Abstract
In an ongoing effort to identify novel drugs that can be used as neurotherapeutic compounds, we have focused on anilino enaminones as potential anticonvulsant agents. Enaminones are organic compounds containing a conjugated system of an amine, an alkene and a ketone. Here, we review the effects of a small library of anilino enaminones on neuronal activity. Our experimental approach employs an olfactory bulb brain slice preparation using whole-cell patch-clamp recording from mitral cells in the main olfactory bulb. The main olfactory bulb is a key integrative center in the olfactory pathway. Mitral cells are the principal output neurons of the main olfactory bulb, receiving olfactory receptor neuron input at their dendrites within glomeruli, and projecting glutamatergic axons through the lateral olfactory tract to the olfactory cortex. The compounds tested are known to be effective in attenuating pentylenetetrazol (PTZ) induced convulsions in rodent models. One compound in particular, KRS-5Me-4-OCF3, evokes potent inhibition of mitral cell activity. Experiments aimed at understanding the cellular mechanism underlying the inhibitory effect revealed that KRS-5Me-4-OCF3 shifts the concentration-response curve for GABA to the left. KRS-5Me-4-OCF3 enhances GABA affinity and acts as a positive allosteric modulator of GABAA receptors. Application of a benzodiazepine site antagonist blocks the effect of KRS-5Me-4-OCF3 indicating that KRS-5Me-4-OCF3 binds at the classical benzodiazepine site to exert its pharmacological action. This anilino enaminone KRS-5Me-4-OCF3 emerges as a candidate for clinical use as an anticonvulsant agent in the battle against epileptic seizures.
1. Enaminones in Drug Discovery and Development
Enaminones are the enamines of β-dicarbonyl compounds that comprised an amino group linked through a carbon-carbon double bond to a keto group (HN-C=C-C=O). [1]. Structure-activity relationship studies are ongoingbecause of the excellent anticonvulsant activity of previously synthesized aromatic-containing enaminones [2]. Specifically, a diverse series of anilino enaminones was recently synthesized and investigated as potential anticonvulsant compounds. These compounds are structurally unique compared to currently available antiepileptic drugs [3,4,5,6,7,8]. Furthermore, this class of enaminones has been shown to exhibit good-to-moderate protection in the traditional preclinical animal models, the subcutaneous pentylenetetrazol test (scPTZ) and the maximal electroshock seizure test. The scPTZ seizure model identifies compounds that inhibit the GABA antagonistic effects of pentylenetetrazol or raise the seizure threshold [9]. In terms of their anticonvulsant activities, anilino enaminones are comparable to some clinically used agents in animal models of seizures. In addition, the compounds have a minimal neurotoxicity side effect profile as well as a wider margin of safety than conventional antiepileptic drugs such as carbamazepine, valproate and phenytoin [10,11,12]. The enaminone system serves as the unique pharmacophoric structure of the anilino analogs. Together with their demonstrated diversity of bioactivities, these features provide an excellent opportunity for drug discovery and development. Here, we review the cellular effects of a small library of anilino enaminones on a key output neuron in a brain slice preparation containing the main olfactory bulb, which was used as an experimental platform to study enaminones.
Previous reports have explored the nucleus accumbens and hippocampus in the rat brain to study enaminone suppression of excitatory synaptic transmission and epileptiform activity [6,13]. The main olfactory bulb is anatomically different from those two brain regions and provides three advantages to test the cellular mechanisms of the effects of anilino enaminones [14,15]. Firstly, epilepsy-related proteins such as GABAA receptors, sodium channels, ionotropic glutamate receptors and metabotropic glutamate receptors are expressed in the key output neuron type of the main olfactory bulb, the mitral cell. Secondly, mitral cells spontaneously generate action potentials with epilepsy-related proteins participating in the regulation of neuronal spiking. Thirdly, the excitability of mitral cells can be regulated by synaptic input. Therefore, mitral cells in an acute slice preparation of the main olfactory bulb serve as a good model for testing the bioactivity of enaminone compounds and to explore the mechanisms underlying their activity.
Anilino enaminones exhibit anticonvulsant activity in vivo [3,12,13]. One anilino enaminone, E139, inhibits EPSCs in the rat nucleus accumbens and hippocampus by increasing extracellular GABA concentrations [6,13], and by inhibiting tetrodotoxin-sensitive sodium currents to regulate action potential firing in neurons [16]. Other studies suggest different mechanisms as the basis for anticonvulsant activity. Benzylamino enaminones have a similar chemical structure to anilino enaminones with benzyl-substitution at the NH-moiety. Certain benzylamino enaminones exhibit anticonvulsant effects in neurons of rats and mice by suppressing glutamate-mediated excitation and action potential firing [17]. Substitutions at the NH-moiety change the target protein to which enaminones bind. Subsequently, enaminones with similar chemical structure may possess different modes of action. Our hypothesis is that the substituted site in enaminones contributes to the mode of action of these compounds. Recent work determined the mechanism of anticonvulsant action of three enaminone compounds that have non-ortho-substituted cyclohexenone [18]. Specifically, the effects of three anilino enaminones on neuronal activity of output neurons, mitral cells, in an olfactory bulb brain slice preparation using whole-cell patch-clamp recording has been examined. We will first review the structural and functional organization of the main olfactory bulb, before describing the modulation of mitral cell activity by anilino enaminones.
2. Organization of the Main Olfactory Bulb
The main olfactory bulb is part of the cerebral cortex, namely the allocortex rather than the neocortex, based on its cytoarchitecture and fetal development. Allocortical structures do not go through a prenatal phase of six-layered structures and instead have three or four layers in the mature brain [19]. In rodents, the main olfactory bulb is a large, layered structure that occupies almost a quarter of the length of the cranial cavity [20] and is dedicated to the processing of odorant information [14,15,21]. Input to the main olfactory bulb comes from two sources, olfactory receptor neurons in the nasal epithelium and centrifugal feedback neurons originating in different higher brain areas. Each of the bipolar olfactory receptor neurons in the nasal epithelium projects a sensory dendrite into the thin layer of nasal mucus covering the nasal epithelium, and an axon to the ipsilateral main olfactory bulb. The axon of each olfactory receptor neuron travels through the most superficial layer of the main olfactory bulb, the olfactory nerve layer, and forms synapses with neurons in one of many spheroidal neuropil structures, the olfactory glomeruli in the glomerular layer (Figure 1) (1800 glomeruli in the mouse main olfactory bulb [22], 8000 in humans [23]).
Deep to the olfactory nerve layer, the glomerular layer contains an array of olfactory glomeruli. Glomeruli are surrounded by neurons and glia [21] and house a diverse array of neuronal elements such as the axon terminals of olfactory receptor neurons. In addition, several types of local interneurons, collectively referred to as juxtaglomerular cells, send dendrites into the glomerular neuropil. Different morphological juxtaglomerular cell types can be distinguished such as external tufted cells, “short axon” cells (which actually have long axons extending through the glomerular layer [24]), and periglomerular cell [14]. Mitral/tufted cells are the main olfactory bulb output neurons. Each of them sends a single apical dendrite into one glomerulus. Olfactory receptor cells form direct synaptic contacts with mitral/tufted cells and with at least some juxtaglomerular cells [25]. Juxtaglomerular cells form synapses with many neuronal elements in the glomerular layer [26]. Periglomerular cells are GABAergic, short-axon cells whereas mitral/tufted cells are glutamatergic [27,28]. Periglomerular cells receive glutamatergic input from olfactory receptor cell axons or dendrodendritic glutamatergic input from mitral/tufted cells [14,27]. Periglomerular cells presynaptically inhibit olfactory receptor neurons through GABAergic transmission [29,30]. Mitral/tufted cells receive spontaneous bursts of inhibitory postsynaptic currents (IPSCs) from periglomerular cells at inhibitory GABAergic synapses [31,32]. In addition, centrifugal axons originating in higher olfactory centers arborize in glomeruli and may provide modulatory feedback to the glomerular layer [21,33].
