
Design and Synthesis of Novel N-Arylsulfonyl-3-(2-yl-ethanone)-6-methylindole Derivatives as Inhibitors of HIV-1 Replication

Abstract
:1. Introduction
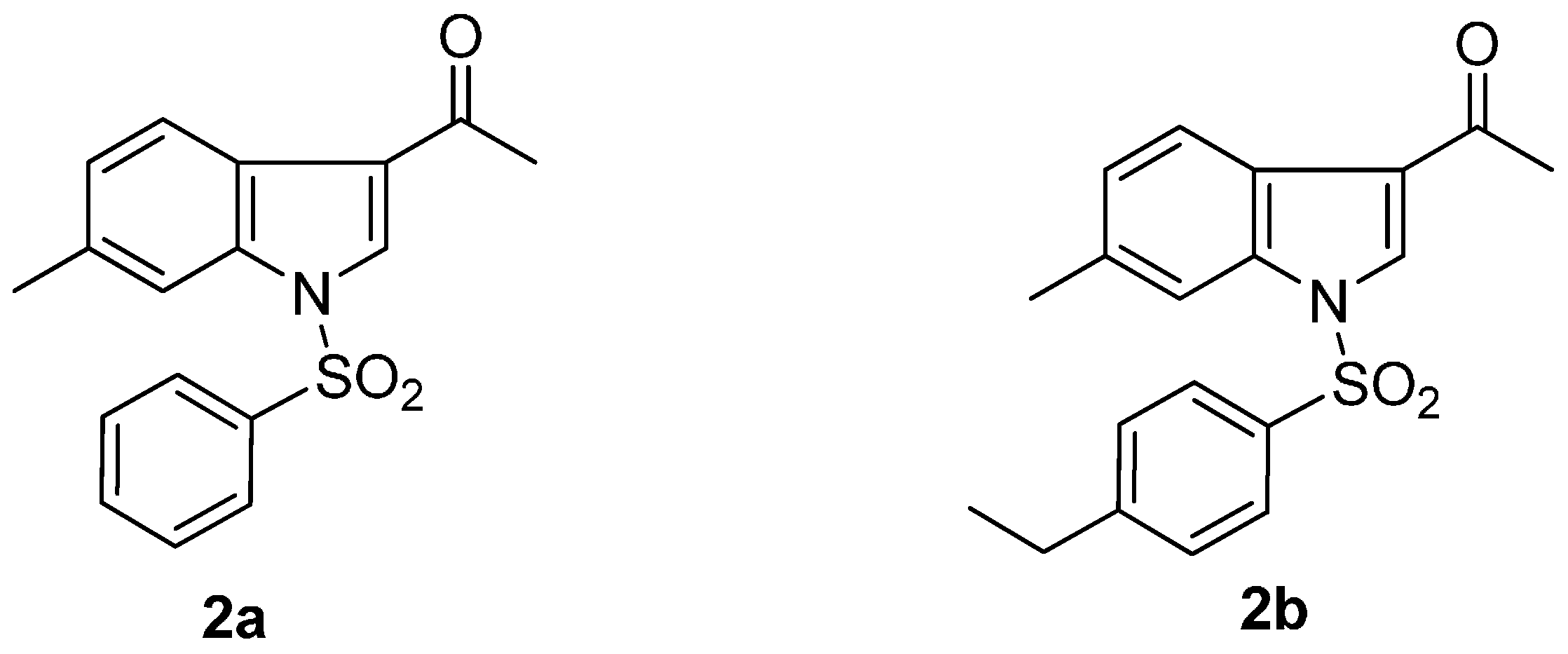
2. Results and Discussion
2.1. Chemistry
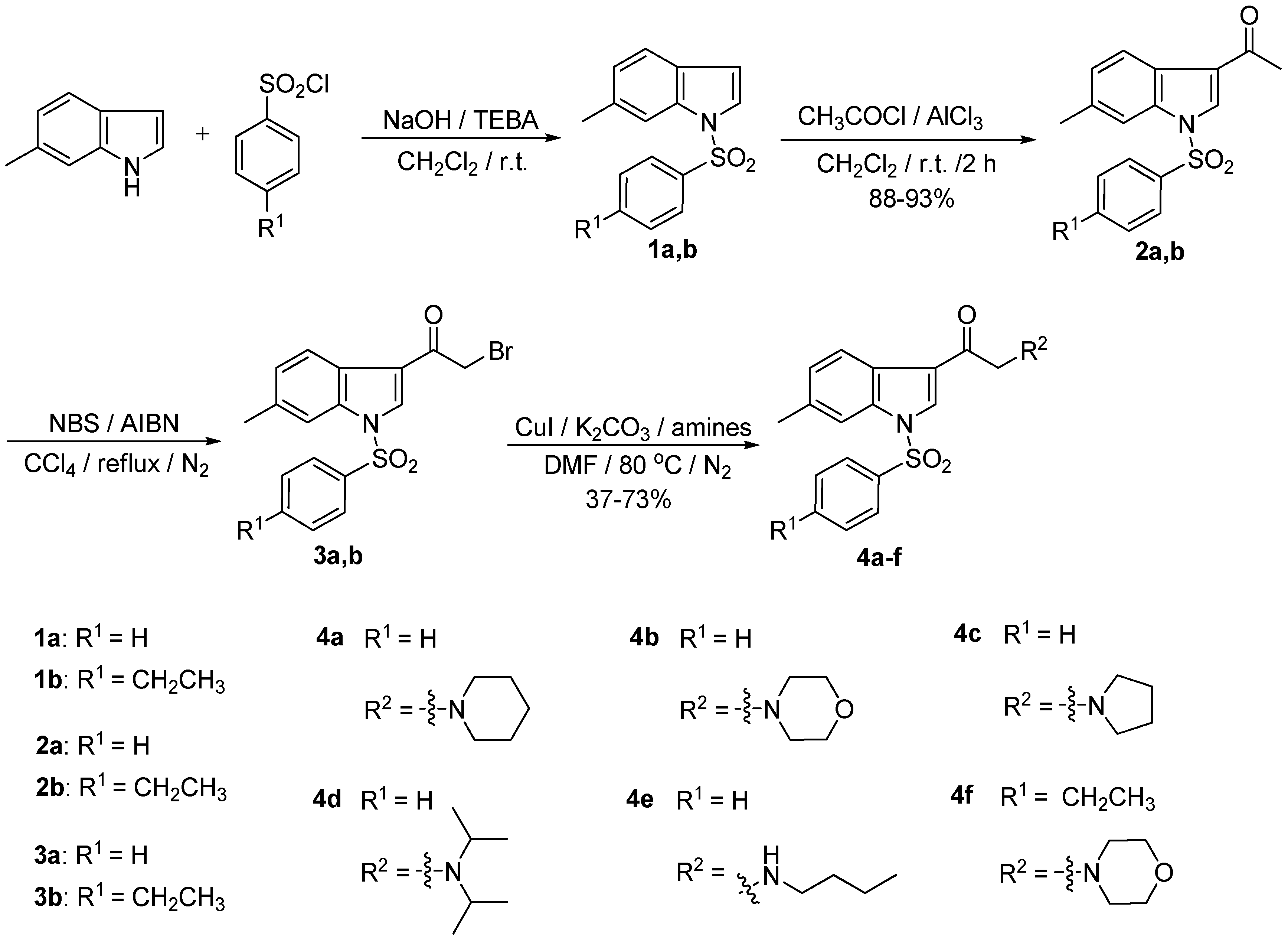
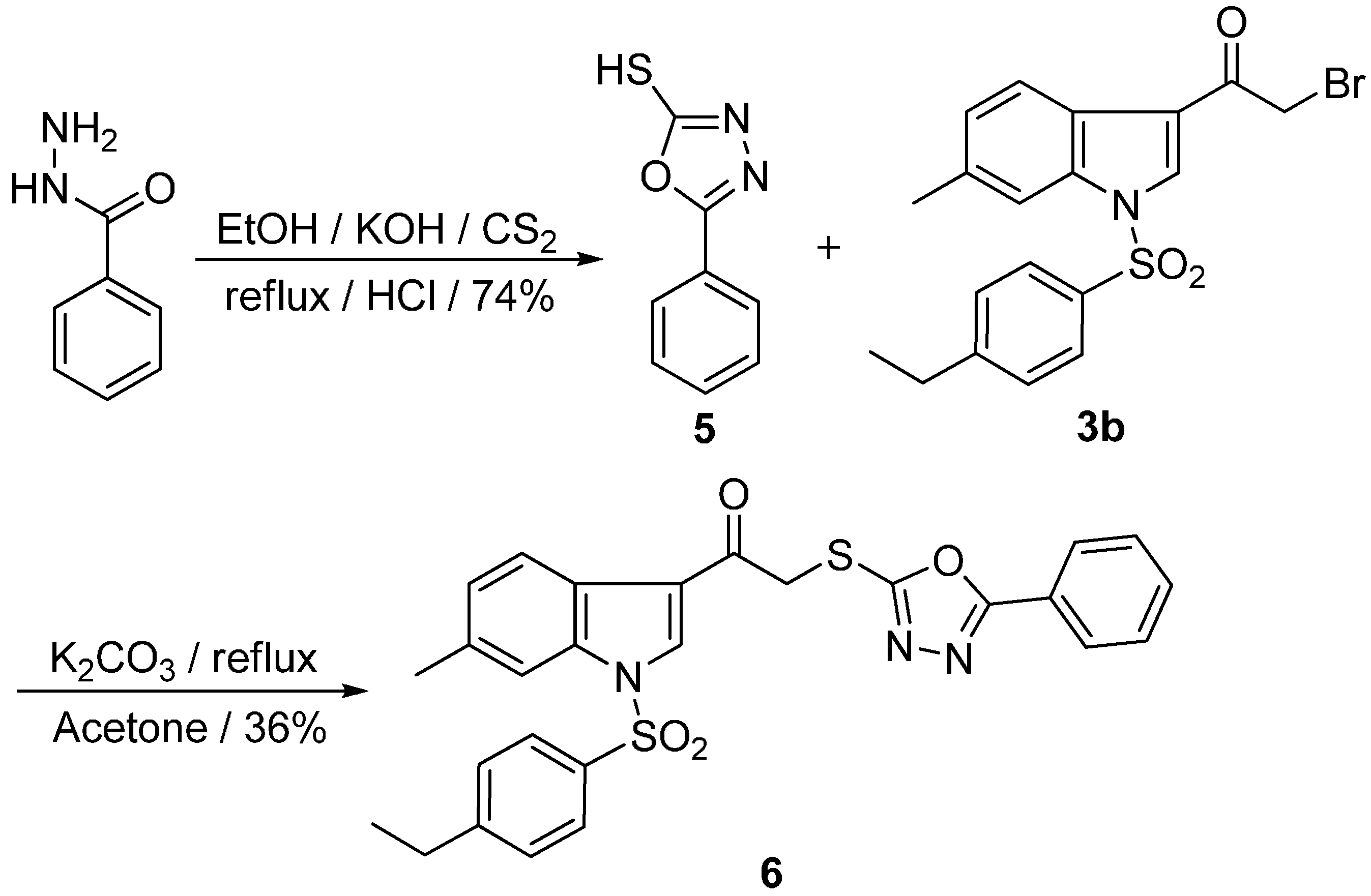
2.2. Biological Activity

{kind=link}
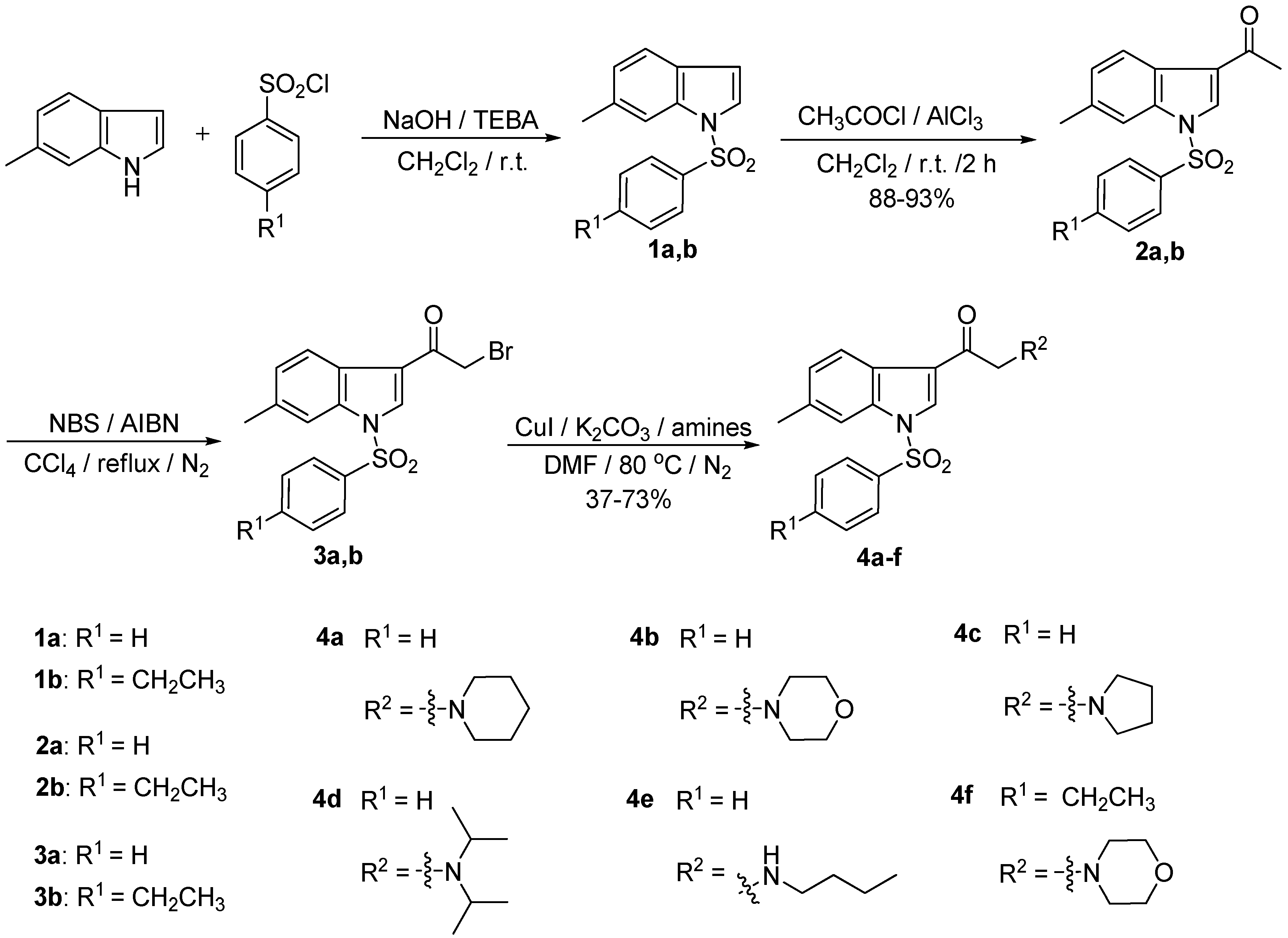{kind=link}
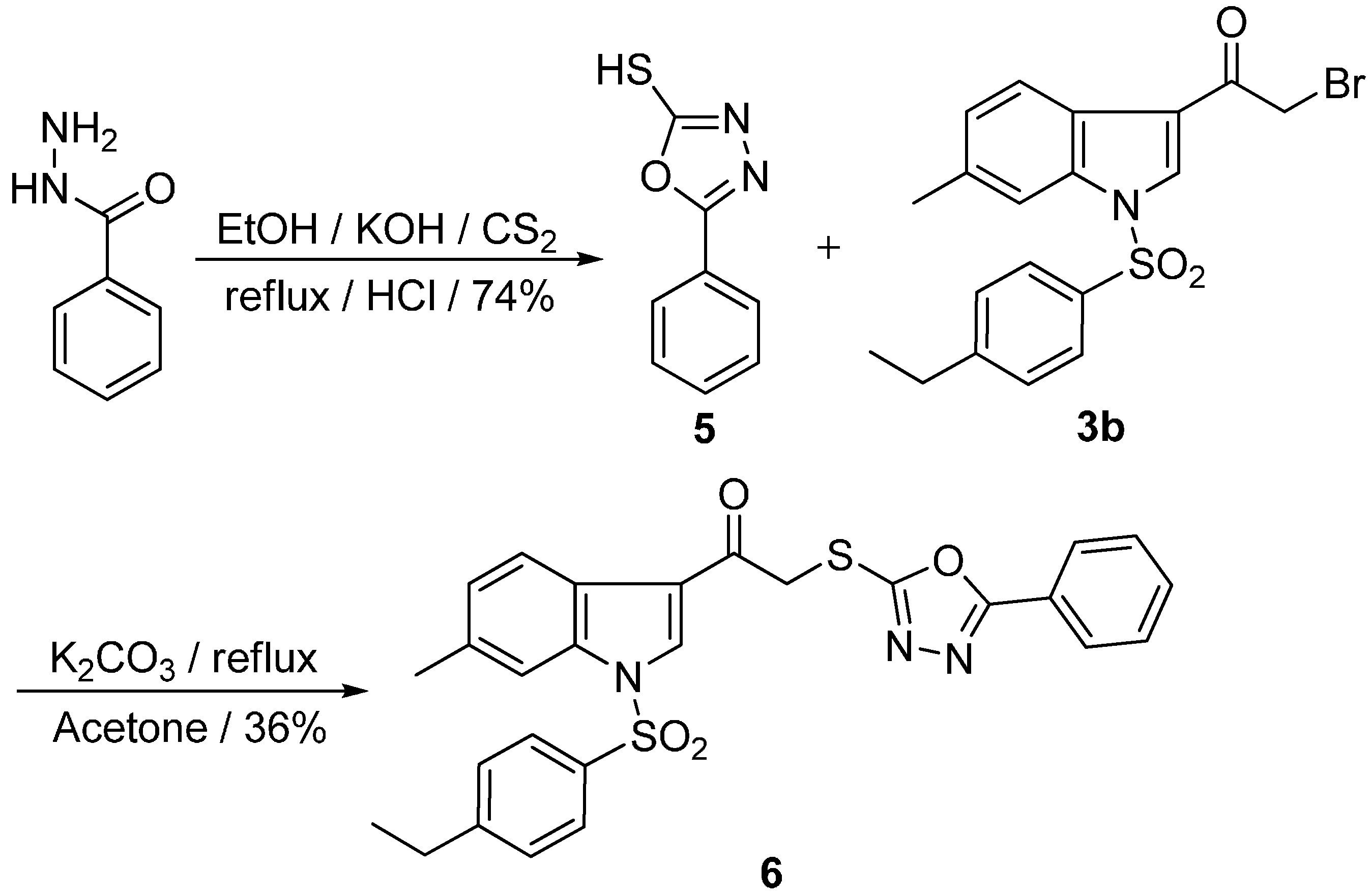{kind=link}
| Compounds | CC50 b(µM) | EC50 c(µM) | TI d |
|---|---|---|---|
| 4a | >504.41 | 32.78 | >15.38 |
| 4b | >501.91 | 56.56 | >8.87 |
| 4c | 225.82 | 106.02 | 2.12 |
| 4d | 220.02 | 17.94 | 12.26 |
| 4e | 43.79 | 29.96 | 1.46 |
| 4f | >468.91 | 9.42 | >49.77 |
| 6 | 309.31 | 4.62 | 66.95 |
| AZT e | 4263.84 | 0.01212 | 351801.98 |
