β-Boomerang Antimicrobial and Antiendotoxic Peptides: Lipidation and Disulfide Bond Effects on Activity and Structure


Abstract
:1. Introduction
2. Experimental Section
3. Results and Discussion
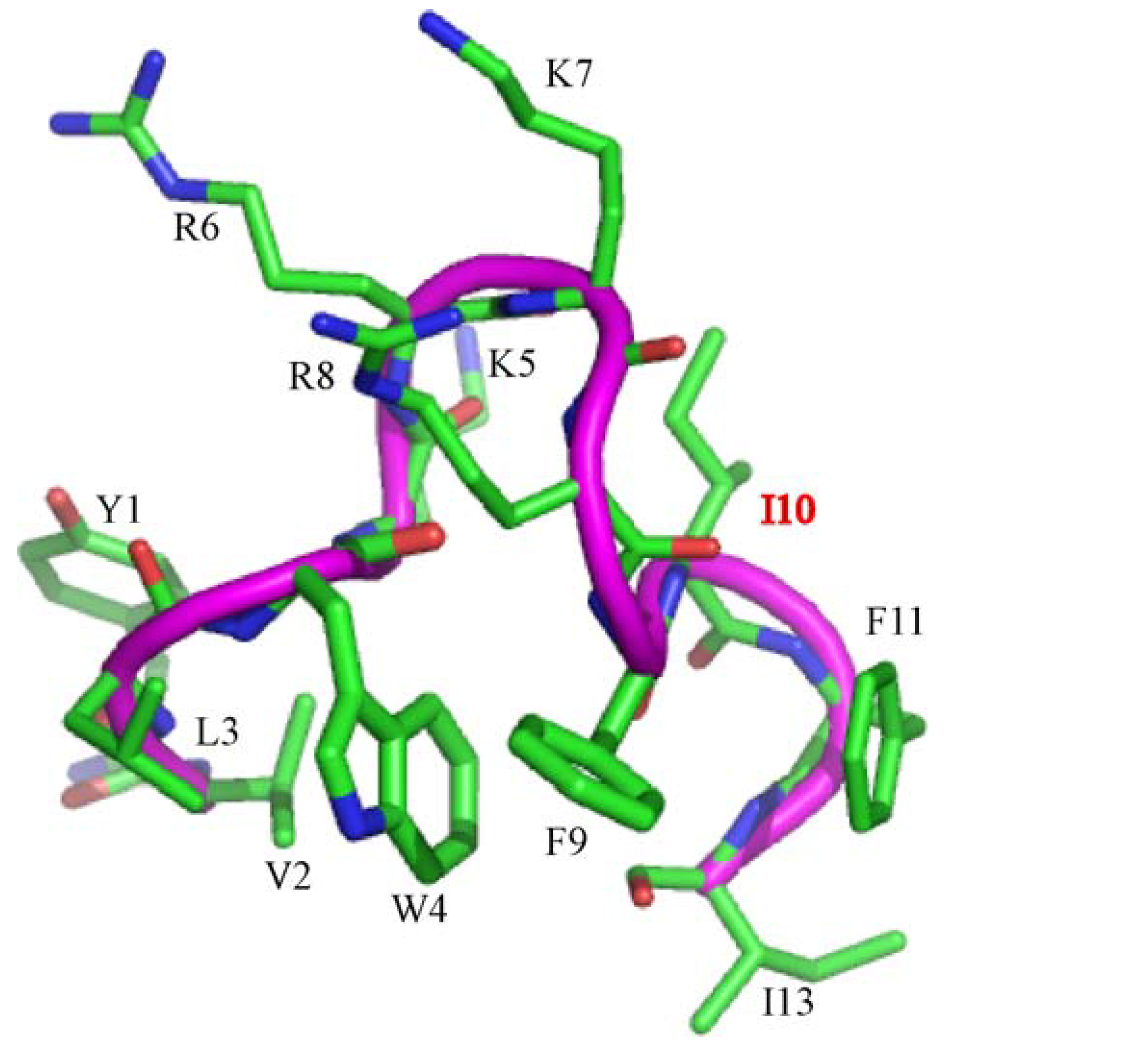
3.1. Peptide Design
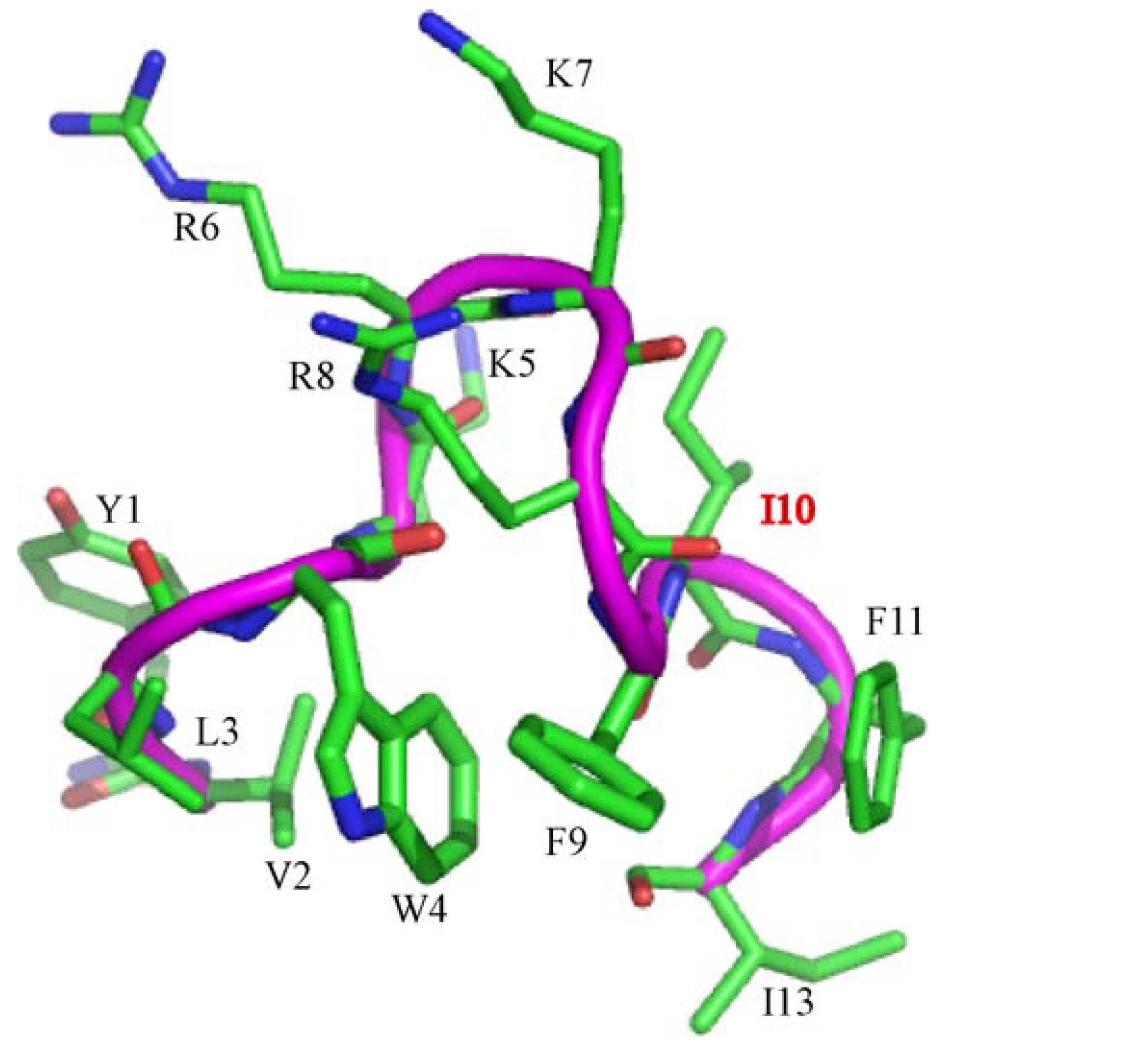

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide designation | Primary structure | Retention time (Rt) |
|---|---|---|
| YI13C | Y-V-L-W-K-R-K-R-K-F-C-F-I | 27.48 |
| C4YI13C | C4- Y-V-L-W-K-R-K-R-K-F-C-F-I | 31.62 |
| C8YI13C | C8- Y-V-L-W-K-R-K-R-K-F-C-F-I | 37.40 |
| C8YI13CAA | C8- Y-V-L-A-K-R-K-R-K-A-C-F-I | 32.38 |
3.2. Antimicrobial Activity
| Bacteria | YI13C | C4YI13C | C8YI13C | C8YI13CAA |
|---|---|---|---|---|
| Gram-negative | ||||
| E.coli (Lab strain) | 12.5 | 10 | 3 | 50 |
| P.aeruginosa (ATCC 27853) | 20 | 15 | 5 | 100 |
| K. pneumoniae (ATCC 13883) | 25 | 8 | 12 | 100 |
| S.enterica (ATCC 14028) | 50 | 50 | 50 | >200 |
| Gram-positive | ||||
| B.subtilis (Lab strain) | 20 | 15 | 5 | 50 |
| S.aureus (ATCC 25923) | 20 | 50 | 5 | 200 |
| S.pyogenes (ATCC 19615) | 50 | 50 | 50 | >200 |
| E.faecalis (ATCC 29212) | 50 | 50 | 4 | >200 |
| % of hemolysis at 50 μM peptide concentration | 21.5 | 14.1 | 21.5 | 30.2 |
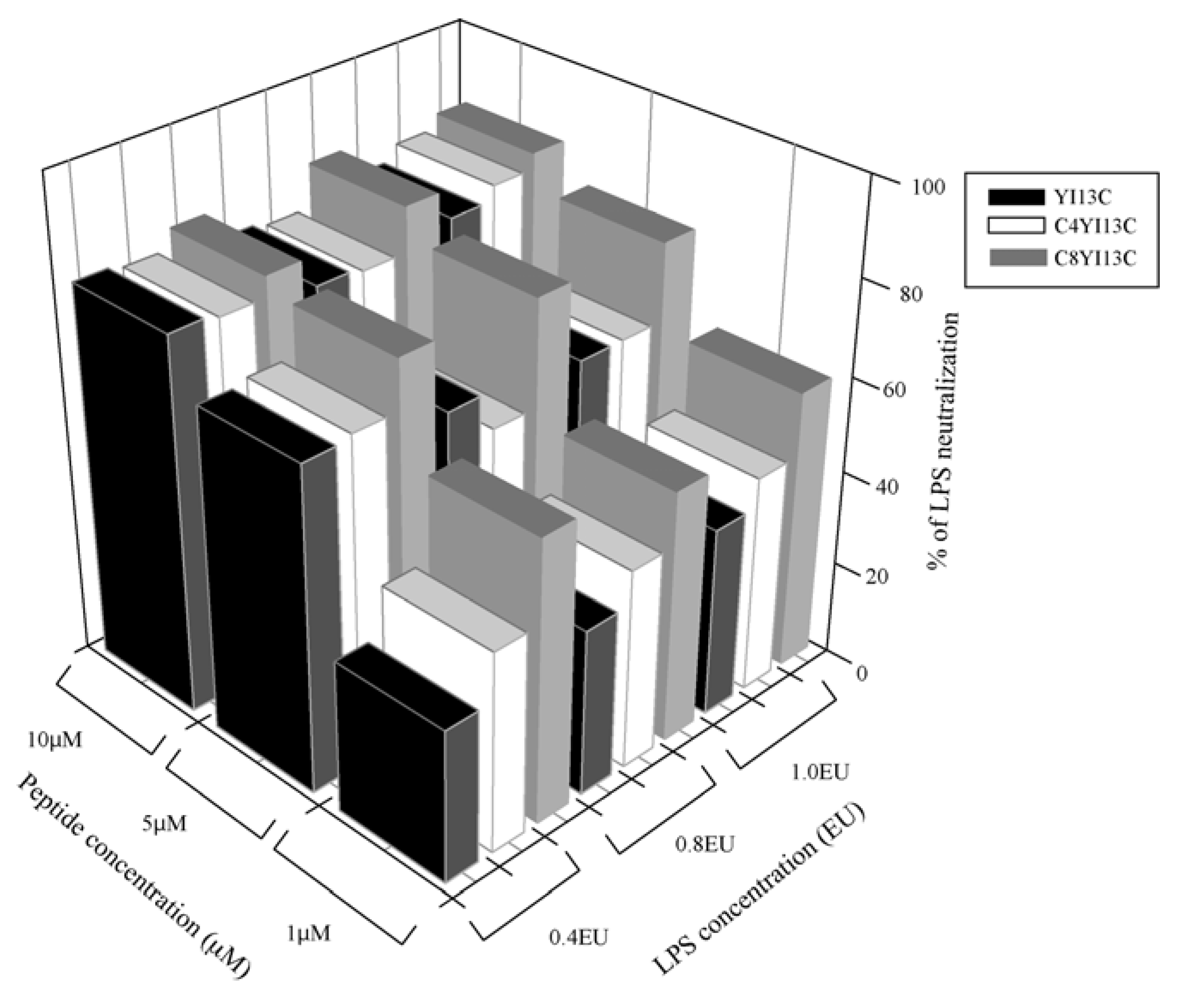
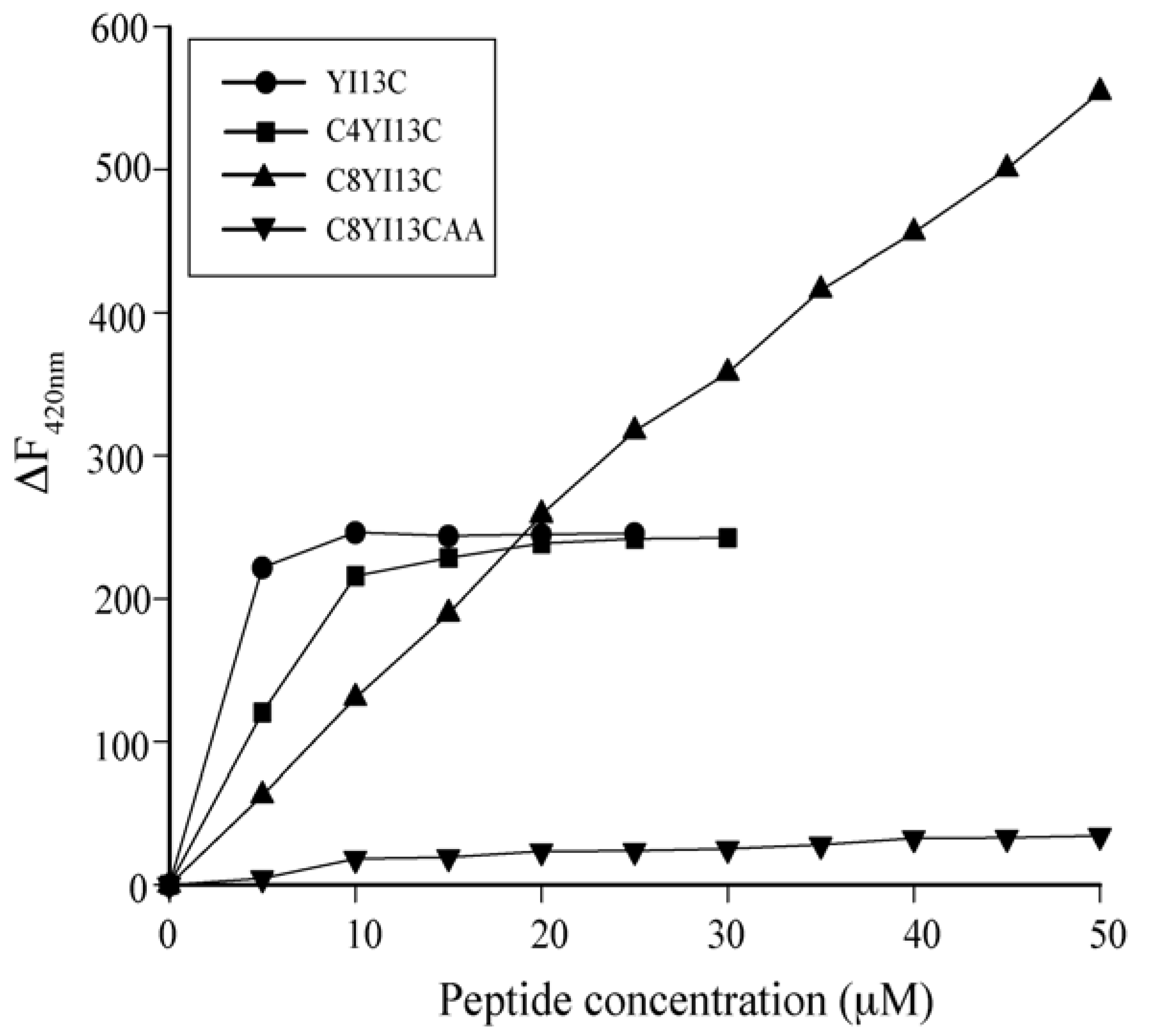
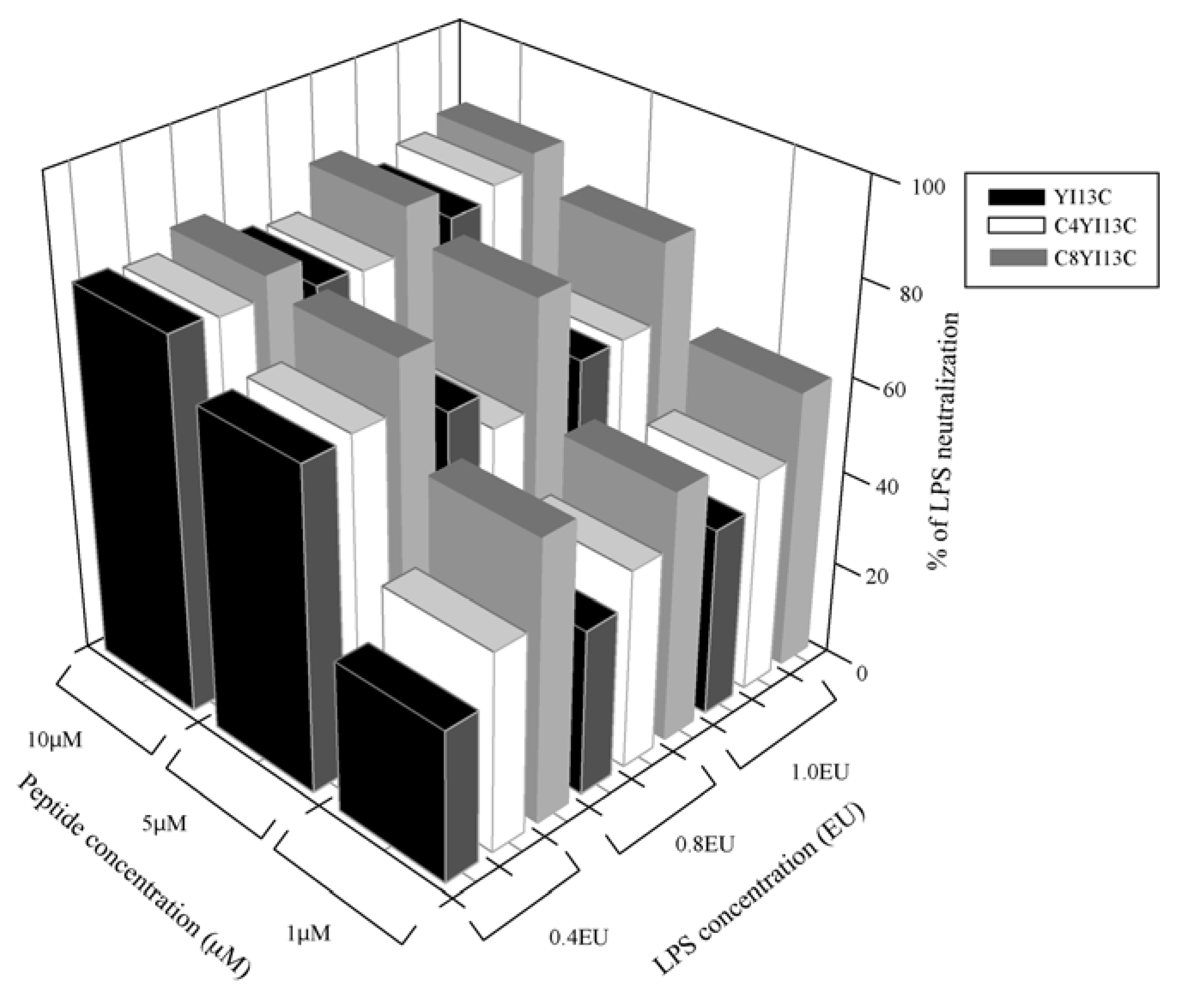
3.3. Neutralization of LPS by LAL Assay
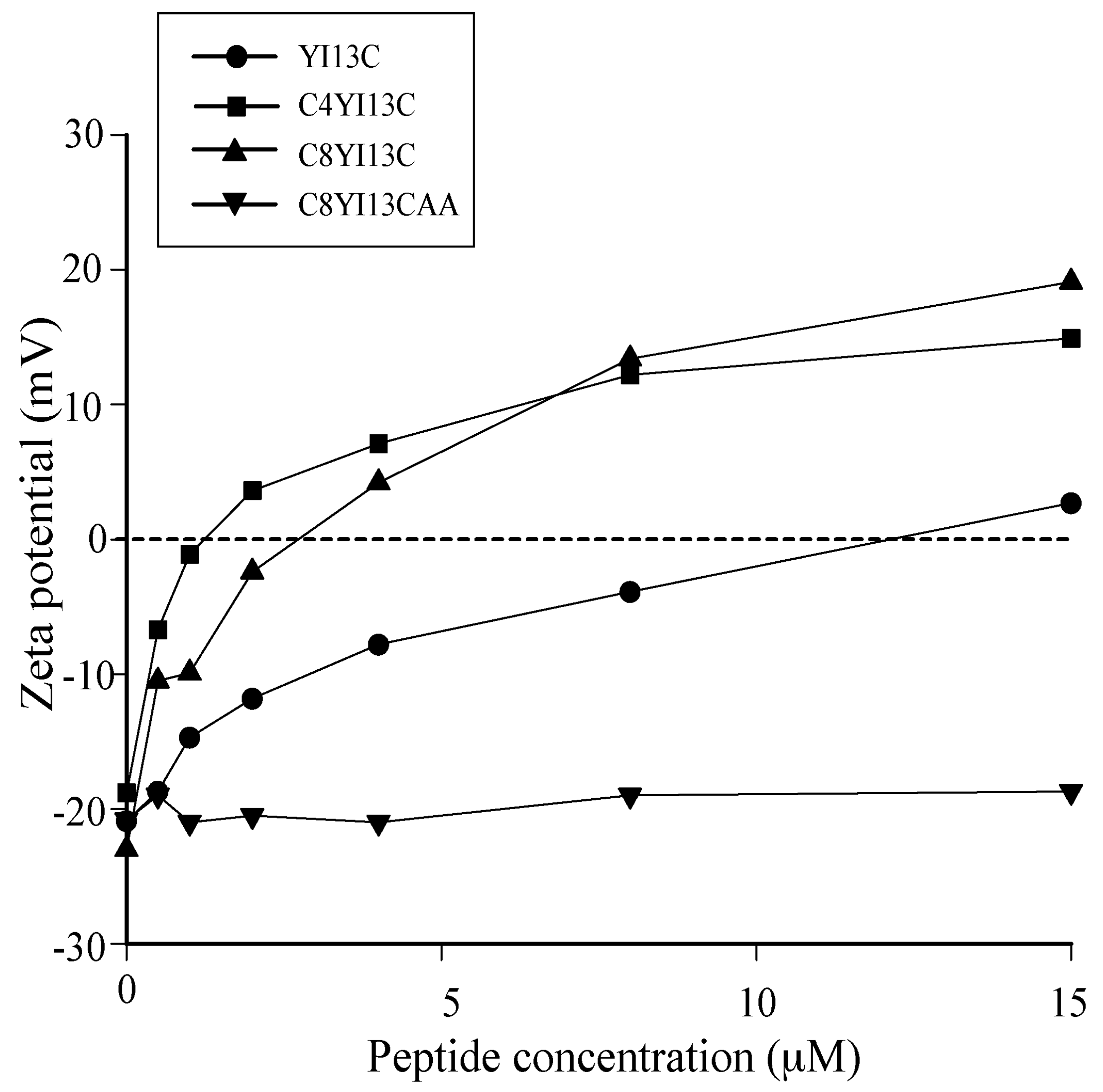
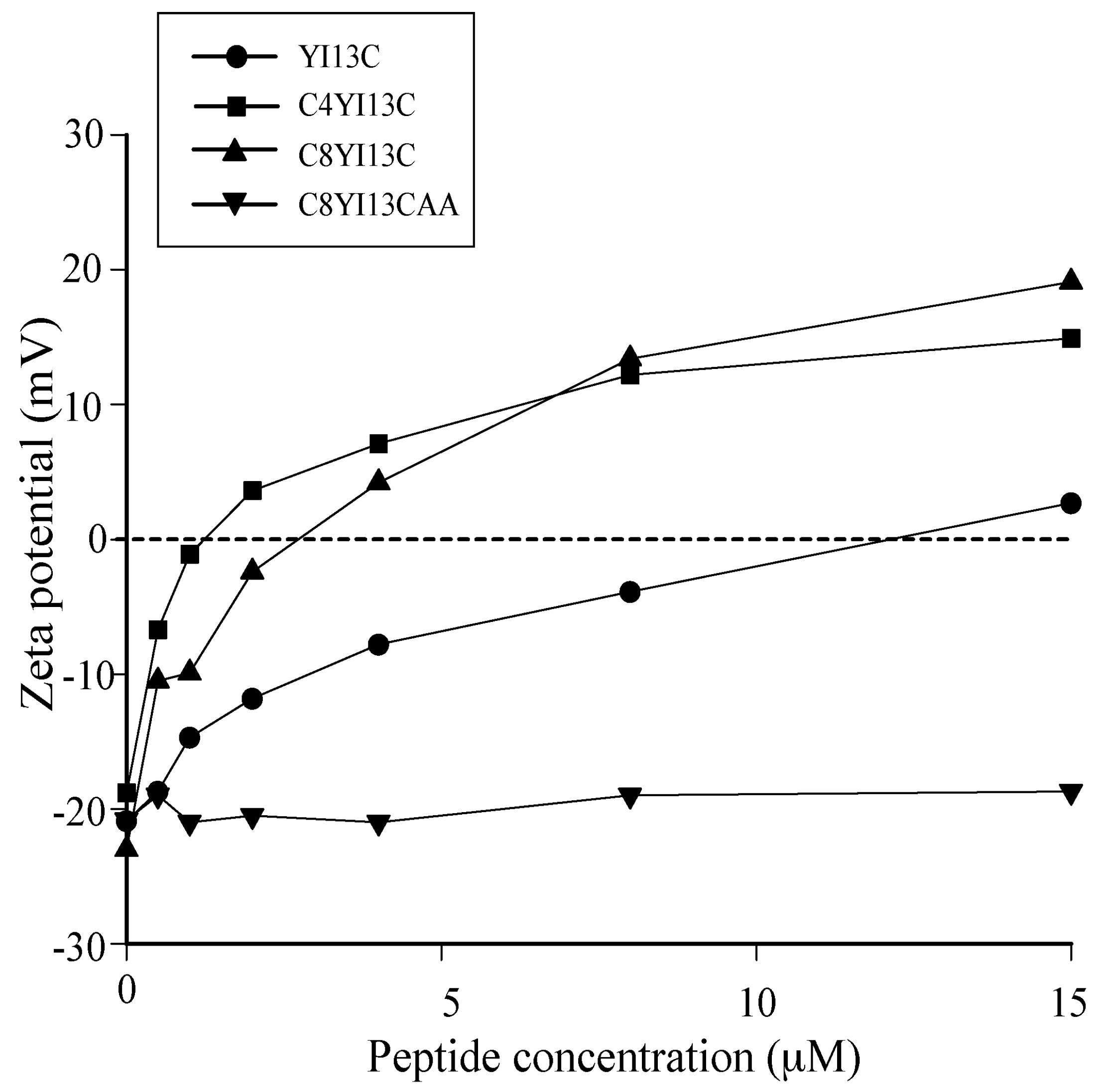
3.4. Surface Charge Neutralization by Zeta Potential Studies
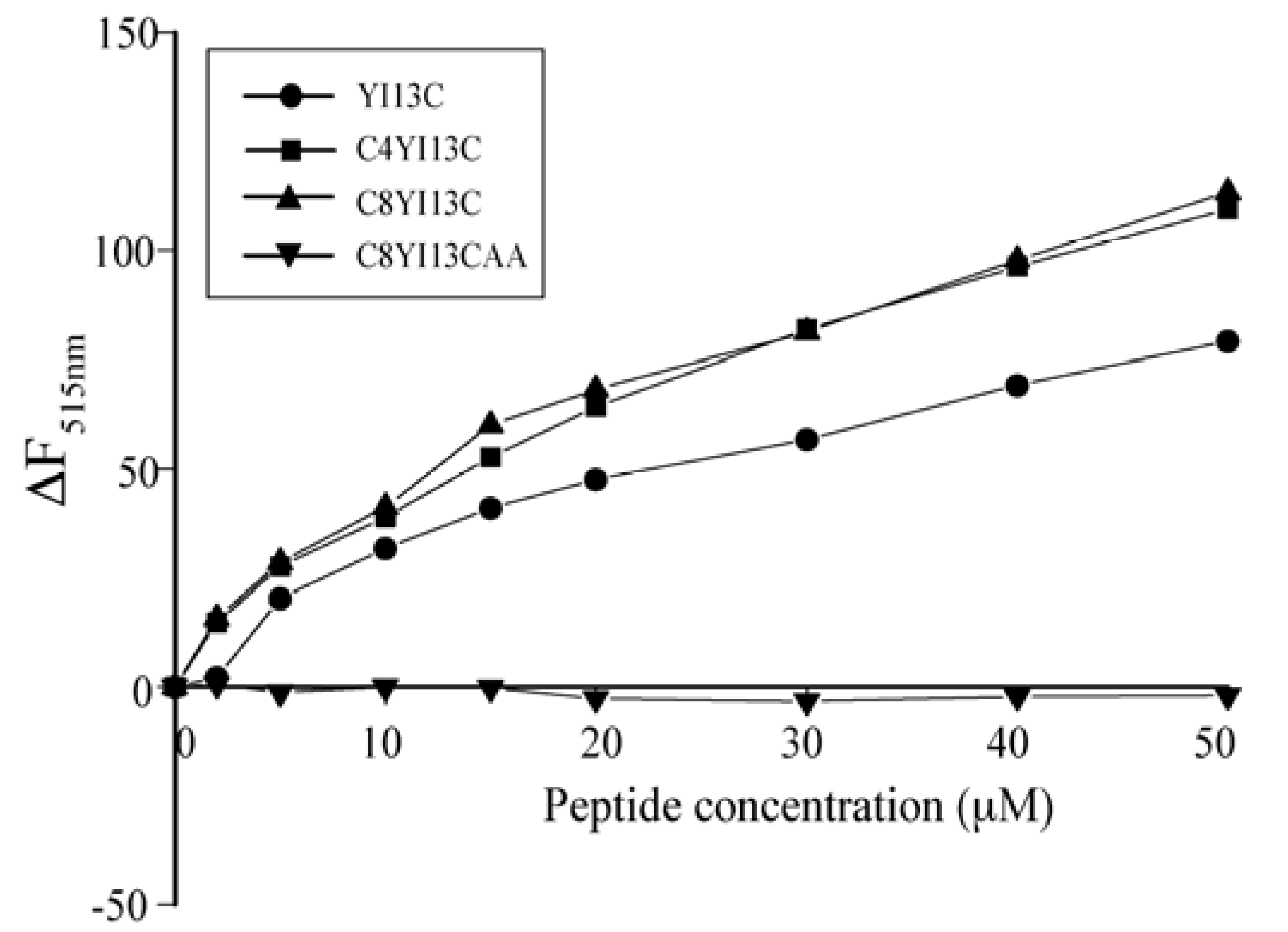
3.5. Outer Membrane Permeability by NPN Assay

3.6. Intrinsic Tryptophan Fluorescence and Acrylamide Quenching
| Peptides | λmax | Stern-Volmer constant (Ksv) | ||||
|---|---|---|---|---|---|---|