The external plexiform layer lies between the glomerular and the mitral cell layer. It consists primarily of the extensive lateral dendrites of mitral/tufted cells, and granule cell dendrites sent from deeper in the main olfactory bulb. The cell bodies of mitral cells form a thin distinct band delineating the mitral cell layer, immediately below the external plexiform layer (Figure 1). Mitral/tufted cells are the principal output neurons of the main olfactory bulb. They receive olfactory receptor cell input at their apical dendrites within glomeruli, and project glutamatergic axons through the lateral olfactory tract to olfactory cortex. Each mitral/tufted cell sends a single apical dendrite into one glomerulus and several lateral dendrites (6–8) that originate at the cell body for up to thousands of micrometers through the external plexiform layer. Lateral dendrites of mitral/tufted cells form reciprocal dendrodendritic synapses with granule cell dendrites and may also receive input from centrifugal fibers and Van Gehuchten cells [34,35]. A large population (~100,000) of granule cells is found in the mitral cell layer [36], outnumbering mitral cells (~40,000) [37]. The apical dendrite of these superficial granule cells as well as the dendrite of deeper granule cells extends into the external plexiform layer to form dendrodendritic synapses with mitral/tufted cells.
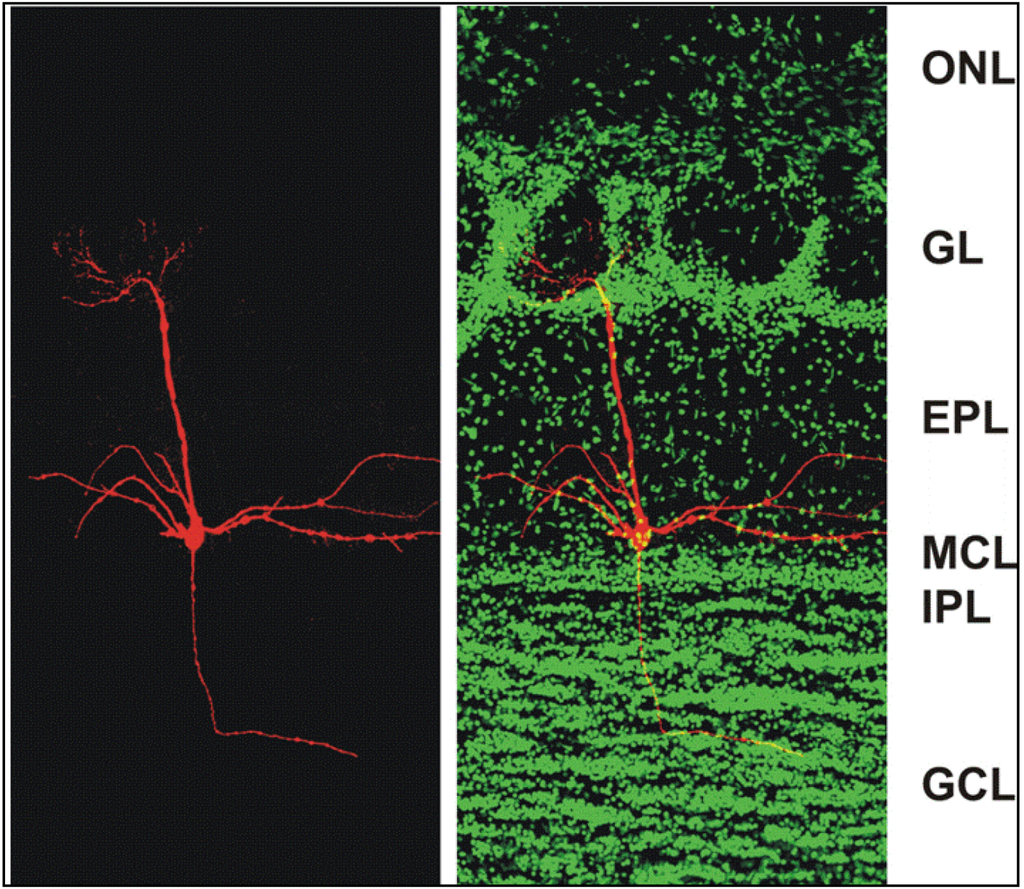
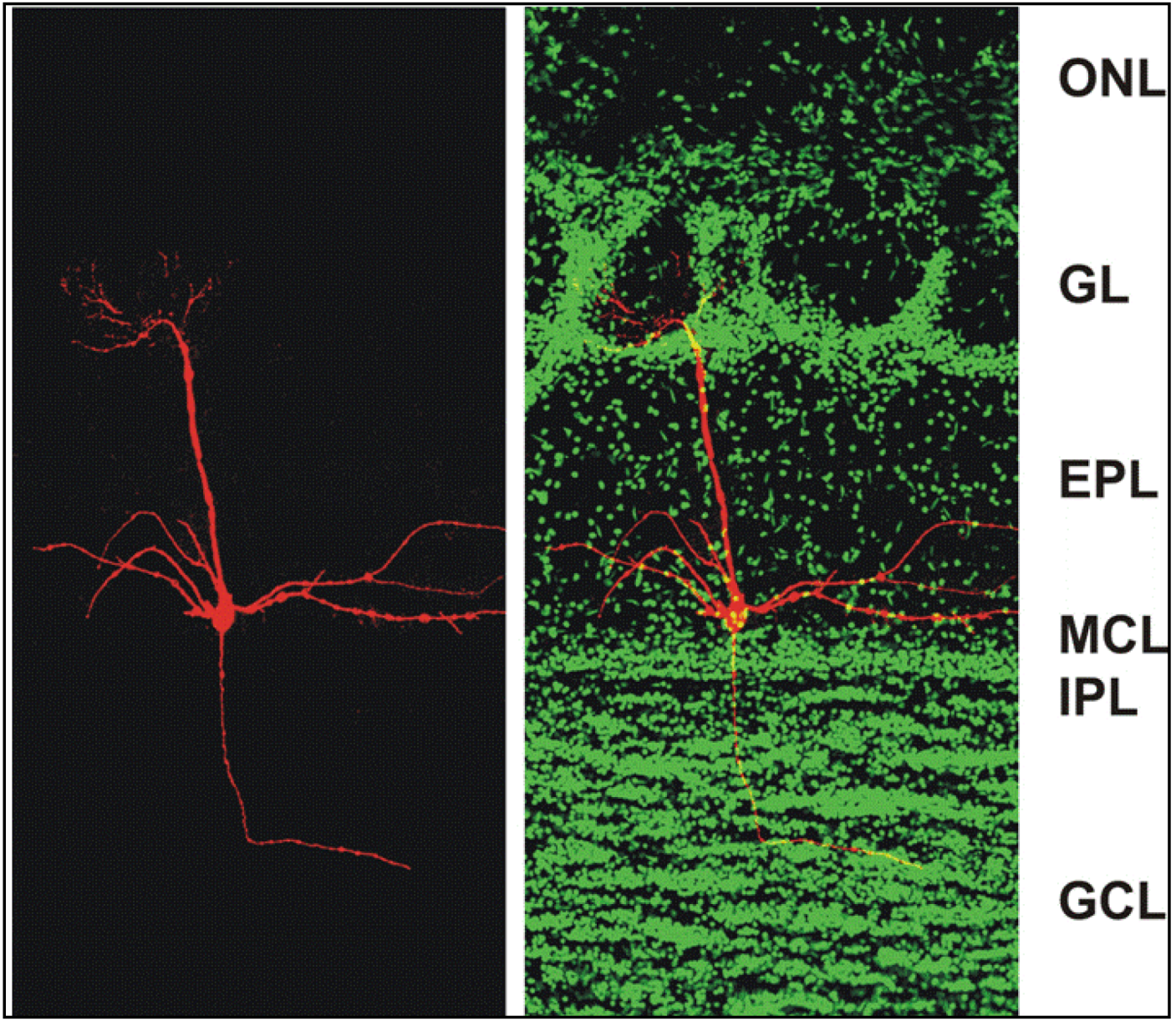
Figure 1.
