Abstract
Seven novel N-arylsulfonyl-3-(2-yl-ethanone)-6-methylindole derivatives 4a–f and 6 were readily synthesized and have been identified as inhibitors of human immunodeficiency virus type-1 (HIV-1) replication. Initial biological studies indicated that among these derivatives, N-(p-ethyl)phenylsulfonyl-3-[2-morpholinoethanone]-6-methylindole (4f) and N-(p-ethyl)phenylsulfonyl-3-[2-(5-phenyl-1,3,4-oxadiazole-2-yl-thio)ethanone]-6-methylindole (6) showed the most promising activity against HIV-1 replication. The effective concentration (EC50) and therapeutic index (TI) values of 4f and 6 were 9.42/4.62 μM, and >49.77/66.95, respectively. The cytotoxicity of these compounds has also been assessed. No significant cytotoxicities were found for any of these compounds.
1. Introduction
The use of chemotherapy to suppress replication of the human immunodeficiency virus (HIV) has transformed the face of acquired immunodeficiency syndrome (AIDS) in the developed world. Pronounced reductions in illness and death have been achieved and healthcare utilization has diminished. HIV therapy has also provided many new insights into the pathogenesis and the viral and cellular dynamics of HIV infection, but challenges remain. Treatment does not suppress HIV replication in all patients, and the emergence of drug-resistant virus hinders subsequent treatment. Chronic therapy can also result in toxicity. These challenges prompt the continued search for new drugs and new therapeutic strategies to control chronic viral replication [1,2,3,4]. Consequently, the design and synthesis of brand new, specific, efficacious, safe chemotherapeutic drugs for the prevention of the spread of HIV-1 infection is imperative. In our previous studies, N-arylsulfonylindoles and N-arylsulfonyl-3-acetylindoles have been demonstrated the significant anti-HIV-1 activity in vitro [5,6]. Particularly, N-phenylsulfonyl-3-acetyl-6-methylindole (2a, Figure 1) and N-(p-ethyl)phenylsulfonyl-3-acetyl-6-methylindole (2b, Figure 1) have shown the most potent anti-HIV-1 activity. This confirmed that introduction of the acetyl group at the 3-position of N-arylsulfonyl-6-methylindoles could generally lead to more potent analogs. As a continuation of the research program of our laboratory searching for anti-HIV-1 agents [7,8,9], we now report the design and synthesis of some novel N-arylsulfonyl-3-(2-yl-ethanone)-6-methylindoles derivatives 4a–f and 6 (Scheme 1 and Scheme 2), which can specifically inhibit HIV-1 replication.
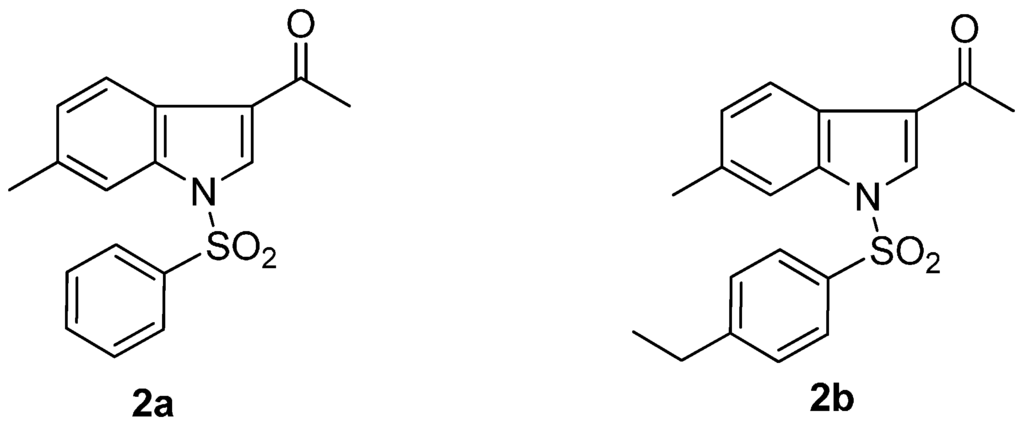
Figure 1.
Structures of N-phenylsulfonyl-3-acetyl-6-methylindole (2a) and N-(p-ethyl)-phenylsulfonyl-3-acetyl-6-methylindole (2b).