| Free | LPS | DPC | Free | LPS | DPC | |
| YI13C | 356 | 339 | 354 | 43.1 | 6.0 | 11.1 |
| C4YI13C | 358 | 338 | 348 | 37.9 | 5.8 | 11.3 |
| C8YI13C | 356 | 334 | 344 | 23.3 | 5.0 | 11.2 |
3.7. Dissociation of FITC-LPS Aggregates
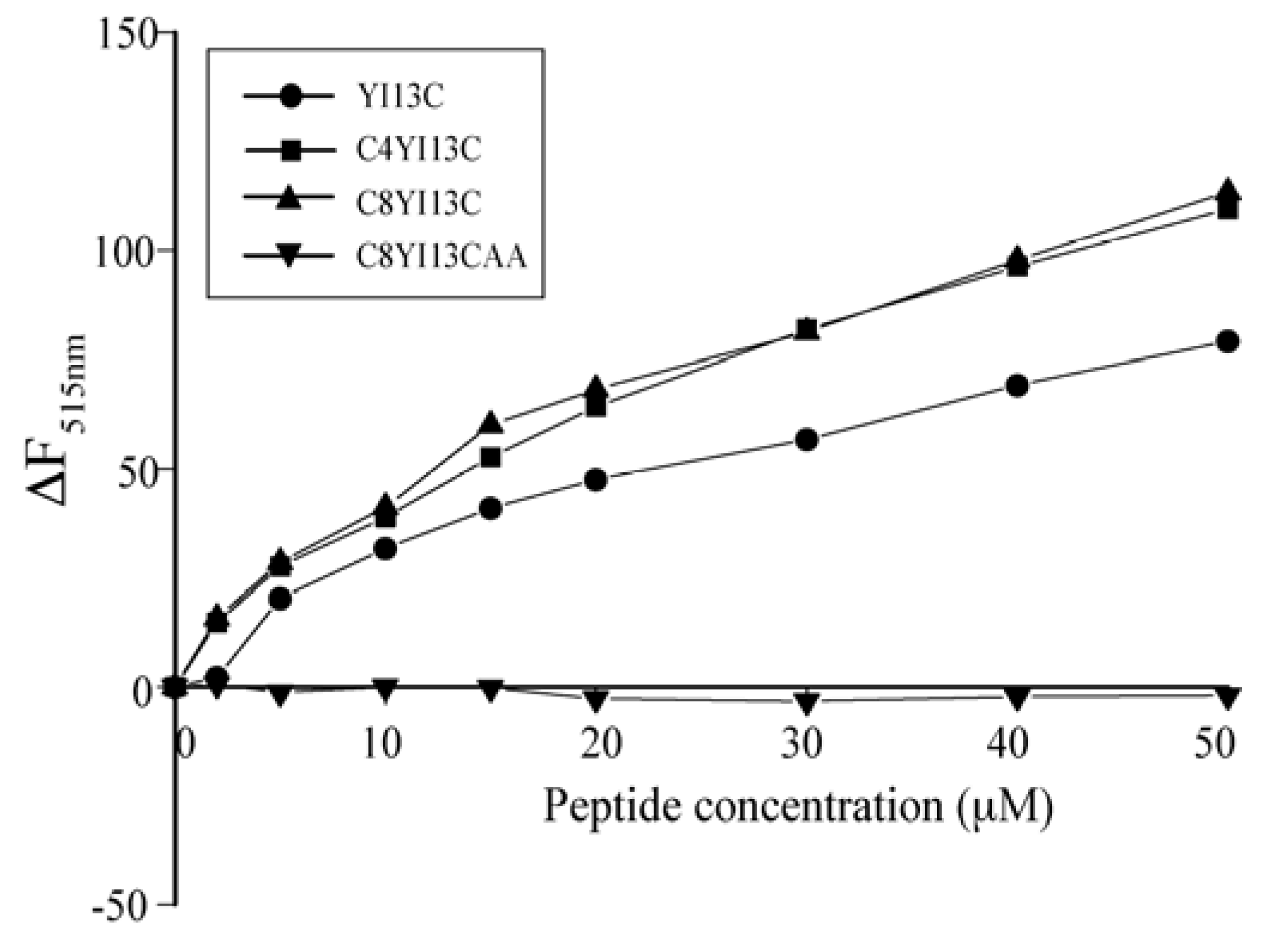
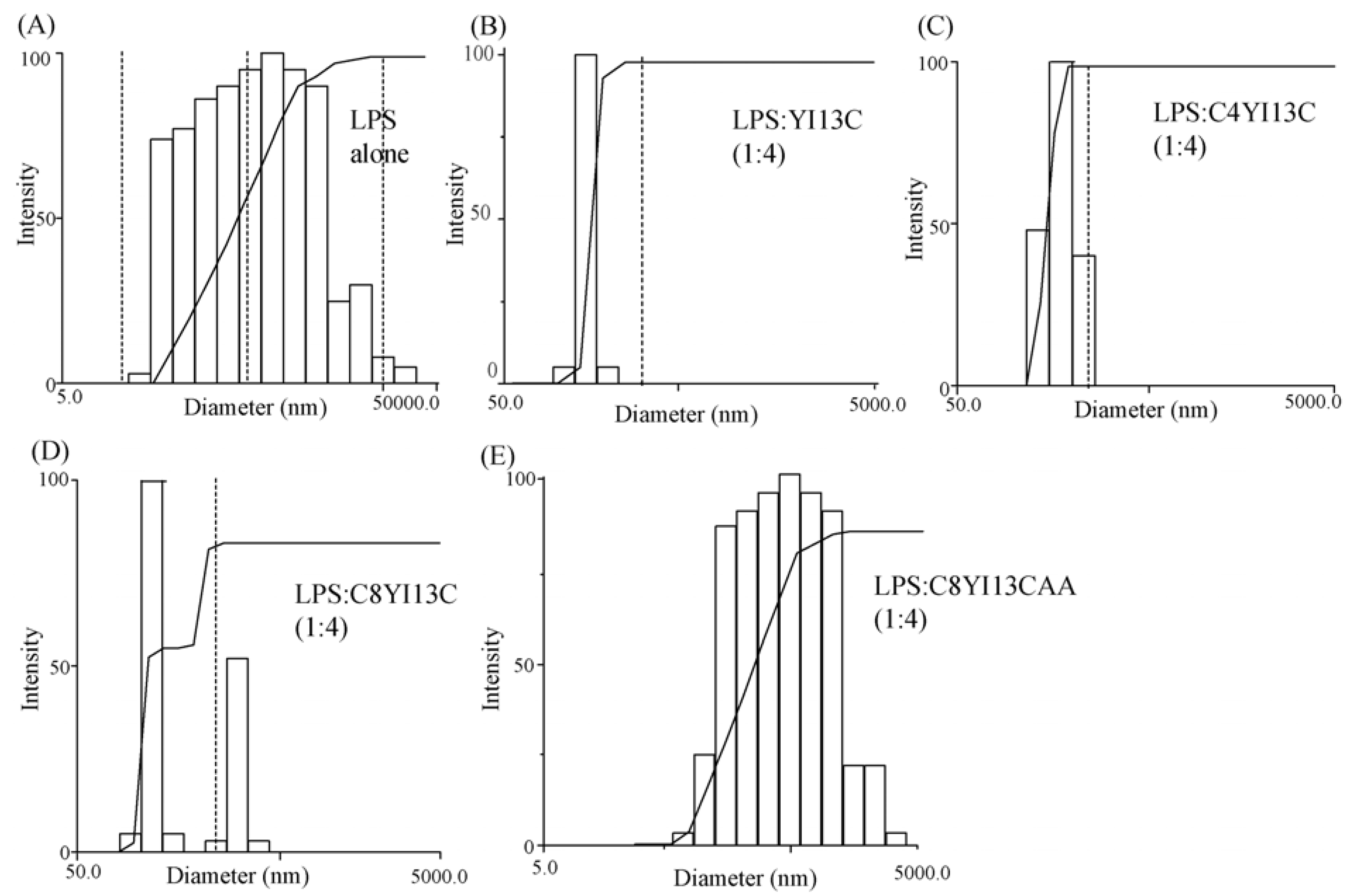
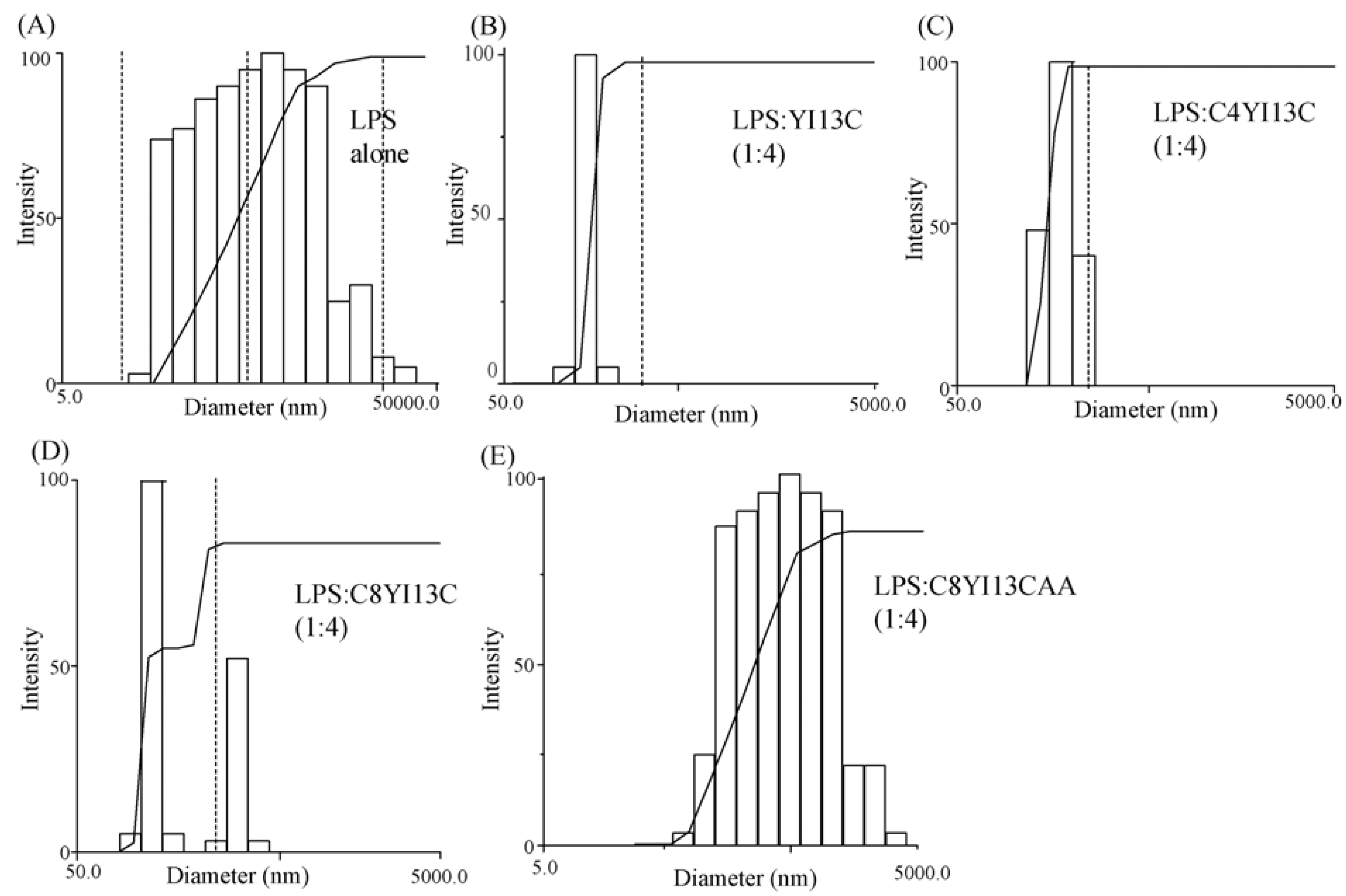
3.8. Dynamic Light Scattering Measurements
| LPS:Peptide | Diameter (nm) |
|---|---|
| LPS | 814 |
| LPS: YI13C (1:4) | 291 |
| LPS: C4YI13C (1:4) | 331 |
| LPS: C8YI13C (1:4) | 385 |
| LPS: C8YI13CAA (1:4) | 630 |
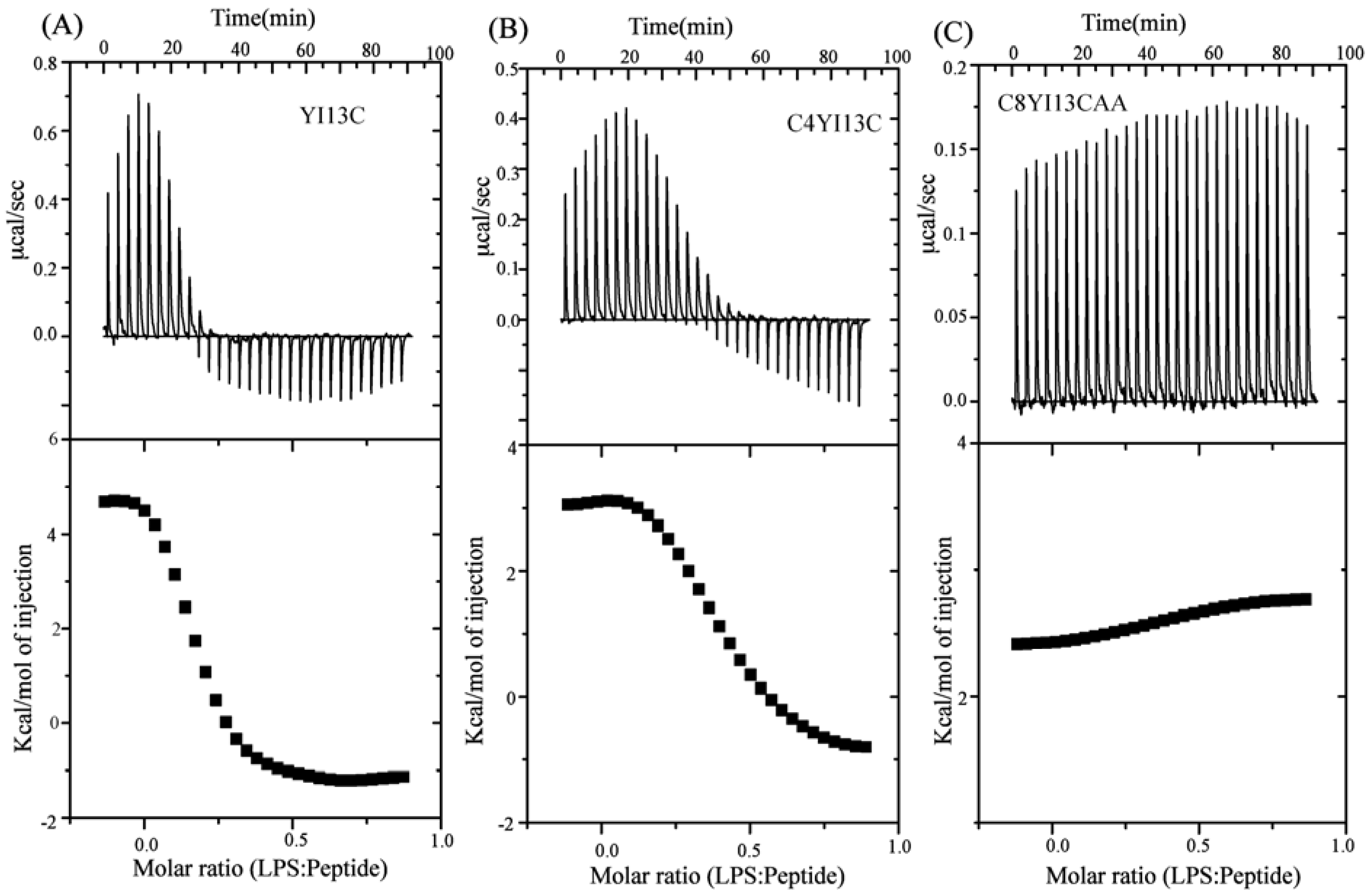
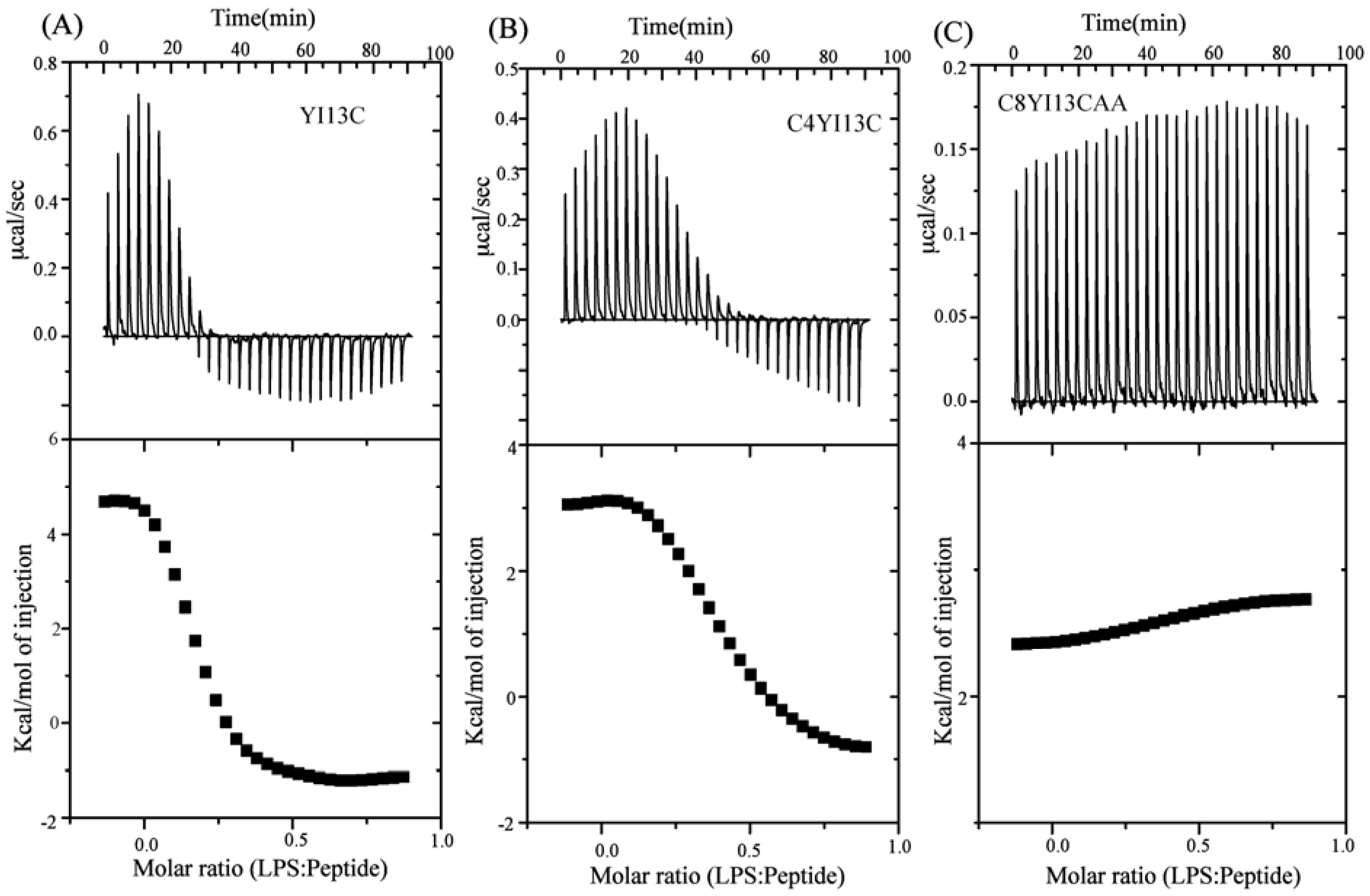
3.9. Isothermal Titration Calorimetry Studies
| Binding Parameters | YI13C | C4WFC |
|---|---|---|
| Ka (µM−1) | 4.2 | 2.2 |
| ΔH (kcal.mol−1) | 4.6 | 3.0 |
| TΔS(kcal.mol−1deg−1) | 13.6 | 11.6 |
| ΔG (kcal.mol−1) | −9.03 | −8.6 |
| Kd (µM) | 0.23 | 0.45 |
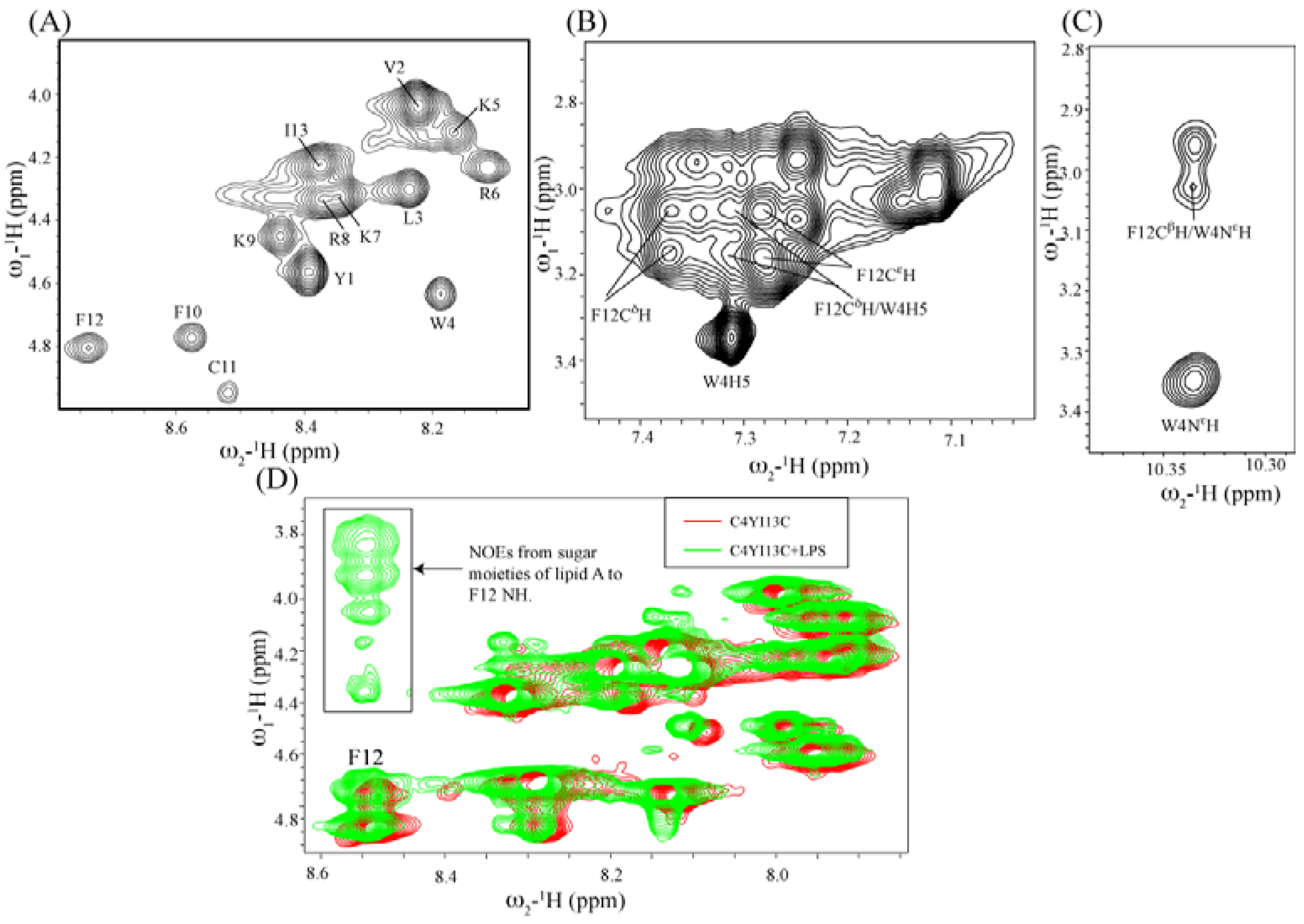
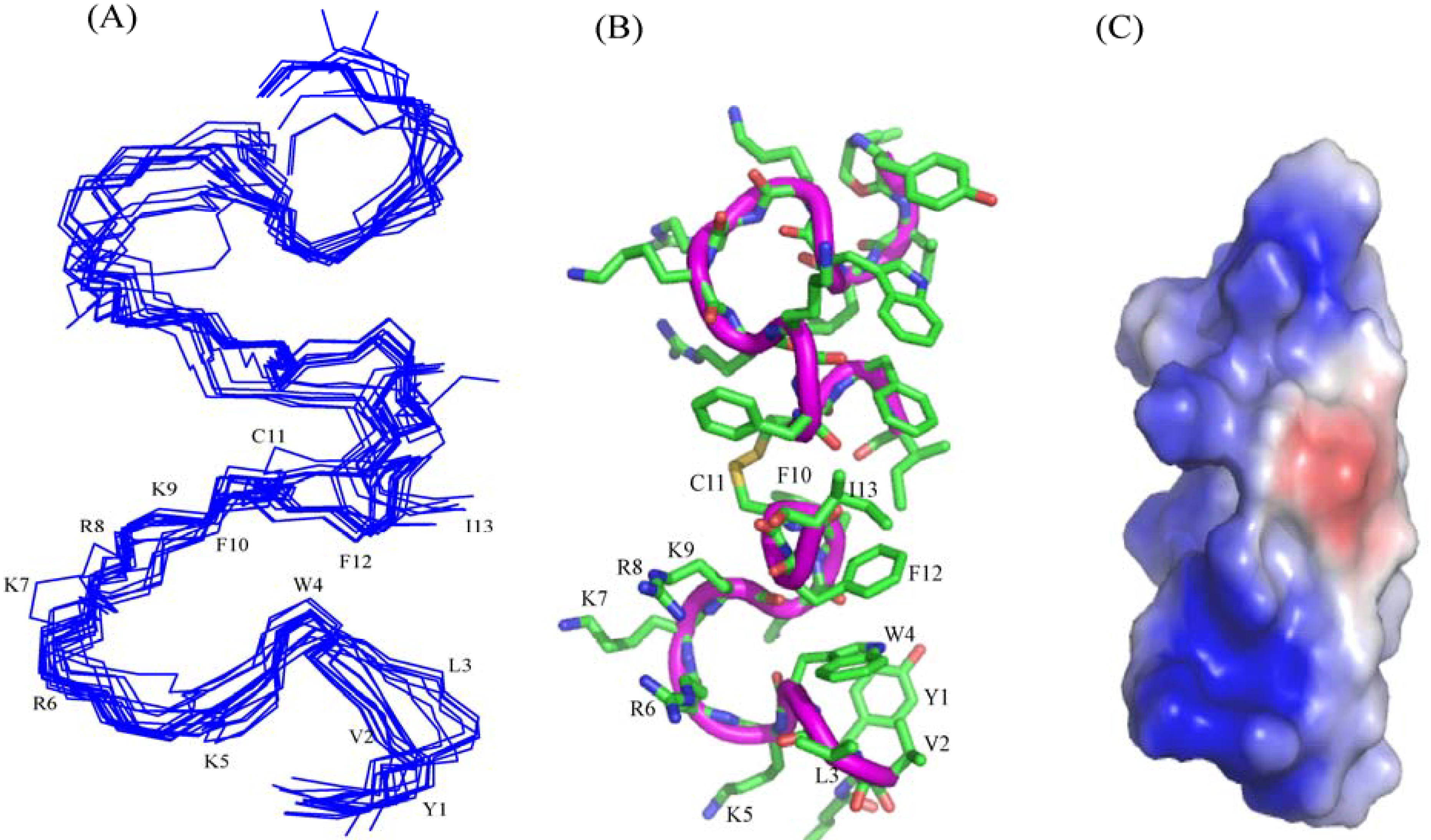
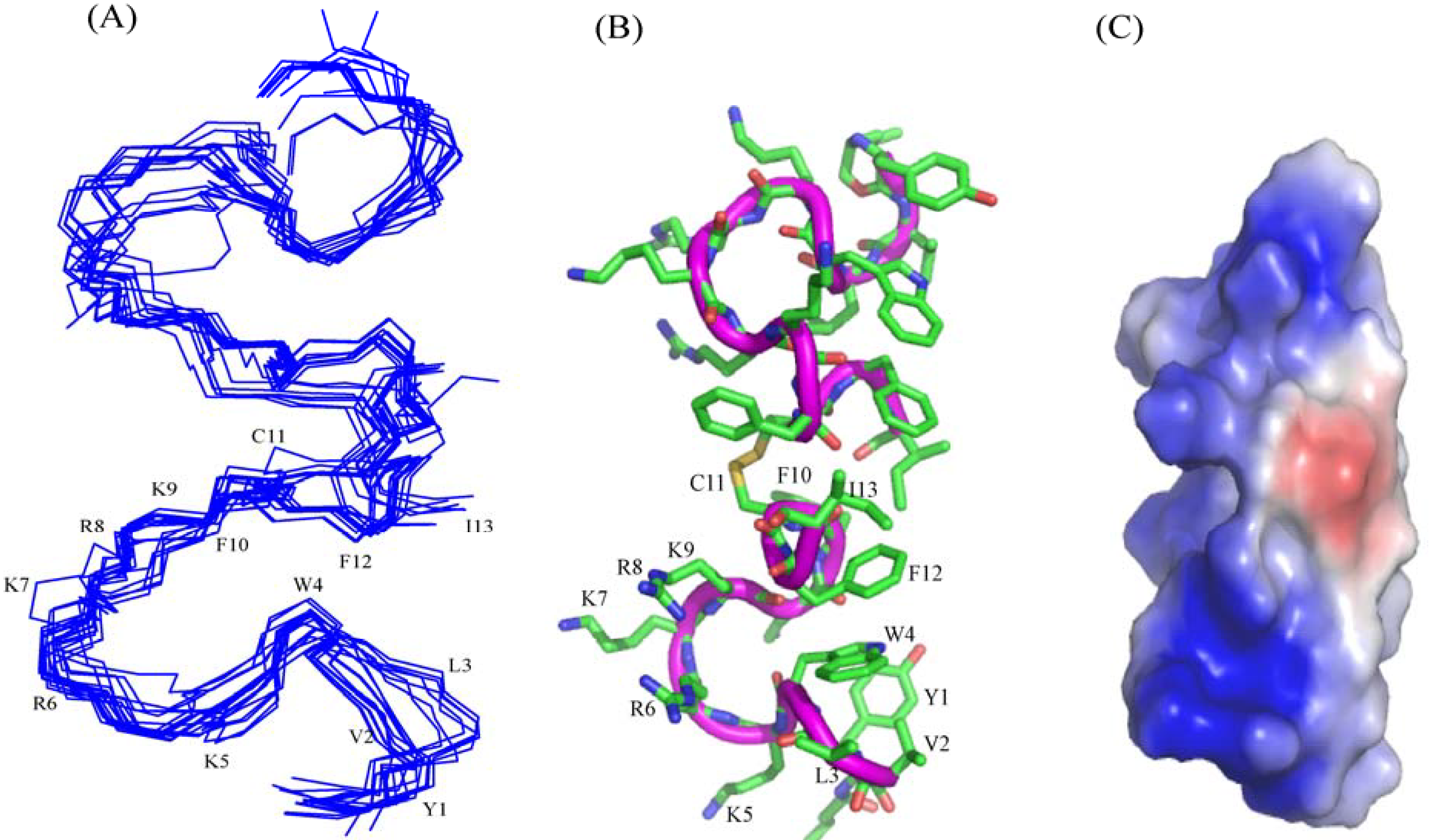
3.10. Structural Characterization by NMR Spectroscopy
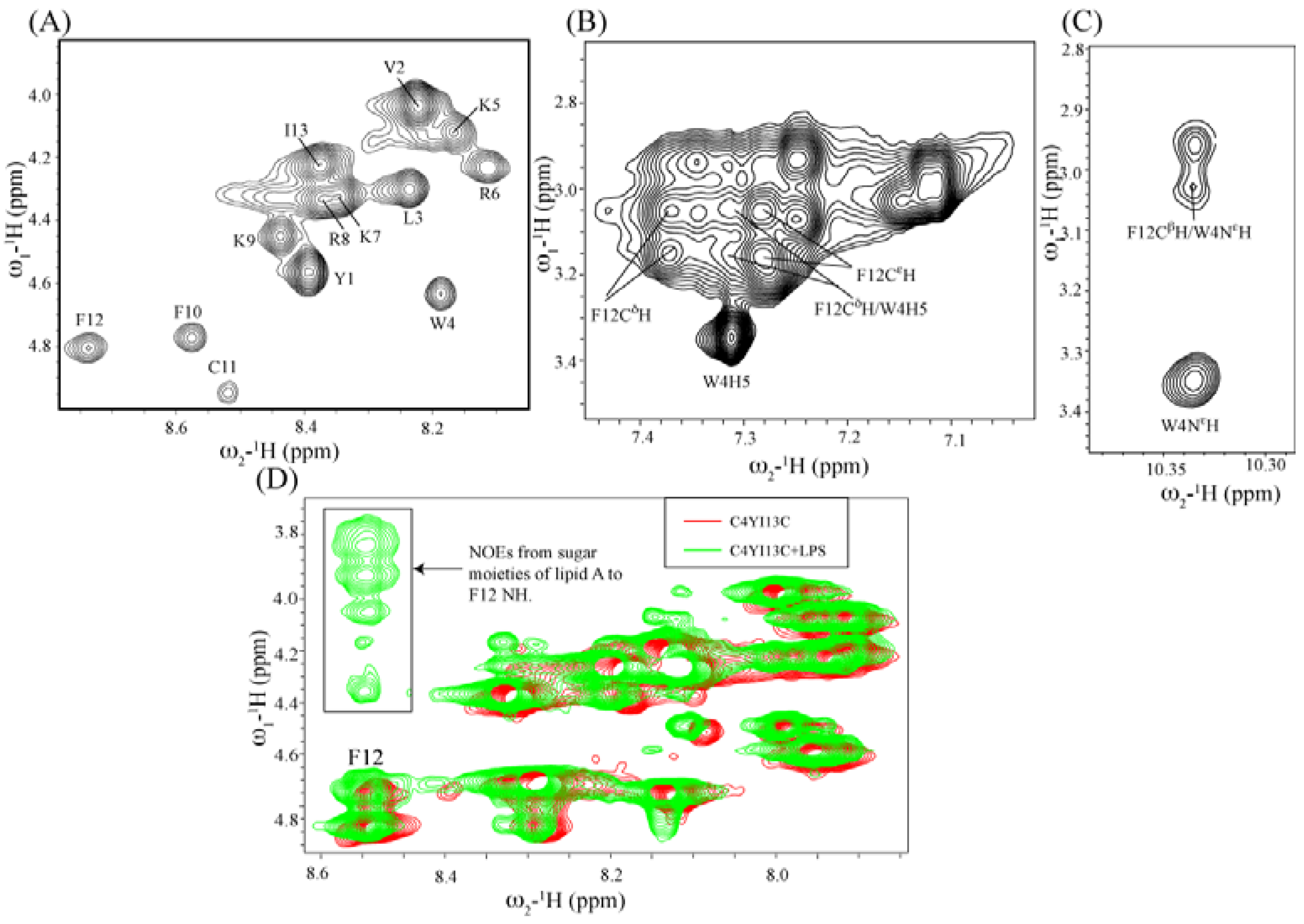
3.11. NMR Studies of YI13C and C4YI13C in LPS
3.12. Structure of C4YI13C Peptide in Aqueous Solution
| Distance restraints | |
| intraresidue (|i − j| =0) | 22 |
| sequential (|i − j| = 1) | 110 |
| medium range (2 ≤ |i − j| ≤ 4) | 48 |
| long range (|i − j| > 5) | 26 |
| total NOE constraints | 207 |
| Deviation from mean structure | |
| backbone atoms (N,Cα, C`) (Å) | 0.76 |
| heavy atoms (Å) | 1.6 |
| Ramachandran plot for the mean structure | |
| % residues in the most favourable and additionally allowed regions | 100 |
| % residues in the generously allowed region | 0 |
| % residues in the disallowed region | 0 |
4. Conclusions
Acknowledgments