Confocal micrographs of a section of the adult mouse main olfactory bulb with a single mitral cell intracellularly filled with biocytin (red) and nuclei stained with Sytox Green (green). The mitral cell soma is located in the mitral cell layer (MCL). One apical dendrite of the mitral cell reaches into one glomerulus and several lateral dendrites span the main olfactory bulb. ONL: olfactory nerve layer; GL: glomerular layer; EPL: external plexiform layer; MCL: mitral cell layer; IPL: internal plexiform layer; GCL: granule cell layer. From [38].
Figure 1.
Confocal micrographs of a section of the adult mouse main olfactory bulb with a single mitral cell intracellularly filled with biocytin (red) and nuclei stained with Sytox Green (green). The mitral cell soma is located in the mitral cell layer (MCL). One apical dendrite of the mitral cell reaches into one glomerulus and several lateral dendrites span the main olfactory bulb. ONL: olfactory nerve layer; GL: glomerular layer; EPL: external plexiform layer; MCL: mitral cell layer; IPL: internal plexiform layer; GCL: granule cell layer. From [38].
The internal plexiform layer is subjacent to the mitral cell layer and contains few cell bodies but is densely packed with neuropil, including mitral/tufted cell axons and granule cell dendrites. Centrifugal axons originating in several brain regions also pass through the internal plexiform layer. The granule cell layer houses several cell types, including the prominent granule cells. These GABAergic local interneurons have no axon. Instead, they send apical dendrites to the external plexiform layer where they interact with mitral/tufted cells, and they have shorter basal dendrites receiving glutamate synapses from axon terminals (reviewed below). Little is known about the function of other neuronal types in this layer such as GABAergic short-axon interneurons, Golgi cells and Blane’s cells [21]. Blane’s cells make inhibitory synaptic contact with granule cells [39].
Immediately below the granule cell layer, in the core of the main olfactory bulb, lies an ependymal layer that is continuous with the ventricular lining in the core of the main olfactory bulb. The main olfactory bulb is a prime example for continuous neuronal generation throughout adult life such that neuronal precursors migrate to the main olfactory bulb through the rostral migratory stream. These precursors enter the granule cell layer through the ependymal layer and differentiate to become functional granule cells or they continue to the glomerular layer to differentiate into periglomerular neurons [40,41].
3. Processing of Sensory and Synaptic Input within the Main Olfactory Bulb
In the main olfactory bulb, glutamate is the major excitatory neurotransmitter and GABA is the main inhibitory neurotransmitter. Most synaptic processing involves serial or reciprocal glutamatergic/GABAergic circuits, principally between the dendrites of output neurons such as mitral cells and local interneurons, with specific stages of processing occurring in discrete layers of the main olfactory bulb [15]. Mitral cell activity is critical for information processing since mitral cells integrate excitatory and inhibitory synaptic input from sensory afferents, local GABAergic interneurons and centrifugal fibers originating in cortical areas.
Sensory input from olfactory receptor cells that express the same odorant receptor protein in the nasal epithelium projects to a single glomerulus and activates mitral cells innervating the glomerulus such that odor-specific patterns of input are distributed across glomeruli. One odorant-specific input channel (i.e., a subset of receptor cells and their connected neurons in the main olfactory bulb such as mitral cells) influences activity in other channels through local inhibitory interneurons to enhance contrast and discriminate between odor-specific patterns of neural activity. These interneuron-mediated lateral interactions take place at two levels of synaptic processing.
Sensory input to olfactory glomeruli leads to widespread depolarization in the glomerular layer at the first level of processing. The depolarization is mediated by glutamate receptors and principally affects periglomerular cells [24]. Subsequently, these inhibitory interneurons release GABA to inhibit mitral cell responses to sensory input resulting in lateral inhibition. Short axon cells are activated by glutamate released by a population of external tufted cells instead of receiving direct glutamatergic input from olfactory afferents. Activation of short axon cells then results in widespread glutamate-mediated depolarization within the glomerular layer. Glutamate release by short axon cells activates periglomerular cells such that activation of one odorant-specific input channel can inhibit the responses of surrounding input channels with different odorant specificities similar to center-surround processing known from the retina [24].
At the second level of processing, in the external plexiform layer, mitral/tufted cell dendrites interact with dendrites of GABAergic granule cells [42]. Mitral cells release glutamate at their long lateral dendrites to activate granule cells which in turn release GABA [43] to inhibit the output of surrounding mitral cells that are part of different odorant-specific input channels. Both ionotropic and metabotropic glutamate receptors (mGluRs) facilitate feedback and feedforward inhibition of mitral cells (Figure 2 and Figure 3) [38,44,45]. Activation of mGluR5 on granule cells increases GABAergic inhibition of mitral cells. In the presence of ionotropic glutamate receptor blockers, the Group I mGluR agonist DHPG increases the frequency of spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs) in mitral cells [44,46]. Mitral cells express mGluR1 and respond to DHPG with an inward current in addition to the IPSCs evoked by GABA release from granule cells.
Inactivation of mGluRs attenuates mitral cell-evoked excitation of granule cells. As previously reported [47], depolarizing current steps applied to mitral cells in the presence of sodium channel blocker TTX and potassium channel blocker TEA are followed by a feedback IPSP in mitral cells. Blockade of mGluRs reduces granule cell-mediated feedback inhibition of mitral cells (Figure 3).
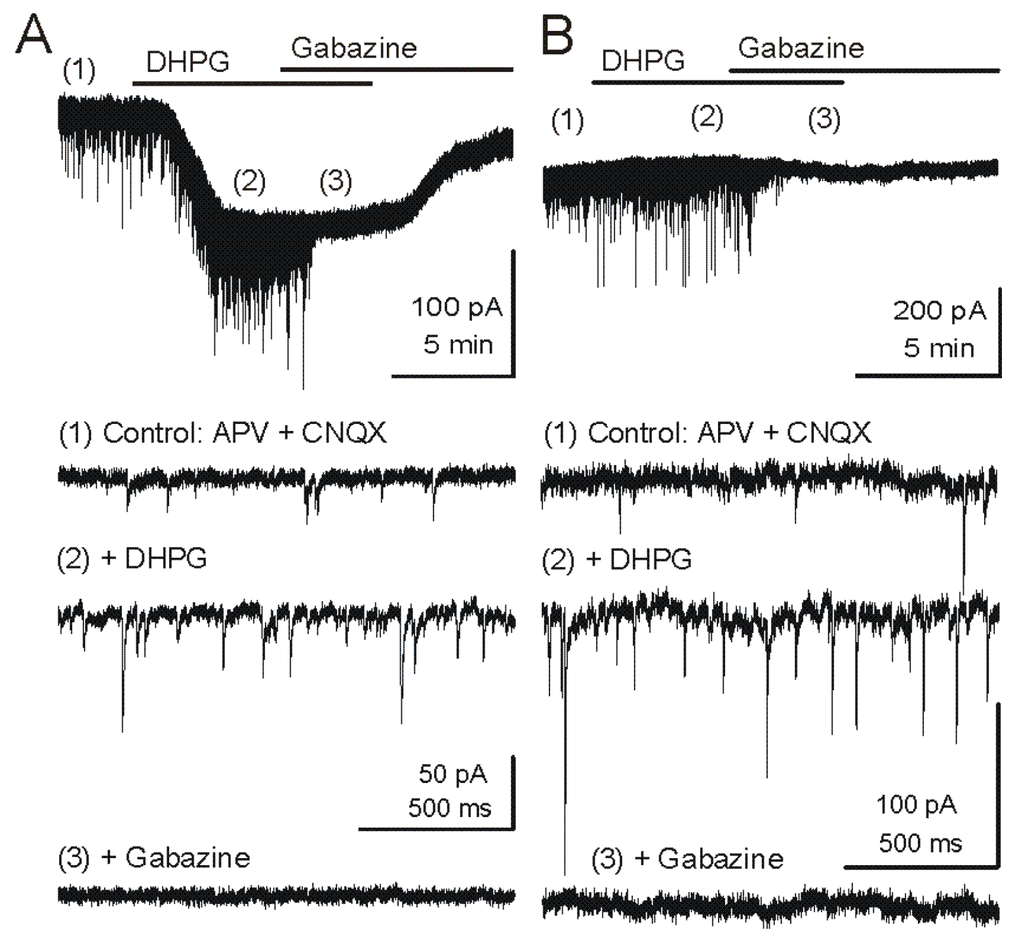
Figure 2.