2. Results and Discussion
2.1. Chemistry
N-Arylsulfonyl-3-(2-yl-ethanone)-6-methylindole derivatives 4a–f were synthesized as shown in Scheme 1. First, 1a,b and 2a,b were obtained as reported in an earlier paper [6]. Subsequently, the key intermediates, N-arylsulfonyl-3-(2-bromoethanone)-6-methylindoles 3a,b were obtained by reaction of N-bromosuccinimide with 2,2'-azobisisobutyronitrile (AIBN) in a CCl4 solution under nitrogen. Compounds 3a,b were used directly in the next step without further purification. Finally, 3a,b reacted with the corresponding amines in the presence of cuprous iodide (CuI) and anhydrous potassium carbonate (K2CO3) at 80 °C to give 4a–f in 37%–73% yields. The compounds were well characterized by 1H-NMR, 13C-NMR, m.p., and MS.
N-(p-Ethyl)phenylsulfonyl-3-[2-(5-phenyl-1,3,4-oxadiazole-2-yl-thio)ethanone]-6-methylindole (6) was synthesized as depicted in Scheme 2. Firstly, intramolecular cyclization of benzoylhydrazine with carbon disulfide and potassium hydroxide in the presence of ethanol resulted in 5-phenyl-1,3,4-oxadiazole-2-thiol (5) [10]. Subsequently, N-(p-ethyl)phenylsulfonyl-3-(2-bromoethanone)-6-methylindole (3b) was reacted with 5 in the presence of K2CO3 to afford 6 in 36% yield.

Scheme 1.
The synthetic route of compounds 4a–f.

Scheme 2.
The synthetic route of compound 6.
2.2. Biological Activity
At the outset, the title compounds of series 4a–f and compound 6 were tested in vitro for their ability to inhibit HIV-1-induced cytopathogenicity, for cytotoxicity and anti-HIV-1 activity in acutely infected C8166 cells in comparison with 3'-azido-3'-deoxythymidine (AZT) used as a positive control (Table 1). All compounds tested could inhibit HIV-1 replication. However, none was as effective as AZT. Among them, compounds 4a, 4d, 4f and 6 exhibited the most potent anti-HIV-1 activity, with EC50 values of 32.78, 17.94, 9.42 and 4.62 μM, and therapeutic index (TI) values of >15.38, 12.26, >49.77 and 66.95, respectively. Maximum activity were obtained with compounds 4f and 6, which were endowed with the highest potency (TI > 49.77 and = 66.95, respectively). However, compounds 4b, 4c, and 4e only showed moderate activity (TI > 8.87, = 2.12 and = 1.46, respectively). The cytotoxicity of these compounds had also been assessed. No significant cytotoxicities were found for any of them.
Meanwhile, some preliminary structure-activity relationships (SAR) of 4a–f and 6 were also observed. We examined the effect of substituents at the C-2 position of the acetyl by use of various amines and 5-phenyl-1,3,4-oxadiazole-2-thiol groups. It can be seen that the piperidinyl-substituted compound 4a was generally more potent than the diisopropylamino-substituted compound 4d, the morpholino-substituted compound 4b, the pyrrolidino-substituted compound 4c and the butylamino-substituted compound 4e, if the R1 substituents at the C-4 position of the arylsulfonyl ring were the same. The cytotoxicity (CC50), anti-HIV-1 activity (EC50) and TI values of 4a, 4d, 4b, 4c, 4e were >504.41/220.02/501.91/225.82/43.79 μM, 32.78/17.94/56.56/106.02/29.96 μM, and 15.38/12.26/8.87/2.12/1.46, respectively. Especially the TI value of 4a was more than 10 times that of 4e. Interestingly, variations at the C-4 position of the arylsulfonyl ring play an important role in HIV-1 replication inhibition. For example, the EC50 and TI values of 4b and 4f were 56.56/9.42 μM, and >8.87/>49.77, respectively.
It is worth noting that the insertion of an ethyl group at the para-position of the arylsulfonyl ring led to compound 4f characterized by both low cytotoxicity (CC50 > 468.91 μM) and antiviral activity (TI > 49.77). A change in activity was observed for compound 4b (EC50 = 56.56 μM, TI > 8.87) when compared with 4f (EC50 = 9.42 μM, TI > 49.77). The results suggest that the substituent at R1 is an important feature for activity. Moreover, when the 5-phenyl-1,3,4-oxadiazole-2-thiol group was introduced at the C-2 position of 3b, the corresponding compound 6 displayed the most potent anti-HIV-1 activity. The EC50 and TI values of 6 were 4.62 μM and 66.95.