Author contribution
Conflict of interests
References
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends. Biotechnol. 2011, 9, 464–472. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar]
- Ganz, T.; Lehrer, R.I. Antimicrobial peptides of vertebrates. Curr. Opin. Immunol. 1998, 10, 41–44. [Google Scholar] [CrossRef]
- Shai, Y. From innate immunity to de-novo designed antimicrobial peptides. Curr. Pharm. Des. 2002, 8, 715–725. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic alpha helical antimicrobial peptides. Biopolym. (Pept. Sci.) 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Boman, H.G. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef]
- Brown, K.L.; Hancock, R.E.W. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Jenssen, H.; Powers, H.; Hancock, R.E.W. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Lehrer, R.I. Cationic peptides: A new source of antibiotics. Trends. Biotechnol. 1998, 16, 82–88. [Google Scholar] [CrossRef]
- Aoki, W.; Ueda, M. Characterisation of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals 2013, 6, 1055–1081. [Google Scholar] [CrossRef]
- Brogden, N.K.; Brogden, K.A. Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 2011, 38, 217–225. [Google Scholar]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar]
- Bhunia, A.; Saravanan, R.; Mohanram, H.; Mangoni, M.L.; Bhattacharjya, S. NMR structures and interactions of temporin-1Tl and temporin-1Tb with lipopolysaccharide micelles. Mechanistic insights into outer membrane permeabilization and synergisitc activity. J. Biol. Chem. 2011, 286, 24394–24406. [Google Scholar] [CrossRef]
- Mohanram, H.; Bhattacharjya, S. Resurrecting inactive antimicrobial peptides from the lipopolysaccharide trap. Antimicrob. Agents Chemother. 2014. [Google Scholar] [CrossRef]