Metabotropic glutamate receptor (mGluR) group I agonist DHPG increases the frequency of GABAergic IPSCs in mitral cells. Voltage clamp recordings from MCs are made with pipettes with a high chloride concentration during blockade of iGluRs. Under these conditions, chloride-mediated IPSCs are reversed in polarity and appear as downward deflections (inward currents). (A) Upper trace - Application of mGluR agonist DHPG (50 μM) evokes an inward current in mitral cells recorded in slices from wildtype mice. Timepoints 1–3 are shown at faster timescale in the three lower tracers, respectively. Note the increase in frequency of fast IPSPs (trace 2), which are completely blocked by the GABAA receptor antagonist gabazine (5 μM) (trace 3). (B) In a mitral cell from an mGluR1−/− mouse, the DHPG-evoked inward current was absent, but DHPG substantially increased the frequency of IPSCs. CNQX (10 μM), APV (50 μM). Recording conditions and labeling as in (A). Modified from [44].
Figure 2.
Metabotropic glutamate receptor (mGluR) group I agonist DHPG increases the frequency of GABAergic IPSCs in mitral cells. Voltage clamp recordings from MCs are made with pipettes with a high chloride concentration during blockade of iGluRs. Under these conditions, chloride-mediated IPSCs are reversed in polarity and appear as downward deflections (inward currents). (A) Upper trace - Application of mGluR agonist DHPG (50 μM) evokes an inward current in mitral cells recorded in slices from wildtype mice. Timepoints 1–3 are shown at faster timescale in the three lower tracers, respectively. Note the increase in frequency of fast IPSPs (trace 2), which are completely blocked by the GABAA receptor antagonist gabazine (5 μM) (trace 3). (B) In a mitral cell from an mGluR1−/− mouse, the DHPG-evoked inward current was absent, but DHPG substantially increased the frequency of IPSCs. CNQX (10 μM), APV (50 μM). Recording conditions and labeling as in (A). Modified from [44].
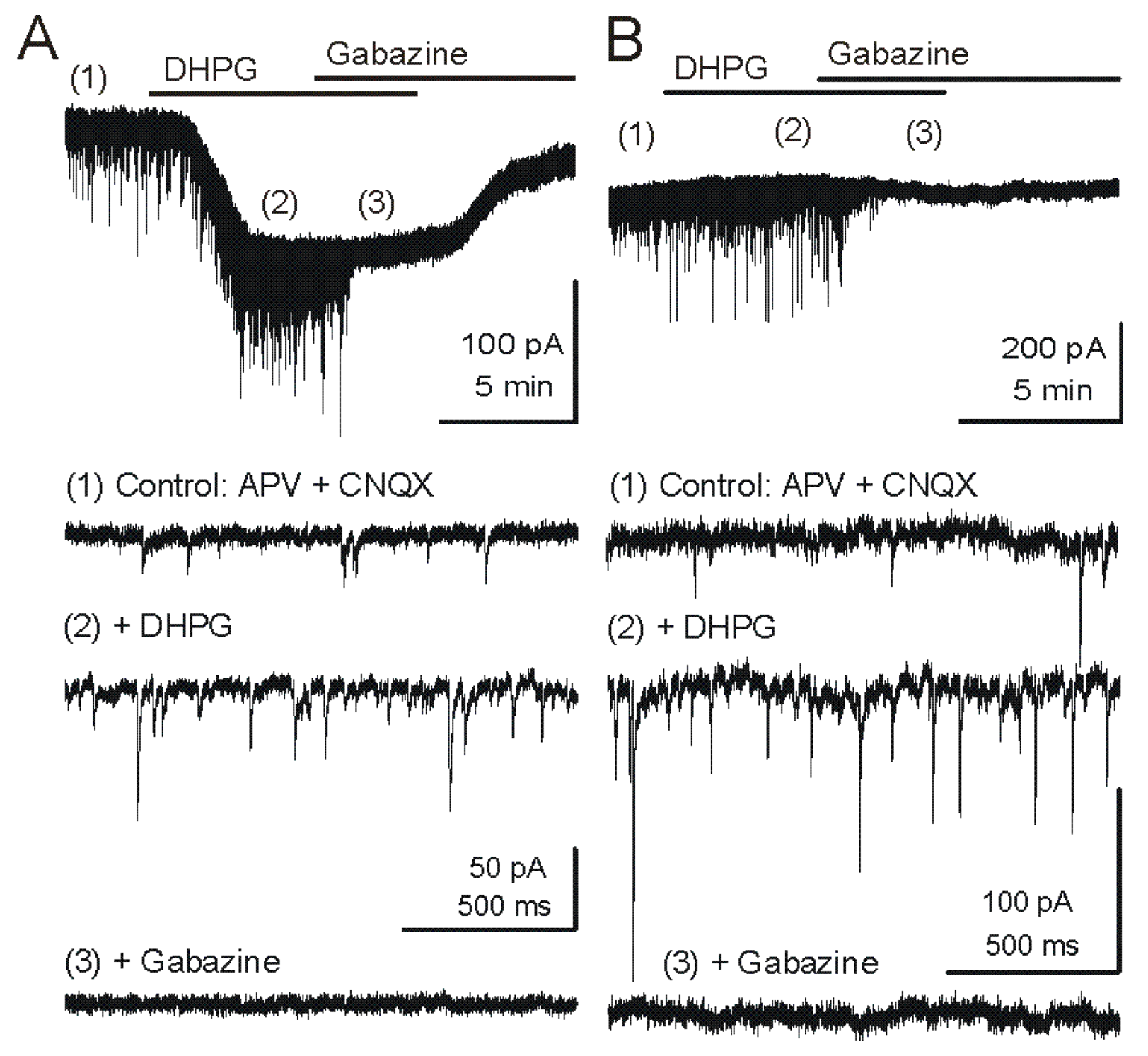
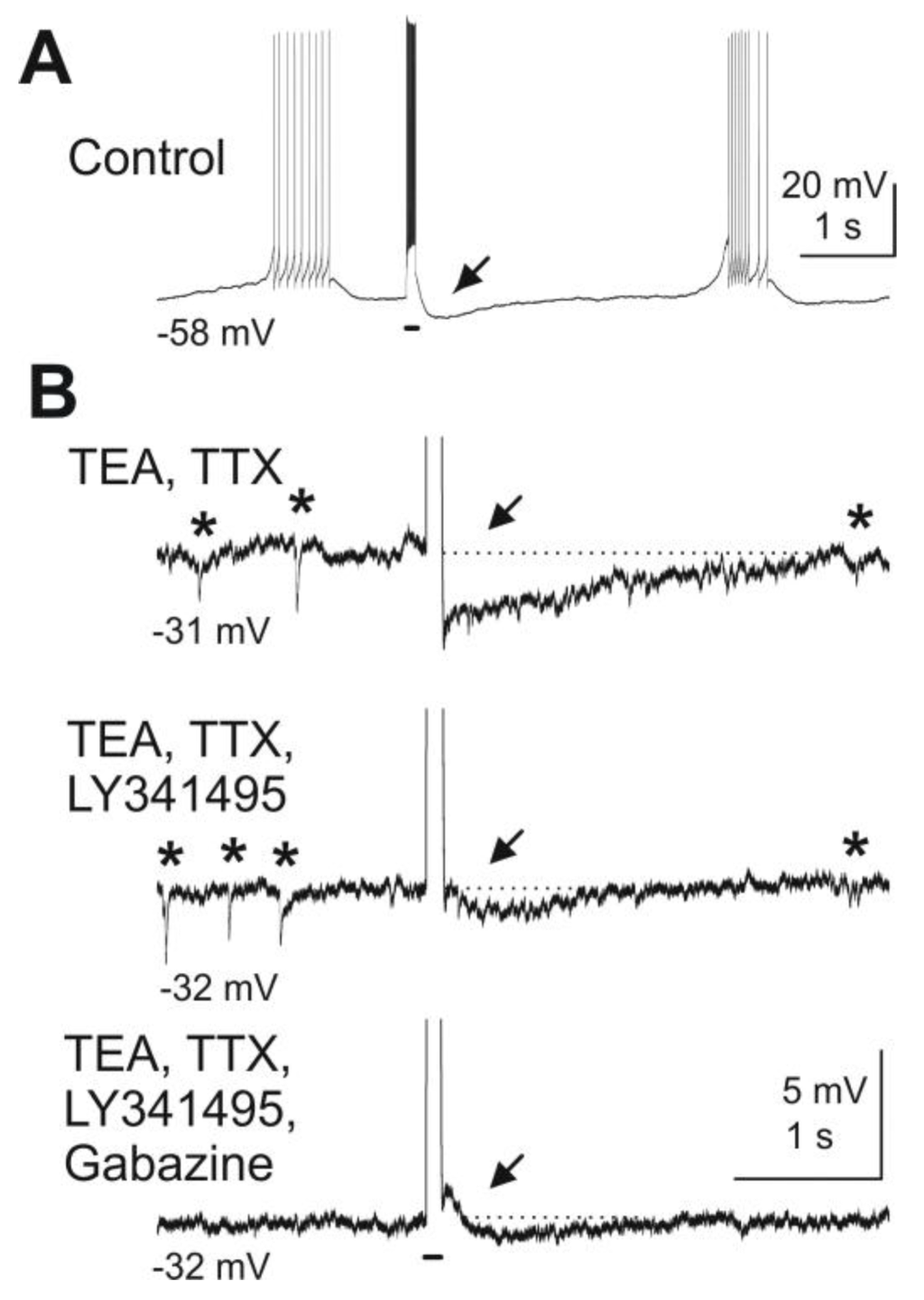
Figure 3.