Table 1.
Anti-HIV-1 activity of N-arylsulfonyl-3-(2-yl-ethanone)-indoles derivatives 4a–f and 6 in vitro a.
| Compounds | CC50 b(µM) | EC50 c(µM) | TI d |
|---|---|---|---|
| 4a | >504.41 | 32.78 | >15.38 |
| 4b | >501.91 | 56.56 | >8.87 |
| 4c | 225.82 | 106.02 | 2.12 |
| 4d | 220.02 | 17.94 | 12.26 |
| 4e | 43.79 | 29.96 | 1.46 |
| 4f | >468.91 | 9.42 | >49.77 |
| 6 | 309.31 | 4.62 | 66.95 |
| AZT e | 4263.84 | 0.01212 | 351801.98 |
a Values are means of two separate experiments. b CC50 (50% cytotoxic concentration), concentration of drug that causes 50% reduction in total C8166 cell number. c EC50 (50% effective concentration), concentration of drug that reduces syncytia formation by 50%. d In vitro therapeutic index (CC50 value/EC50 value). eAZT was used as a positive control.
3. Experimental Section
3.1. General Information
All reagents and solvents were of reagent grade or purified according to standard methods before use. Analytical thin-layer chromatography (TLC) and preparative thin-layer chromatography (PTLC) were performed with silica gel plates using silica gel 60 GF254 (Qingdao Haiyang Chemical Co., Ltd., Qingdao, China). Melting points were determined on a digital melting-point apparatus and were uncorrected. 1H-NMR and 13C-NMR spectra were recorded at 300 MHz or 400 MHz or 500 MHz and 125 MHz respectively on a Bruker Avance DMX 300 MHz or 400 MHz or 500 MHz spectrophotometer in CDCl3 with tetra methyl silane (TMS) as an internal standard. Electrospray iontrap mass spectrometry (ESI-TRAP-MS) and mass spectra (EI-MS) were carried out with Bruker ESI-TRAP Esquire 3000 plus mass spectrometry instrument and a HP 5988 instrument, respectively. High-resolution mass spectra (HR-MS) were recorded with an IonSpec 4.7 T FTMS instrument.
3.2. General Procedure for the Synthesis of 4a–f
Compounds 1a,b and 2a,b were prepared as reported in the earlier paper [6]. The structures of the compounds were well characterized by 1H-NMR, m.p., and MS.
N-Phenylsulfonyl-6-methylindole (1a): White solid, m.p.70–72 °C. 1H-NMR (300 MHz, CDCl3) δ: 7.81–7.88 (m, 3H), 7.41–7.49 (m, 5H), 7.04 (d, J = 6.9 Hz, 1H), 6.61 (s, 1H), 2.47 (s, 3H). EI-MS m/z: 271 (M+, 48).
N-(p-Ethyl)phenylsulfonyl-6-methylindole (1b): White solid, m.p. 84–86 °C. 1H-NMR (300 MHz, CDCl3) δ: 7.76–7.81 (m, 3H), 7.49 (s, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.03–7.25 (m, 3H), 6.59 (s, 1H), 2.61 (q, J = 7.2 Hz, 2H), 2.47 (s, 3H), 1.17 (t, J = 7.2 Hz, 3H). EI-MS m/z: 299 (M+, 42).
N-Phenylsulfonyl-3-acetyl-6-methylindole (2a): White solid, m.p. 220–221 °C. 1H-NMR (300 MHz, CDCl3) δ: 8.16 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H), 7.93 (d, J = 7.5 Hz, 2H), 7.73 (s, 1H), 7.58–7.63 (m, 1H), 7.48–7.53 (m, 2H), 7.15 (d, J = 8.1 Hz, 1H), 2.56 (s, 3H, COCH3), 2.47 (s, 3H, CH3). ESI-TRAP-MS m/z: 314 ([M+H]+, 100).
N-(p-Ethyl)phenylsulfonyl-3-acetyl-6-methylindole (2b): White solid, m.p. 118–120 °C. 1H-NMR (400 MHz, CDCl3) δ: 8.17 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.74 (s, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 8.0 Hz, 1H), 2.66 (q, J = 7.6 Hz, 2H, CH2CH3), 2.55 (s, 3H, COCH3), 2.48 (s, 3H, CH3), 1.18 (t, J = 8.0 Hz, 3H, CH2CH3). EI-MS m/z: 341 (M+, 29).