- Barnickel, G.; Bradaczek, H.; Naumann, D.; Rietschel, E.T.; Giesbrecht, P.; Labischinski, H. High state of order of isolated bacterial lipopolysaccharide and its possible contribution to the permeation barrier property of the outer membrane. J. Bacteriol. 1985, 162, 9–13. [Google Scholar]
- Allende, D.; McIntosh, T.J. Lipopolysaccharides in bacterial membranes act like cholesterol in eukaryotic plasma membranes in providing protection against melittin-induced bilayer lysis. Biochemistry 2003, 42, 1101–1108. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Epand, R.F.; Rosenfeld, Y.; Peleg, A.; Barra, D.; Epand, R.M.; Shai, Y. Lipopolysaccharide, a key molecule involved in the synergism between temporins in inhibiting bacterial growth and in endotoxin neutralization. J. Biol. Chem. 2008, 283, 22907–22917. [Google Scholar] [CrossRef]
- Marra, M.N.; Wilde, C.G.; Griffith, J.E.; Snable, J.L.; Scott, R.W. Bactericidal/permeability increasing protein has endotoxin neutralizing ability. J. Immunol. 1990, 144, 662–666. [Google Scholar]
- Bhattacharjya, S. De novo Designed lipopolysaccharide binding peptides: structure based development of antiendotoxic and antimicrobial drugs. Curr. Med. Chem. 2010, 17, 3080–3093. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Xu, J.; Kochanek, K.D.; Tejada-vera, B. Deaths: Preliminary data for 2007. National Vital Statistics Report 2009, 58, 1–52. [Google Scholar]
- David, S.A. Towards a rational development of anti-endotoxin agents: Novel approaches to sequestration of bacterial endotoxins with small molecules. J. Mol. Recognit. 2001, 14, 370–387. [Google Scholar] [CrossRef]