Blockade of mGluRs reduces granule cell-mediated feedback inhibition of mitral cells. Depolarizing current pulses injected into a mitral cell evoked granule cell-mediated feedback IPSPs. (A) A depolarizing current pulse (100 ms, 300 pA, indicated by bar beneath the trace) evoked a burst of action potentials followed by an IPSP (at arrow). (B) After wash-in of TEA (5 mM) and TTX (1 μM) and current clamp of the mitral cell at ca. −30 mV, spontaneous IPSPs are visible (marked by asterisks above the trace) and current pulses revealed a feedback IPSP (first trace). The mGluR antagonist LY341495 (100 μM) reduces the IPSP in amplitude (second trace). Addition of the GABAA receptor blocker gabazine (10 μM) completely abolishes any remaining feedback IPSP as well as spontaneous IPSPs (third trace). Modified from [44].
Figure 3.
Blockade of mGluRs reduces granule cell-mediated feedback inhibition of mitral cells. Depolarizing current pulses injected into a mitral cell evoked granule cell-mediated feedback IPSPs. (A) A depolarizing current pulse (100 ms, 300 pA, indicated by bar beneath the trace) evoked a burst of action potentials followed by an IPSP (at arrow). (B) After wash-in of TEA (5 mM) and TTX (1 μM) and current clamp of the mitral cell at ca. −30 mV, spontaneous IPSPs are visible (marked by asterisks above the trace) and current pulses revealed a feedback IPSP (first trace). The mGluR antagonist LY341495 (100 μM) reduces the IPSP in amplitude (second trace). Addition of the GABAA receptor blocker gabazine (10 μM) completely abolishes any remaining feedback IPSP as well as spontaneous IPSPs (third trace). Modified from [44].

In summary, the main olfactory bulb employs as a serial, two-stage, and lateral inhibitory network. The first stage is found at the level of input to mitral cells and the second stage takes place at the level of output from mitral cells and is possibly regulated by odorant-specific feedback from the olfactory cortex [24,48,49]. The purpose of this serial processing might lie in a reduction of cellular noise while enhancing contrast between odorant-specific input channels to sharpen odorant representation. Inhibitory interneurons such as periglomerular cells and granule cells act locally to provide feedback and lateral inhibition through dendrodenditic interactions with mitral cells [50].
4. Epilepsy and the Balance of Excitation and Inhibition
Epilepsy is a relatively frequent, long-term neurological disorder that affects 1% of the world population [51]. It is characterized by excessive temporary neuronal discharges resulting in uncontrolled convulsions referred to as epileptic seizures. Epileptic seizures can be brief (seconds) or last for a long time and are convulsive or non-convulsive. The underlying cause of epilepsy is typically unknown even though several pathological conditions can result in epilepsy such a brain injury, stroke or drug abuse. It is known that epileptic seizures result from excessive and abnormal cortical neuronal activity [52]. During an epileptic seizure, neuronal activity at a seizure focus is poorly controlled and subsequent electrical excitation spreads in brain circuits [53]. While epilepsy cannot be cured, most cases of epilepsy are treatable with medication such that patients can lead a normal life, possibly with some restrictions regarding driving a motor vehicle [54]. Despite the optimal use of available antiepileptic drugs, 25%–40% of patients are still considered to be refractory to existing drugs while others experience seizure control at the expense of acute adverse effects [55]. Therefore, discovery of novel antiepileptic agents with improved efficacy and side effect profiles is a key milestone in the battle against epilepsies. Not surprisingly, most anti-seizure medications inhibit neuronal excitability by modulating the function of several types of proteins such as sodium channels, NMDA receptors and GABA receptors [56]. Neuronal excitability in the brain is the integral of intrinsic membrane conductances and synaptic inputs. Neuronal excitability at rest is regulated by both excitatory and inhibitory inputs [57]. Mitral cells in the main olfactory bulb exhibit spontaneous action potential firing as a pattern of neuronal activity [15], which can be modulated by intrinsic membrane receptors as well as synaptic inputs [14,50]. Mitral cells express high levels of different receptors such as GABA receptors (GABAA, GABAB), ionotropic and metabotropic glutamate receptors (NMDA, AMPA, mGluR1, kainate), and serotonin receptors (5-HT1A, 5-HT2A/C). Most of these receptor proteins have been implicated in neurological disorders such as epilepsy [14,58,59,60]). Therefore, modulating any of these receptor proteins can change neuronal excitability as well as synaptic input-output relations and by extension can be critical for excitation-inhibition balance in neurons. The focus of this review is on the effects of a small library of enaminones on neuronal activity of mitral cells and the mechanisms underlying the inhibitory or excitatory actions of these enaminone compounds. The analogs are non-ortho-substituted cyclic enaminones and suppress neuronal excitability through activation of GABAA receptors and display the characteristics of a positive allosteric modulator.
5. Synthesis of Anilino Enaminones
The anilino enaminones reviewed here, KRS-5Me-4-OCF3 (5-Methyl-3-(4-trifluoromethoxy-phenylamino)-cyclohex-2-enone), KRS-5Me-4-F (3-(4-Fluoro-phenylamino)-5-methyl-cyclohex-2-enone) and KRS-5Me-3-Cl (3-(3-Chloro-phenylamino)-5-methyl-cyclohex-2-enone), were recently synthesized via a condensation reaction of the 5-methyl diketone with the corresponding substituted aniline derivatives (Figure 4). Their chemical structures are shown in Figure 5. More specifically, the mono methyl anilino enaminones (3) were prepared from the 5-methylcyclohexane-1,3-dione (2) form by the decarboxylation of 4-carbo-tert-butoxy-5-methylcyclohexane-1,3-dione (1), and were refluxed with appropriate substituted aniline derivatives under standard conditions [11]. The β-hydroxy keto tert-butoxy ester was prepared as previously reported [3,61,62]. The enaminone structures were confirmed via NMR analyses at 400 MHz. The analogs were subsequently evaluated for anticonvulsant activity by the National Institute of Neurological Disorders and Stroke Anticonvulsant Screening Program, National Institutes of Health. These enaminones have shown maximal electroshock seizure and scPTZ rodent activity.