A solution of N-arylsulfonyl-3-acetyl-6-methylindoles 2a,b (1 mmol) in CCl4 (5 mL) was heated to reflux after which N-bromosuccinimide (1.1 mmol) and AIBN (8 mg) were admixed carefully and added in three portions under nitrogen. The mixture was refluxed for 20 min. Another portion of AIBN (4 mg) was then added. The mixture was kept at reflux for 3 h and then allowed to cool. The precipitated succinimide was filtered off and washed with hexane (4 × 10 mL). The combined filtrates were concentrated under reduced pressure to afford 3a,b. The crude residues of 3a,b were flash evaporated under reduced pressure with dry THF three times and used directly in the next step without further purification. These compounds are unstable on silica gel. Subsequently, to a mixture of 3a,b (1 mmol), the corresponding amines (1 mmol), CuI (0.2 mmol) and anhydrous K2CO3 (2 mmol) in DMF (5 mL) were added with stirring at 80 °C under nitrogen. When the reaction was complete according to TLC analysis, the reaction mixture was cooled to room temperature, poured into ice water (20 mL), and extracted with EtOAc (30 mL × 3). Finally, the combined organic phase was washed by brine, dried over anhydrous Na2SO4, concentrated in vacuo and purifed by silica gel column chromatography to give the pure compounds 4a–f. The structures of the compounds were well characterized by 1H-NMR, 13C-NMR, m.p., and MS.
N-Phenylsulfonyl-3-[2-piperidinylethanone]-6-methylindole (4a): Yellow solid, yield 73%, m.p. 132–134 °C. 1H-NMR (400 MHz, CDCl3) δ: 8.23 (d, J = 8.4 Hz, 1H), 8.18 (s, 1H), 7.95–7.97 (m, 2H), 7.91 (s, 1H), 7.58–7.60 (m, 1H), 7.47–7.51 (m, 2H), 7.31 (d, J = 8.4 Hz, 1H), 3.59 (s, 2H), 2.57 (s, 3H), 2.34 (s, 4H), 1.55–1.60 (m, 4H), 1.43–1.44 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ: 193.4, 137.5, 137.0, 135.1, 134.5, 132.0, 129.5, 127.1, 126.4, 126.3, 122.5, 121.8, 113.3, 63.7, 54.3, 27.8, 26.0, 24.3. MS (ESI-TRAP), m/z (%): 397 ([M+H]+, 100). HRMS (ESI): Calcd for C22H25N2O3S ([M+H]+), 397.1580; found, 397.1571.
N-Phenylsulfonyl-3-[2-morpholinoethanone]-6-methylindole (4b): White solid, yield 66%, m.p. 174–176 °C. 1H-NMR (500 MHz, CDCl3) δ: 8.25 (d, J = 8.0 Hz, 1H), 8.19 (s, 1H), 7.94–7.96 (m, 2H), 7.90 (s, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.48–7.51 (m, 2H), 7.33 (dd, J = 8.0 Hz, 1.0 Hz, 1H), 3.71 (t, J = 4.5 Hz, 4H), 3.61 (s, 2H), 2.57 (s, 3H), 2.41 (t, J = 4.0 Hz, 4H). 13C-NMR (125 MHz, CDCl3) δ: 193.4, 137.5, 136.2, 135.1, 134.5, 132.1, 129.5, 127.1, 126.7, 126.2, 122.8, 121.7, 113.3, 67.0, 63.3, 53.4, 27.8. MS (ESI-TRAP), m/z (%): 399 ([M+H]+, 100). HRMS (ESI): Calcd for C21H23N2O4S ([M+H]+), 399.1373; found, 399.1361.
N-Phenylsulfonyl-3-[2-pyrrolidinoethanone]-6-methylindole (4c): Yellow solid, yield 57%, m.p. 124–126 °C. 1H-NMR (500 MHz, CDCl3) δ: 8.24 (d, J = 8.5 Hz, 1H), 8.18 (s, 1H), 7.95–7.97 (m, 2H), 7.91 (s, 1H), 7.58–7.61 (m, 1H), 7.47–7.50 (m, 2H), 7.33–7.35 (m, 1H), 3.75 (s, 2H), 2.56 (s, 3H), 2.49 (s, 4H), 1.78–1.80 (m, 4H). 13C-NMR (125 MHz, CDCl3) δ: 193.3, 137.5, 135.1, 134.5, 132.0, 129.5, 127.1, 126.5, 126.1, 122.7, 121.8, 113.2, 60.5, 53.8, 27.7, 23.5. MS (ESI-TRAP), m/z (%): 383 ([M+H]+, 100). HRMS (ESI): Calcd for C21H23N2O3S ([M+H]+), 383.1424; found, 383.1417.