- Bowdish, D.M.E.; Hancock, R.E.W. Anti-endotoxin properties of cationic host defence peptides and proteins. J. Endotoxin. Res. 2005, 11, 230–236. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Lichtenstein, A.K.; Ganz, T. Defensins: antimicrobial and cytotoxic peptides of mammlian cells. Annu. Rev. Immunol. 1993, 11, 105–128. [Google Scholar] [CrossRef]
- Kokryakov, V.N.; Harwig, S.S.L.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Harwig, S.S.L.; Waring, A.; Yang, H.J.; Cho, Y.; Tan, L.; Lehrer, R.I. Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur. J. Biochem. 1996, 240, 352–357. [Google Scholar]
- Mani, R.; Waring, A.J.; Lehrer, R.I.; Hong, M. Membrane disruptive abilities of beta hairpin antimicrobial peptides correlate with conformation and activity: A 31P and 1H NMR study. Biochim. Biophys. Acta 2005, 1716, 11–18. [Google Scholar]
- Varkey, J.; Nagaraj, R. Antibacterial activity of human neutrophil defensin HNP-1 analogs without cysteines. Antimicrob. Agents Chemother. 2005, 49, 4561–4566. [Google Scholar] [CrossRef]
- Haney, E.F.; Vogel, H.J. NMR of antimicrobial peptides. Annu. Rep. NMR 2009, 65, 1–51. [Google Scholar]
- Findlay, B.; Zhanel, G.G.; Schweizer, F. Cationic amphiphiles, a new generation of antimicrobials inspired by the natural antimicrobial peptide scaffold. Antimicrob. Agents Chemother. 2010, 54, 4049–4058. [Google Scholar] [CrossRef]
- Andra, J.; Lohner, K.; Blondelle, S.E.; Jerala, R.; Moriyon, I.; Koch, M.H.; Garidel, P.; Brandenburg, K. Enhancement of endotoxin neutralization by coupling of a C12-alkyl chain to a lactoferricin derived peptide. Biochem. J. 2005, 385, 135–143. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: Similarities and differences in cell specificities and modes of action. Cell. Mol. Life Sci. 2011, 68, 2267–2280. [Google Scholar] [CrossRef]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. N-terminal modifications of Polymyxin B nonapeptide and their effect on antibacterial activity. Peptides 2001, 22, 1675–1681. [Google Scholar] [CrossRef]