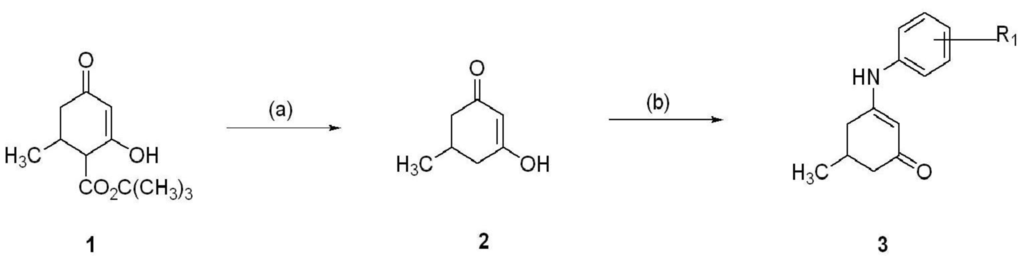
Figure 4.
Synthesis of aniline enaminones. From [18].
Figure 4.
Synthesis of aniline enaminones. From [18].

Conditions: (a) H2SO4, Δ; (b) Δ, substituted amine.

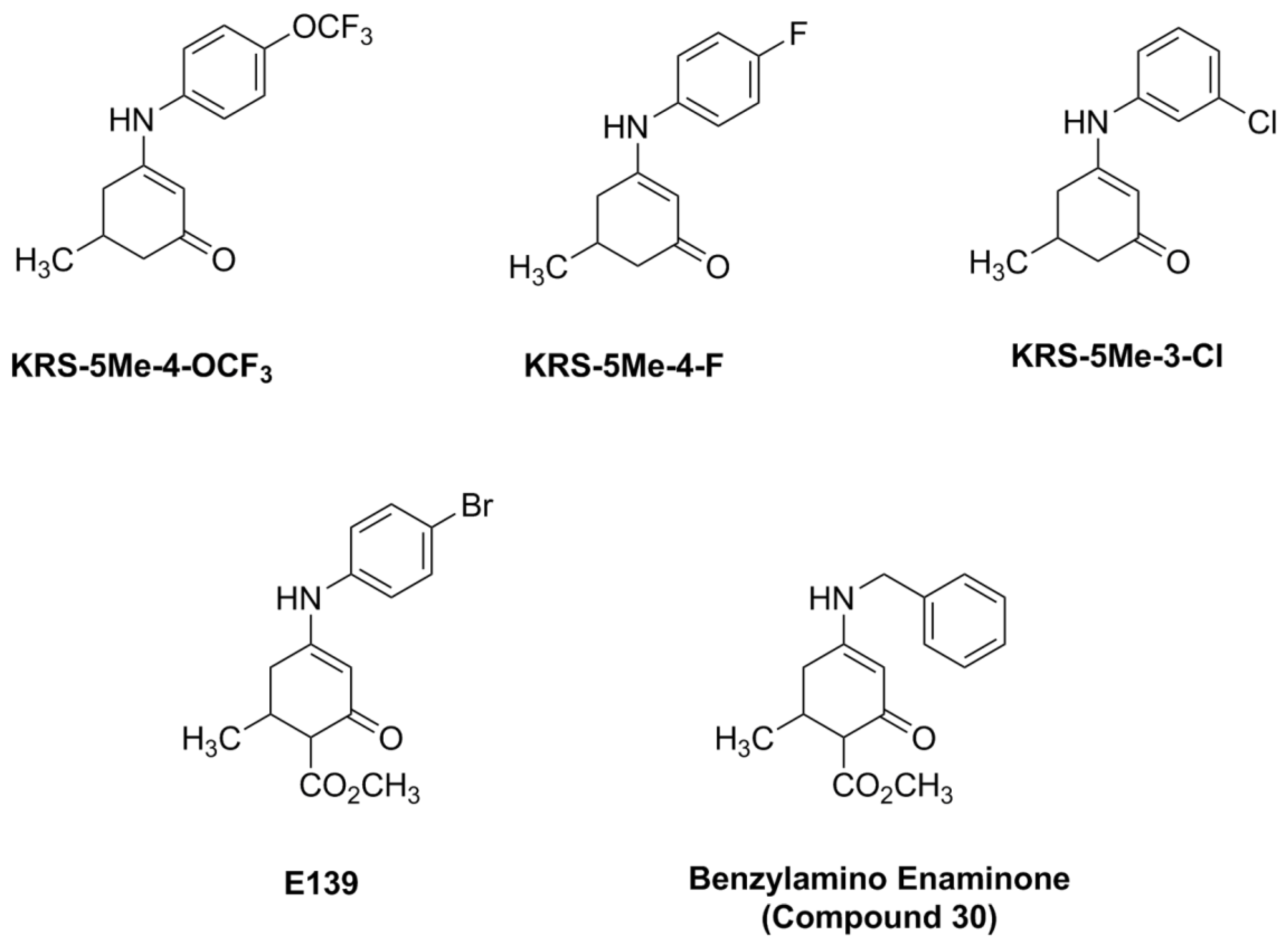
Figure 5.
Chemical structure of aniline enaminone analogs. The difference among the three mono methyl compounds is the anilino substitution represented by meta-Chloro; para-Fluoro, and para-trifluoro. E139 is a para-bromo anilino enaminone derivative with a methyl ester substituted moiety at cyclohexenone and Compound 30 is the benzylamino analog. From [18].
Figure 5.
Chemical structure of aniline enaminone analogs. The difference among the three mono methyl compounds is the anilino substitution represented by meta-Chloro; para-Fluoro, and para-trifluoro. E139 is a para-bromo anilino enaminone derivative with a methyl ester substituted moiety at cyclohexenone and Compound 30 is the benzylamino analog. From [18].
6. Anilino Enaminones Inhibit Activity of Mitral Cells
In brain slices, mitral cells spontaneously generate action potentials at 1 to 6 Hz [14]. Bath application of either KRS-5Me-3Cl or KRS-5Me-4F modulates the spike rate and membrane potential of mitral cells (Figure 6) [18]. KRS-5Me-4-OCF3 shows a more potent inhibition of mitral cell activity compared to the other two compounds (Figure 6A,B). KRS-5Me-4-OCF3 (20 μM) reversibly decreases mitral cell firing accompanied by hyperpolarization of the membrane potential. Between these three anilino enaminones, KRS-5Me-4-OCF3 is the most potent compound in terms of reducing spiking activity of mitral cells. Therefore, enaminone KRS-5Me-4-OCF3 is potentially a more potent candidate as an anticonvulsant and has been used to determine the mechanism underlying its inhibitory effect on neuronal activity [18]. Anilino enaminone KRS-5Me-4-OCF3 and its analogs display inhibitory effects on neuronal activity with different potencies depending on the chemical structure and concentration of the enaminone. This is consistent with previous in vivo results that KRS-5Me-4-OCF3 is the most potent anticonvulsant agent [11].
