61338. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Sommermeyer, D.; Hudecek, M.; Berger, M.; Balakrishnan, A.; Paszkiewicz, P.J.; Kosasih, P.L.; Rader, C.; Riddell, S.R. Safety of targeting ror1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 2015, 3, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Dutour, A.; Marin, V.; Pizzitola, I.; Valsesia-Wittmann, S.; Lee, D.; Yvon, E.; Finney, H.; Lawson, A.; Brenner, M.; Biondi, A.; et al. In vitro and in vivo antitumor effect of anti-CD33 chimeric receptor-expressing ebv-ctl against cd33 acute myeloid leukemia. Adv. Hematol. 2012, 2012, 683065. [Google Scholar]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. Cd44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.C.; Bretzlaff, W.; et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013, 122, 3138–3148. [Google Scholar]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. Pd-1- and ctla-4-based inhibitory chimeric antigen receptors (icars) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of scid-x1 patients. J. Clin. Invest. 2008, 118, 3143–3150. [Google Scholar]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra153. [Google Scholar]
- Ciceri, F.; Bonini, C.; Marktel, S.; Zappone, E.; Servida, P.; Bernardi, M.; Pescarollo, A.; Bondanza, A.; Peccatori, J.; Rossini, S.; et al. Antitumor effects of hsv-tk-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood 2007, 109, 4698–4707. [Google Scholar]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.L.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the tk007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar]
- Tiraby, M.; Cazaux, C.; Baron, M.; Drocourt, D.; Reynes, J.P.; Tiraby, G. Concomitant expression of E. Coli cytosine deaminase and uracil phosphoribosyltransferase improves the cytotoxicity of 5-fluorocytosine. FEMS Microbiol. Lett. 1998, 167, 41–49. [Google Scholar]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. New Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar]
- Clackson, T.; Yang, W.; Rozamus, L.W.; Hatada, M.; Amara, J.F.; Rollins, C.T.; Stevenson, L.F.; Magari, S.R.; Wood, S.A.; Courage, N.L.; et al. Redesigning an fkbp-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 10437–10442. [Google Scholar]
- Griffioen, M.; van Egmond, E.H.; Kester, M.G.; Willemze, R.; Falkenburg, J.H.; Heemskerk, M.H. Retroviral transfer of human CD20 as a suicide gene for adoptive t-cell therapy. Haematologica 2009, 94, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Introna, M.; Barbui, A.M.; Bambacioni, F.; Casati, C.; Gaipa, G.; Borleri, G.; Bernasconi, S.; Barbui, T.; Golay, J.; Biondi, A.; et al. Genetic modification of human T cells with cd20: A strategy to purify and lyse transduced cells with anti-CD20 antibodies. Human Gene Ther. 2000, 11, 611–620. [Google Scholar]
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-transduced t lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Human Gene Ther. 2004, 15, 63–76. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Kieback, E.; Charo, J.; Sommermeyer, D.; Blankenstein, T.; Uckert, W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc. Natl .Acad. Sci. USA 2008, 105, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. Hsv-tk gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar]
- Tiberghien, P.; Ferrand, C.; Lioure, B.; Milpied, N.; Angonin, R.; Deconinck, E.; Certoux, J.M.; Robinet, E.; Saas, P.; Petracca, B.; et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a t-cell-depleted allogeneic marrow graft. Blood 2001, 97, 63–72. [Google Scholar]
- Zhou, X.; Di Stasi, A.; Tey, S.K.; Krance, R.A.; Martinez, C.; Leung, K.S.; Durett, A.G.; Wu, M.F.; Liu, H.; Leen, A.M.; et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014, 123, 3895–3905. [Google Scholar]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar]
- Moolten, F.L. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: Paradigm for a prospective cancer control strategy. Cancer Res. 1986, 46, 5276–5281. [Google Scholar] [PubMed]