- Farnaud, S.; Evans, R.W. Lactoferrin-a multifunctional protein with antimicrobial properties. Mol. Immunol. 2003, 40, 395–405. [Google Scholar] [CrossRef]
- Majerle, A.; Kidric, J.; Jerala, R. Enhancement of antibacterial and lipopolysacchride binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemother. 2003, 51, 1159–1165. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Lev, N.; Shai, Y. Effect of the hydrophobicity to net positive charge ratio on antibacterial and anti-endotoxin activities of structurally similar antimicrobial peptides. Biochemistry 2010, 49, 853–861. [Google Scholar] [CrossRef]
- Lockwood, N.A.; Haseman, J.R.; Tirrell, M.V.; Mayo, K.H. Acylation of SC4 dodecapeptide increases bactericidal potency against Gram positive bacteria, including drug resistant strains. Biochem. J. 2004, 378, 93–103. [Google Scholar] [CrossRef]
- Etzerodt, T.; Henriksen, J.R.; Rasmussen, P.; Clausen, M.H.; Andresen, T.L. Selective acylation enhances membrane charge sensitivity of the antimicrobial peptide Mastoporan-X. Biophys. J. 2011, 100, 399–409. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Domadia, P.N.; Bhunia, A.; Malladi, S.; David, S.A. High-resolution solution structure of a designed peptide bound to lipopolysaccharide: Transferred nuclear Overhauser effects, micelle selectivity, and anti-endotoxic activity. Biochemistry 2007, 46, 5864–5874. [Google Scholar] [CrossRef]
- Bhunia, A.; Mohanram, H.; Domadia, P.N.; Torres, J.; Bhattacharjya, S. Designed beta-boomerang antiendotoxic and antimicrobial peptides: sturctures and activities in lipopolysaccharide. J. Bio. Chem. 2009, 284, 21991–22004. [Google Scholar] [CrossRef]