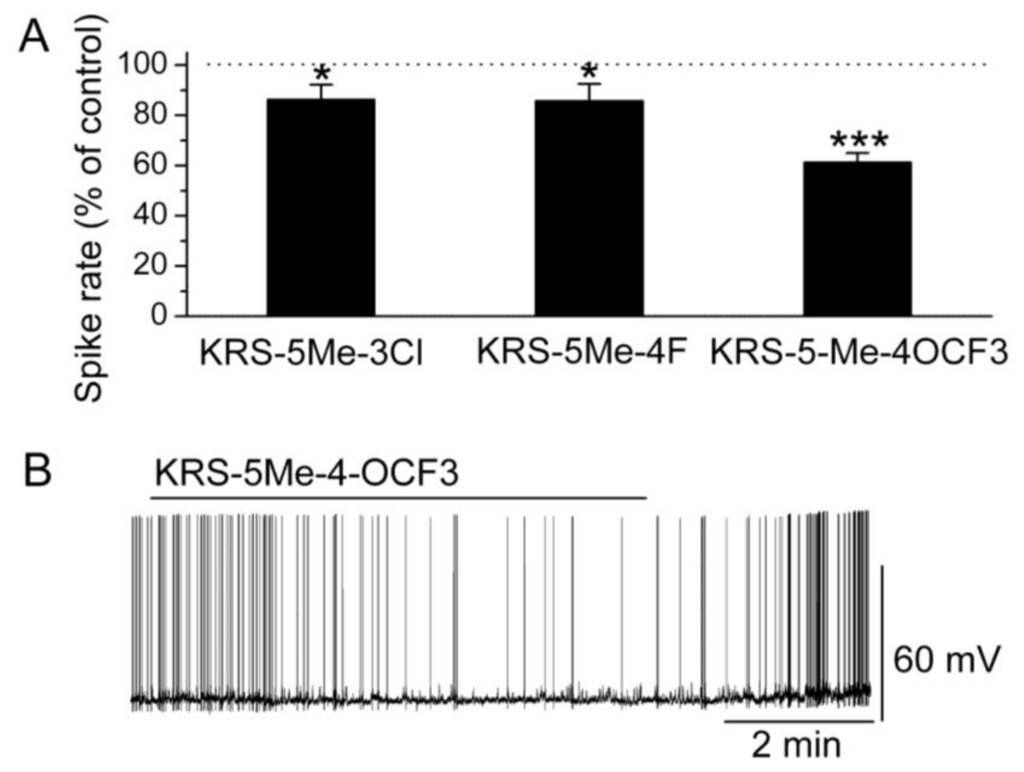
Figure 6.
Anilino enaminones depress the spiking activity of mitral cells. (A) Normalized bar graph shows inhibition of spiking of mitral cells in response to bath application of KRS-5Me-4-OCF3 (20 μM), KRS-5Me-3Cl (20 μM), and KRS-5Me-4F (20 μM). Responses to enaminones are normalized with respect to control conditions (* p < 0.05, *** p < 0.001). (B) Original recording from a representative mitral cell illustrates the inhibition in firing rate and hyperpolarization following application of KRS-5Me-4-OCF3. From [18].
Figure 6.
Anilino enaminones depress the spiking activity of mitral cells. (A) Normalized bar graph shows inhibition of spiking of mitral cells in response to bath application of KRS-5Me-4-OCF3 (20 μM), KRS-5Me-3Cl (20 μM), and KRS-5Me-4F (20 μM). Responses to enaminones are normalized with respect to control conditions (* p < 0.05, *** p < 0.001). (B) Original recording from a representative mitral cell illustrates the inhibition in firing rate and hyperpolarization following application of KRS-5Me-4-OCF3. From [18].
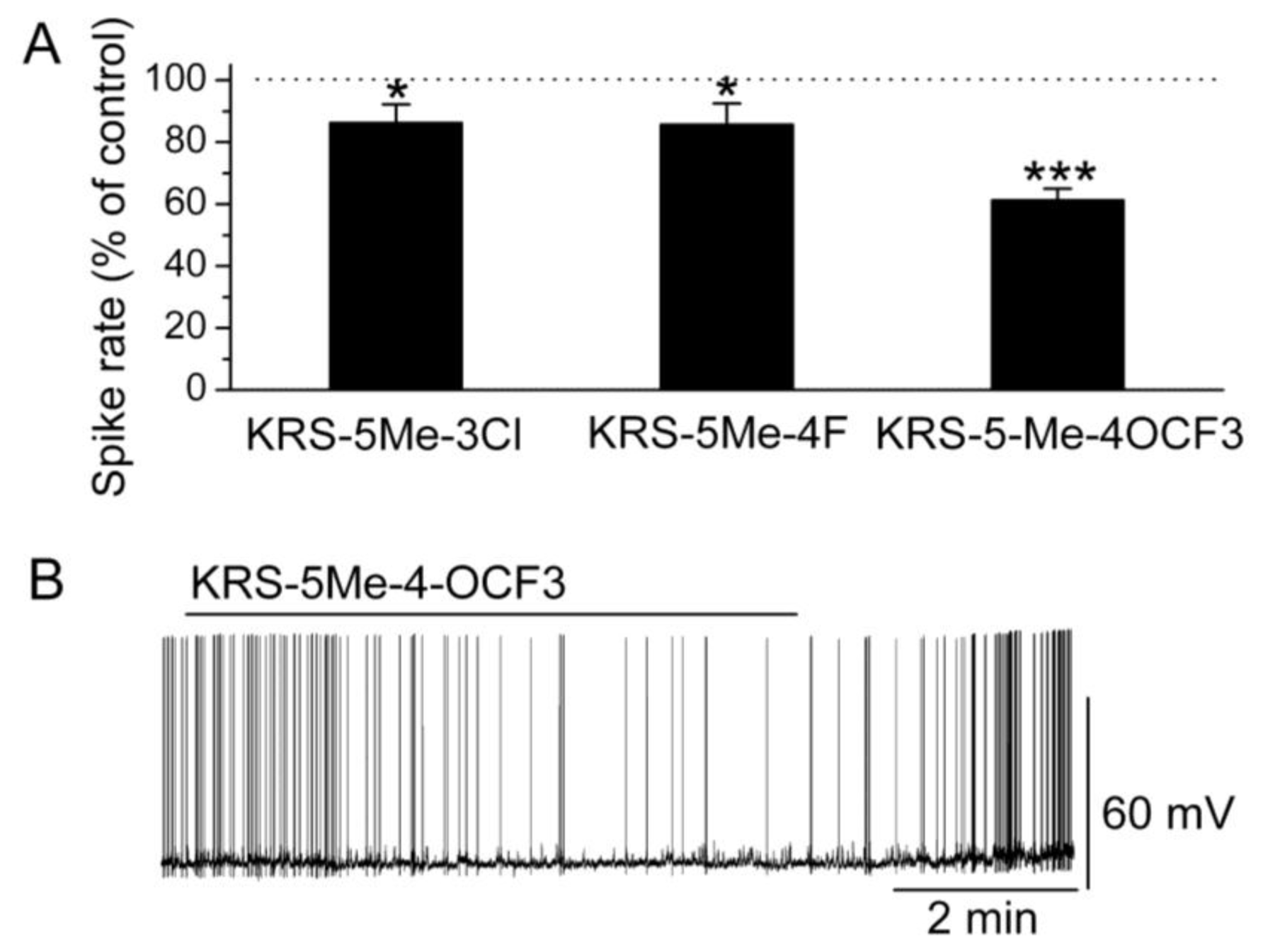
A specific substituted site in the chemical structure of enaminones may be required for receptor targeting and for conferring anticonvulsant activity. A para-substitution of the phenyl group with –OCF3 evokes the most potent suppression of neuronal activity. In contrast, a para-, or meta-substitution of the phenyl group with fluoro, chloro decreases the potency of the inhibitory activity. Possibly, a substitution in the phenyl group influences the potency of the inhibitory action of enaminones most strongly. Similarly, the significance of the substitution group has been demonstrated in benzylamino enaminones in which the unsubstituted benzylamine analog compound 30 (Figure 5) shows the most potent activity in protecting rodents from seizure onset and excitatory synaptic depression [17].