N-Phenylsulfonyl-3-[2-diisopropylaminoethanone]-6-methylindole (4d): Tan liquid, yield 49%. 1H-NMR (500 MHz, CDCl3) δ: 8.17–8.19 (m, 2H), 8.02 (s, 1H), 7.97 (d, J = 8.0 Hz, 2H), 7.57–7.60 (m, 1H), 7.46–7.49 (m, 2H), 7.32 (d, J = 8.0 Hz, 1H), 3.74 (s, 2H), 2.99–3.04 (m, 2H), 2.56 (s, 3H), 1.03 (d, J = 6.5 Hz, 12H). 13C-NMR (125 MHz, CDCl3) δ: 193.4, 142.0, 137.6, 135.3, 134.4, 131.6, 129.5, 127.2, 126.0, 124.9, 122.2, 121.8, 112.1, 49.0, 48.0, 27.7, 20.8. MS (ESI-TRAP), m/z (%): 413 ([M+H]+, 100). HRMS (ESI): Calcd for C23H29N2O3S ([M+H]+), 413.1893; found, 413.1890.
N-Phenylsulfonyl-3-[2-butylaminoethanone]-6-methylindole (4e): Yellow liquid, yield 51%. 1H-NMR (500 MHz, CDCl3) δ: 8.26 (d, J = 8.5 Hz, 1H), 8.18 (s, 1H), 7.95–7.97 (m, 2H), 7.90 (s, 1H), 7.59–7.62 (m, 1H), 7.48–7.51 (m, 2H), 7.31–7.33 (m, 1H), 3.91 (s, 2H), 2.75 (s, 1H), 2.58–2.61 (m, 2H), 2.56 (s, 3H), 1.48–1.51 (m, 2H), 1.32–1.36 (m, 2H), 0.89–0.92 (m, 3H). 13C-NMR (125 MHz, CDCl3) δ: 193.4, 138.7, 137.6, 135.2, 134.5, 132.0, 129.6, 127.0, 126.4, 125.4, 122.9, 121.8, 112.4, 53.9, 48.8, 32.1, 27.7, 20.4, 14.0. MS (ESI-TRAP), m/z (%): 385 ([M+H]+, 100). HRMS (ESI): Calcd for C21H25N2O3S ([M+H]+), 385.1580; found, 385.1572.
N-(p-Ethyl)phenylsulfonyl-3-[2-morpholinoethanone]-6-methylindole (4f): White solid, yield 37%, m.p. 108–110 °C. 1H-NMR (500 MHz, CDCl3) δ: 8.25 (d, J = 8.5 Hz, 1H), 8.19 (s, 1H), 7.92 (s, 1H), 7.87 (d, J = 8.0 Hz, 2H), 7.29–7.33 (m, 3H), 3.71 (s, 4H), 3.62 (s, 2H), 2.68(q, J = 7.5 Hz, 2H), 2.56 (s, 3H), 2.42 (s, 4H), 1.21 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ: 193.4, 151.9, 135.1, 134.7, 132.2, 129.0, 127.3, 126.7, 126.2, 122.8, 121.5, 113.4, 67.0, 63.3, 53.4, 28.8, 27.7, 14.8. MS (ESI-TRAP), m/z (%): 427 ([M+H]+, 100). HRMS (ESI): Calcd for C23H27N2O4S ([M+H]+), 427.1686; found, 427.1677.
3.3. General Procedure for the Synthesis of 6
Firstly, intramolecular cyclization of benzoylhydrazine with carbon disulfide and potassium hydroxide in the presence of ethanol resulted in 5-phenyl-1,3,4-oxadiazole-2-thiol (5) [10]. Subsequently, a mixture of 5 (0.5 mmol), 3b (0.6 mmol) and K2CO3 (1 mmol) in acetone (5 mL) was reacted at reflux, and the reaction process was checked by TLC. When the reaction was complete after 9 h, the organic solvent was removed, and the residue was directly purified by preparative TLC to give 6. The structure of the compound was well characterized by 1H-NMR, 13C-NMR, m.p., and MS.