- Bhunia, A.; Geoklin, C.; Domadia, P.N.; Warshakoon, H.; Cromer, J.R.; David, S.A.; Bhattacharjya, S. Interactions of a designed peptide with lipopolysaccharide: Bound conformation and anti-endotoxic activity. Biochem. Biophys. Res. Commun. 2008, 369, 853–857. [Google Scholar] [CrossRef]
- Bhunia, A.; Mohanram, H.; Bhattacharjya, S. Lipopolysacchride bound structures of the active fragments of fowlicidin-1, a cathelicidin family of antimicrobial and antiendotoxic peptide from chicken, determined by transferred nuclear Overhauser effect spectroscopy. Biopolym. (Pept. Sci.) 2008, 92, 9–22. [Google Scholar]
- Bhunia, A.; Domadia, P.N.; Bhattacharjya, S. Structural and thermodynamic analyses of the interaction between melittin and lipopolysaccharide. Biochim. Biophys. Acta 2007, 1768, 3282–3291. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Ramamoorthy, A. Multifunctional host defense peptides: Functional and mechanistic insights from NMR structures of potent antimicrobial peptides. FEBS J. 2009, 276, 6465–6473. [Google Scholar] [CrossRef]
- Guntert, P. Automated NMR protein structure calculation with CYANA. Meth. Mol. Biol. 2004, 278, 353–378. [Google Scholar]
- Laskowski, R.A.; Rullmann, J.A.C.; MacAruthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–496. [Google Scholar]
- Domingues, M.M.; Castanho, M.A.R.B.; Santos, N.C. rBPI21 promotes lipopolysaccharide aggregation and exerts its antimicrobial effects by (hemi)fusion of PG-containing membranes. PLoS One 2009, 4, 8385. [Google Scholar] [CrossRef]
- Srimal, S.; Surolia, N.; Balasubramanian, S.; Surolia, A. Titration calorimetric studies to elucidate the specificity of the interactions of polymyxin B with lipopolysaccharides and lipidA. Biochem. J. 1996, 315, 679–686. [Google Scholar]