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Synthesis of 4a–f
3.3. General Procedure for the Synthesis of 6
3.4. Anti-HIV-1 Activity Assay
3.4.1. Cells and Virus
3.4.2. MTT-based Cytotoxicity Assay
3.4.3. Syncytia Assay
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Richman, D.D. HIV chemotherapy. Nature 2001, 410, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Clavel, F.; Hance, A.J. HIV Drug resistance. N. Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar]
- Gupta, R.; Hill, A.; Sawyer, A.W.; Pillay, D. Emergence of drug resistance in HIV type 1-infected patients after receipt of first-line highly active antiretroviral therapy: A systematic review of clinical trials. Clin. Infect. Dis. 2008, 47, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lv, M. Developments of indoles as anti-HIV-1 inhibitors. Curr. Pharm. Des. 2009, 15, 2120–2148. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Liu, W.Q.; Xu, H.; Yang, L.M.; Lv, M.; Zheng, Y.T. Anti human immunodeficiency virus-1 (HIV-1) agents 3: Synthesis and in vitro anti-HIV-1 activity of some N-arylsulfonylindoles. Chem. Pharm. Bull. 2009, 57, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.Q.; Huang, N.; Xu, H.; Yang, L.M.; Lv, M.; Zheng, Y.T. Anti HIV-1 agents 5: Synthesis and anti-HIV-1 activity of some N-arylsulfonyl-3-acetylindoles in vitro. Bioorg. Med. Chem. Lett. 2010, 20, 3534–3536. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, W.Q.; Fan, L.L.; Chen, Y.; Yang, L.M.; Lv, L.; Zheng, Y.T. Synthesis and HIV-1 integrase inhibition activity of some N-arylindoles. Chem. Pharm. Bull. 2008, 56, 720–722. [Google Scholar]