- Beltinger, C.; Fulda, S.; Kammertoens, T.; Meyer, E.; Uckert, W.; Debatin, K.M. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc. Natl. Acad. Sci. USA 1999, 96, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.M.; Wandless, T.J.; Schreiber, S.L.; Crabtree, G.R. Controlling signal transduction with synthetic ligands. Science 1993, 262, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Iuliucci, J.D.; Oliver, S.D.; Morley, S.; Ward, C.; Ward, J.; Dalgarno, D.; Clackson, T.; Berger, H.J. Intravenous safety and pharmacokinetics of a novel dimerizer drug, ap1903, in healthy volunteers. J. Clin. Pharmacol. 2001, 41, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Greco, R.; Lupo-Stanghellini, M.T.; Vago, L.; Bonini, C. Use of tk-cells in haploidentical hematopoietic stem cell transplantation. Curr. Opin. Hematol. 2012, 19, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Perna, S.K.; Zanussi, M.; Mazzi, B.; Barlassina, C.; Stanghellini, M.T.; Perrelli, N.F.; Cosentino, C.; Torri, F.; Angius, A.; et al. Loss of mismatched hla in leukemia after stem-cell transplantation. New Engl. J. Med. 2009, 361, 478–488. [Google Scholar]
- Traversari, C.; Marktel, S.; Magnani, Z.; Mangia, P.; Russo, V.; Ciceri, F.; Bonini, C.; Bordignon, C. The potential immunogenicity of the tk suicide gene does not prevent full clinical benefit associated with the use of tk-transduced donor lymphocytes in hsct for hematologic malignancies. Blood 2007, 109, 4708–4715. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering cd19-specific t lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Budde, L.E.; Berger, C.; Lin, Y.; Wang, J.; Lin, X.; Frayo, S.E.; Brouns, S.A.; Spencer, D.M.; Till, B.G.; Jensen, M.C.; et al. Combining a cd20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of t cell adoptive immunotherapy for lymphoma. PloS ONE 2013, 8, e82742. [Google Scholar]
- Cornelissen, J.J.; Gratwohl, A.; Schlenk, R.F.; Sierra, J.; Bornhauser, M.; Juliusson, G.; Racil, Z.; Rowe, J.M.; Russell, N.; Mohty, M.; et al. The european leukemianet aml working party consensus statement on allogeneic hsct for patients with aml in remission: An integrated-risk adapted approach. Nat. Rev. Clin. Oncol. 2012, 9, 579–590. [Google Scholar]
- Amrolia, P.J.; Muccioli-Casadei, G.; Yvon, E.; Huls, H.; Sili, U.; Wieder, E.D.; Bollard, C.; Michalek, J.; Ghetie, V.; Heslop, H.E.; et al. Selective depletion of donor alloreactive T cells without loss of antiviral or antileukemic responses. Blood 2003, 102, 2292–2299. [Google Scholar] [PubMed]
- Melenhorst, J.J.; Leen, A.M.; Bollard, C.M.; Quigley, M.F.; Price, D.A.; Rooney, C.M.; Brenner, M.K.; Barrett, A.J.; Heslop, H.E. Allogeneic virus-specific T cells with hla alloreactivity do not produce gvhd in human subjects. Blood 2010, 116, 4700–4702. [Google Scholar] [CrossRef] [PubMed]
- Van Loenen, M.M.; de Boer, R.; van Liempt, E.; Meij, P.; Jedema, I.; Falkenburg, J.H.; Heemskerk, M.H. A good manufacturing practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica 2014, 99, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived cd19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar]
- Rubio, M.T.; Moreira-Teixeira, L.; Bachy, E.; Bouillie, M.; Milpied, P.; Coman, T.; Suarez, F.; Marcais, A.; Sibon, D.; Buzyn, A.; et al. Early posttransplantation donor-derived invariant natural killer t-cell recovery predicts the occurrence of acute graft-versus-host disease and overall survival. Blood 2012, 120, 2144–2154. [Google Scholar]
- Lamb, L.S., Jr.; Lopez, R.D. Gammadelta T cells: A new frontier for immunotherapy? Biol. Blood Marrow Transpl. 2005, 11, 161–168. [Google Scholar] [CrossRef]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific t-cells expressing polyclonal repertoire of endogenous gammadelta t-cell receptors and introduced cd19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing t cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minagawa, K.; Zhou, X.; Mineishi, S.; Di Stasi, A. Seatbelts in CAR therapy: How Safe Are CARS? Pharmaceuticals 2015, 8, 230-249. https://doi.org/10.3390/ph8020230
Minagawa K, Zhou X, Mineishi S, Di Stasi A. Seatbelts in CAR therapy: How Safe Are CARS? Pharmaceuticals. 2015; 8(2):230-249. https://doi.org/10.3390/ph8020230
Chicago/Turabian StyleMinagawa, Kentaro, Xiaoou Zhou, Shin Mineishi, and Antonio Di Stasi. 2015. "Seatbelts in CAR therapy: How Safe Are CARS?" Pharmaceuticals 8, no. 2: 230-249. https://doi.org/10.3390/ph8020230
APA StyleMinagawa, K., Zhou, X., Mineishi, S., & Di Stasi, A. (2015). Seatbelts in CAR therapy: How Safe Are CARS? Pharmaceuticals, 8(2), 230-249. https://doi.org/10.3390/ph8020230