N-(p-Ethyl)phenylsulfonyl-3-[2-(5-phenyl-1,3,4-oxadiazole-2-yl-thio)ethanone]-6-methylindole (6): White solid, yield 36%, m.p. 122–124°C. 1H-NMR (500 MHz, CDCl3) δ: 8.29 (d, J = 8.0 Hz, 1H), 8.18 (s, 1H), 8.15 (s, 1H), 7.98–8.00 (m, 2H), 7.90 (d, J = 8.5 Hz, 2H), 7.49–7.52 (m, 3H), 7.43 (d, J = 7.5 Hz, 1H), 7.29–7.41 (m, 2H), 4.64 (s, 2H), 2.58–2.59 (m, 2H), 2.54 (s, 3H), 1.13 (t, J = 7.5 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ: 193.2, 165.8, 163.6, 151.9, 134.9, 134.4, 133.7, 132.5, 131.7, 129.2, 129.1, 129.0, 127.3, 127.2, 126.6, 125.9, 123.6, 123.4, 121.4, 114.0, 37.0, 28.8, 27.7, 14.6. MS (ESI-TRAP), m/z (%): 518 ([M+H]+, 100). HRMS (ESI): Calcd for C27H24N3O4S2 ([M+H]+), 518.1203; found, 518.1211.
3.4. Anti-HIV-1 Activity Assay
3.4.1. Cells and Virus
Cell line (C8166) and the laboratory-derived virus (HIV-1 IIIB) were obtained from the MRC, AIDS Reagent Project, London, UK. C8166 was maintained in RPMI-1640 supplemented with 10% heat-inactivated new born calf serum (Gibco, USA). The cells used in all experiments were in log-phase growth. The 50% HIV-1 IIIB tissue culture infectious dose (TCID50) in C8166 cells was determined and calculated by the Reed and Muench method. Virus stocks were stored in small aliquots at −70 °C [11].
3.4.2. MTT-based Cytotoxicity Assay
Cellular toxicity of compounds 4a–f and 6 on C8166 cells was assessed by the MTT method as described previously [12]. Briefly, cells were seeded on 96-well microtiter plate in the absence or presence of various concentrations of compounds in triplicate and incubated at 37 °C in a humid atmosphere of 5% CO2 for 3 days. The supernatants were discarded and MTT reagent (5 mg/mL in PBS) was added to each wells, then incubated for 4 h, 100 μL of 50% DMF-20% SDS was added. After the formazan was dissolved completely, the plates were read on a Bio-TekElx800 ELISA reader at 595/630 nm. The cytotoxic concentration that caused the reduction of viable C8166 cells by 50% (CC50) was determined from dose-response curve.
3.4.3. Syncytia Assay
In the presence of 100 μL of various concentrations of compounds, C8166 cells (4 × 105/mL) were infected with virus HIV-1IIIB at a multiplicity of infection (M.O.I) of 0.06. The final volume per well was 200 μL. Control assays were performed without the testing compounds in HIV-1IIIB infected and uninfected cultures. After 3 days of culture, the cytopathic effect (CPE) was measured by counting the number of syncytia. Percentage inhibition of syncytia formation was calculated and 50% effective concentration (EC50) was calculated. AZT (Sigma, USA) was used as a positive control. Therapeutic index (TI) = CC50/EC50 [13].
4. Conclusions
In summary, a series of N-arylsulfonyl-3-(2-yl-ethanone)-6-methylindolederivatives 4a–f and 6 were easily synthesized and have been identified as inhibitors of HIV-1 replication. Initial biological studies indicated that N-(p-ethyl)phenylsulfonyl-3-[2-morpholinoethanone]-6-methylindole (4f) and N-(p-ethyl)phenylsulfonyl-3-[2-(5-phenyl-1,3,4-oxadiazole-2-ylthio)-ethanone]-6-methylindole (6) show promising activity against HIV-1 replication. Our structure-based design has led to the discovery that the introduction of an ethyl group at the para-position of the arylsulfonyl ring generated a novel class of highly potent anti-HIV-1 compounds. Moreover, the preliminary SAR showed that the morpholino group or the 5-phenyl-1,3,4-oxadiazole-2-thiol group at the C-2 position on the acetyl of N-arylsulfonyl-3-(2-yl-ethanone)-6-methylindoles was very important for potent anti-HIV-1 activity. The EC50 and TI values of 4f and 6 were 9.42/4.62 μM, and >49.77/66.95, respectively. The application of this novel and potent structure modification is expected to provide the foundation for the rational modification of N-arylsulfonyl-3-acetylindoles and accelerate the discovery of more potent anti-HIV-1 compounds.