- Fan, L.L.; Huang, N.; Yang, R.G.; He, S.Z.; Yang, L.M.; Xu, H.; Zheng, Y.T. Discovery of 5,6-dihydro-indolo[1,2-a]quinoxaline derivatives as new HIV-1 inhibitors in vitro. Lett. Drug. Des. Discov. 2012, 9, 44–47. [Google Scholar] [CrossRef]
- Yang, R.G.; Yang, L.M.; Ke, Y.Z.; Huang, N.; Zhang, R.; Zheng, Y.T.; Xu, H. Synthesis of new 2-(N-arylsulfonylindol-3-yl)-3-aryl-1,3-thiazolidin-4-ones as HIV-1 inhibitors in vitro. Lett. Drug. Des. Discov. 2012, 9, 415–420. [Google Scholar] [CrossRef]
- Patel, M.B.; Modi, N.R.; Raval, J.P.; Menon, S.K. Calix[4]arene based 1,3,4-oxadiazole and thiadiazole derivatives: Design, synthesis, and biological evaluation. Org. Biomol. Chem. 2012, 10, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Wang, Q.; Chen, J.J.; Zhang, X.M.; Tam, S.C.; Zheng, Y.T. The anti-HIV-1 effect of scutellarin. Biochem. Biophys. Res. Commun. 