- Bhattacharjya, S.; David, S.A.; Mathan, V.I.; Balaram, P. Polymyxin B nonapeptide: Conformations in water and in the lipopolysaccharide bound state determined by two-dimensional NMR and molecular dynamics. Biopolymers 1997, 41, 251–265. [Google Scholar] [CrossRef]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Vasil, M.L.; Hodges, R.S. “Specificity determinants” improve therapeutic indices of two antimicrobial peptides piscidin 1 and dermaseptin S4 against the gram-negative pathogens Acinetobacter baumannii and Pseudomonas aeruginosa. Pharmaceuticals 2014, 7, 366–391. [Google Scholar] [CrossRef]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Wang, G. Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs. Pharmaceuticals (Basel) 2013, 6, 728–758. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mohanram, H.; Bhattacharjya, S. β-Boomerang Antimicrobial and Antiendotoxic Peptides: Lipidation and Disulfide Bond Effects on Activity and Structure. Pharmaceuticals 2014, 7, 482-501. https://doi.org/10.3390/ph7040482
Mohanram H, Bhattacharjya S. β-Boomerang Antimicrobial and Antiendotoxic Peptides: Lipidation and Disulfide Bond Effects on Activity and Structure. Pharmaceuticals. 2014; 7(4):482-501. https://doi.org/10.3390/ph7040482
Chicago/Turabian StyleMohanram, Harini, and Surajit Bhattacharjya. 2014. "β-Boomerang Antimicrobial and Antiendotoxic Peptides: Lipidation and Disulfide Bond Effects on Activity and Structure" Pharmaceuticals 7, no. 4: 482-501. https://doi.org/10.3390/ph7040482
APA StyleMohanram, H., & Bhattacharjya, S. (2014). β-Boomerang Antimicrobial and Antiendotoxic Peptides: Lipidation and Disulfide Bond Effects on Activity and Structure. Pharmaceuticals, 7(4), 482-501. https://doi.org/10.3390/ph7040482