Acknowledgements
This work was financially supported in part by grants from the program for Henan University of Science and Technology Doctoral Scientific Research Fund project (09001763), and the Youth Science Foundation of Henan University of Science and Technology (13580038). We would like to acknowledge the MRC AIDS Research Project and the NIH AIDS Research and Reference Reagent Program for providing cell lines and viruses.
Author Contributions
Che Z.P. and Chen G.Q. conceived and designed the experiments; Che Z.P., Liu S.M., Tian Y.E., Hu Z.J. and Chen Y.W. performed the experiments; Che Z.P. and Tian Y.E. analyzed the data; Che Z.P. and Tian Y.E. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Richman, D.D. HIV chemotherapy. Nature 2001, 410, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Clavel, F.; Hance, A.J. HIV Drug resistance. N. Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar]
- Gupta, R.; Hill, A.; Sawyer, A.W.; Pillay, D. Emergence of drug resistance in HIV type 1-infected patients after receipt of first-line highly active antiretroviral therapy: A systematic review of clinical trials. Clin. Infect. Dis. 2008, 47, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lv, M. Developments of indoles as anti-HIV-1 inhibitors. Curr. Pharm. Des. 2009, 15, 2120–2148. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Liu, W.Q.; Xu, H.; Yang, L.M.; Lv, M.; Zheng, Y.T. Anti human immunodeficiency virus-1 (HIV-1) agents 3: Synthesis and in vitro anti-HIV-1 activity of some N-arylsulfonylindoles. Chem. Pharm. Bull. 2009, 57, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Ran, J.Q.; Huang, N.; Xu, H.; Yang, L.M.; Lv, M.; Zheng, Y.T. Anti HIV-1 agents 5: Synthesis and anti-HIV-1 activity of some N-arylsulfonyl-3-acetylindoles in vitro. Bioorg. Med. Chem. Lett. 2010, 20, 3534–3536. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, W.Q.; Fan, L.L.; Chen, Y.; Yang, L.M.; Lv, L.; Zheng, Y.T. Synthesis and HIV-1 integrase inhibition activity of some N-arylindoles. Chem. Pharm. Bull. 2008, 56, 720–722. [Google Scholar]
- Fan, L.L.; Huang, N.; Yang, R.G.; He, S.Z.; Yang, L.M.; Xu, H.; Zheng, Y.T. Discovery of 5,6-dihydro-indolo[1,2-a]quinoxaline derivatives as new HIV-1 inhibitors in vitro. Lett. Drug. Des. Discov. 2012, 9, 44–47. [Google Scholar] [CrossRef]
- Yang, R.G.; Yang, L.M.; Ke, Y.Z.; Huang, N.; Zhang, R.; Zheng, Y.T.; Xu, H. Synthesis of new 2-(N-arylsulfonylindol-3-yl)-3-aryl-1,3-thiazolidin-4-ones as HIV-1 inhibitors in vitro. Lett. Drug. Des. Discov. 2012, 9, 415–420. [Google Scholar] [CrossRef]
- Patel, M.B.; Modi, N.R.; Raval, J.P.; Menon, S.K. Calix[4]arene based 1,3,4-oxadiazole and thiadiazole derivatives: Design, synthesis, and biological evaluation. Org. Biomol. Chem. 2012, 10, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Wang, Q.; Chen, J.J.; Zhang, X.M.; Tam, S.C.; Zheng, Y.T. The anti-HIV-1 effect of scutellarin. Biochem. Biophys. Res. Commun. 2005, 334, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.T.; Zhang, W.F.; Ben, K.L.; Wang, J.H. In vitro immunotoxicity and cytotoxicity of trichosanthin against human normal immunocytes and lekeumia-lymphoma cells. Immunopharmacol. Immunotoxicol. 1995, 17, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ding, Z.H.; Liu, J.K.; Zheng, Y.T. Xanthohumol, a novel anti-HIV-1 agent purified from Hop Humuluslupulus. Antiviral Res. 2004, 64, 189–194. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).