2005, 334, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.T.; Zhang, W.F.; Ben, K.L.; Wang, J.H. In vitro immunotoxicity and cytotoxicity of trichosanthin against human normal immunocytes and lekeumia-lymphoma cells. Immunopharmacol. Immunotoxicol. 1995, 17, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ding, Z.H.; Liu, J.K.; Zheng, Y.T. Xanthohumol, a novel anti-HIV-1 agent purified from Hop Humuluslupulus. Antiviral Res. 2004, 64, 189–194. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, Z.; Liu, S.; Tian, Y.; Hu, Z.; Chen, Y.; Chen, G. Design and Synthesis of Novel N-Arylsulfonyl-3-(2-yl-ethanone)-6-methylindole Derivatives as Inhibitors of HIV-1 Replication. Pharmaceuticals 2015, 8, 221-229. https://doi.org/10.3390/ph8020221
Che Z, Liu S, Tian Y, Hu Z, Chen Y, Chen G. Design and Synthesis of Novel N-Arylsulfonyl-3-(2-yl-ethanone)-6-methylindole Derivatives as Inhibitors of HIV-1 Replication. Pharmaceuticals. 2015; 8(2):221-229. https://doi.org/10.3390/ph8020221
Chicago/Turabian StyleChe, Zhiping, Shengming Liu, Yuee Tian, Zhenjie Hu, Yingwu Chen, and Genqiang Chen. 2015. "Design and Synthesis of Novel N-Arylsulfonyl-3-(2-yl-ethanone)-6-methylindole Derivatives as Inhibitors of HIV-1 Replication" Pharmaceuticals 8, no. 2: 221-229. https://doi.org/10.3390/ph8020221
APA StyleChe, Z., Liu, S., Tian, Y., Hu, Z., Chen, Y., & Chen, G. (2015). Design and Synthesis of Novel N-Arylsulfonyl-3-(2-yl-ethanone)-6-methylindole Derivatives as Inhibitors of HIV-1 Replication. Pharmaceuticals, 8(2), 221-229. https://doi.org/10.3390/ph8020221