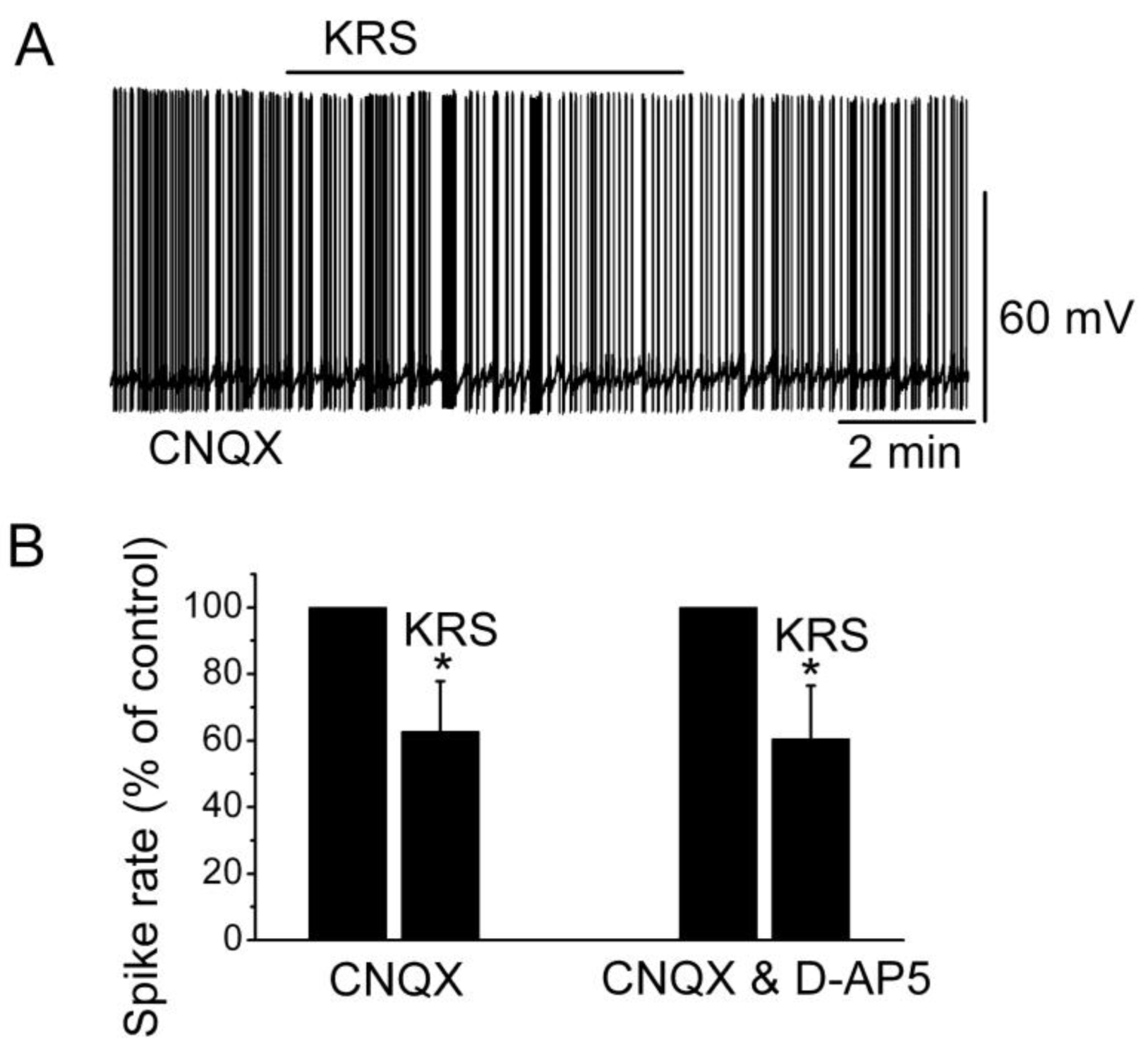
Ionotropic glutamate receptors are involved in regulating neuronal excitability in the main olfactory bulb [14]. Blockade of ionotropic glutamate receptors may result in neuronal inhibition. To determine if the inhibitory effect of KRS-5Me-4-OCF3 is mediated through interaction with ionotropic glutamate receptors, neuronal inhibition evoked by KRS-5Me-4-OCF3 can be tested in the presence of AMPA/kainate and NMDA receptor inhibitors, namely, CNQX, a potent AMPA/kainate receptor antagonist, and D-AP5, a potent NMDA receptor antagonist. In the presence of both CNQX and D-AP5, KRS-5Me-4-OCF3 reduces the firing rate and hyperpolarizes mitral cells not significantly different from the values of KRS-5Me-4-OCF3-induced suppression recorded in ACSF control condition [18]. Therefore, ionotropic glutamate receptors are not involved in the KRS-5Me-4-OCF3-induced MC inhibition (Figure 7).
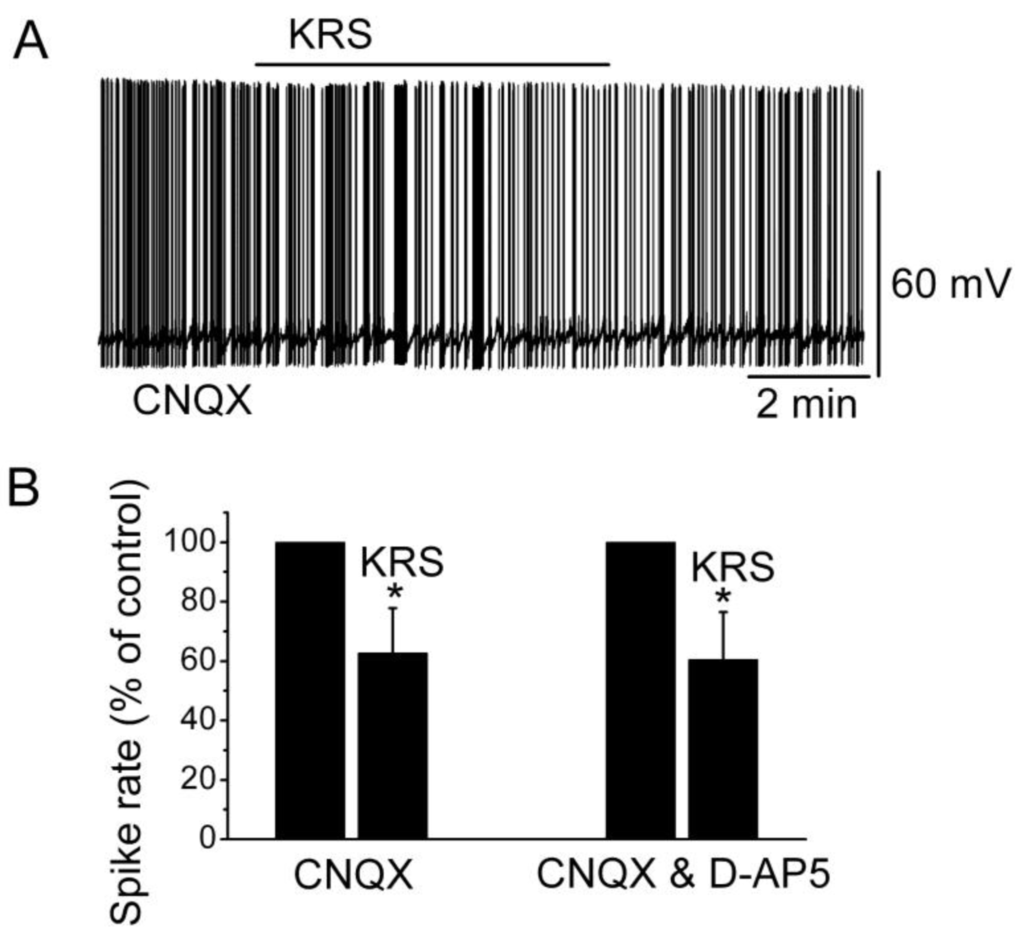
Figure 7.
Neither AMPA/kainate nor NMDA receptor antagonists blocks the KRS-5Me-4-OCF3-induced inhibition of MCs. (A) KRS-5Me-4-OCF3-induced suppression of neuronal firing recorded in a representative mitral cell in the presence of CNQX (10 μM). (B) The normalized and averaged results showed the persistence of KRS-5Me-4-OCF3-induced inhibitory effects on spontaneous spiking of mitral cells in the presence of either CNQX or CNQX plus D-AP5 (50 μM) (* p < 0.05). From [18].
Figure 7.
Neither AMPA/kainate nor NMDA receptor antagonists blocks the KRS-5Me-4-OCF3-induced inhibition of MCs. (A) KRS-5Me-4-OCF3-induced suppression of neuronal firing recorded in a representative mitral cell in the presence of CNQX (10 μM). (B) The normalized and averaged results showed the persistence of KRS-5Me-4-OCF3-induced inhibitory effects on spontaneous spiking of mitral cells in the presence of either CNQX or CNQX plus D-AP5 (50 μM) (* p < 0.05). From [18].
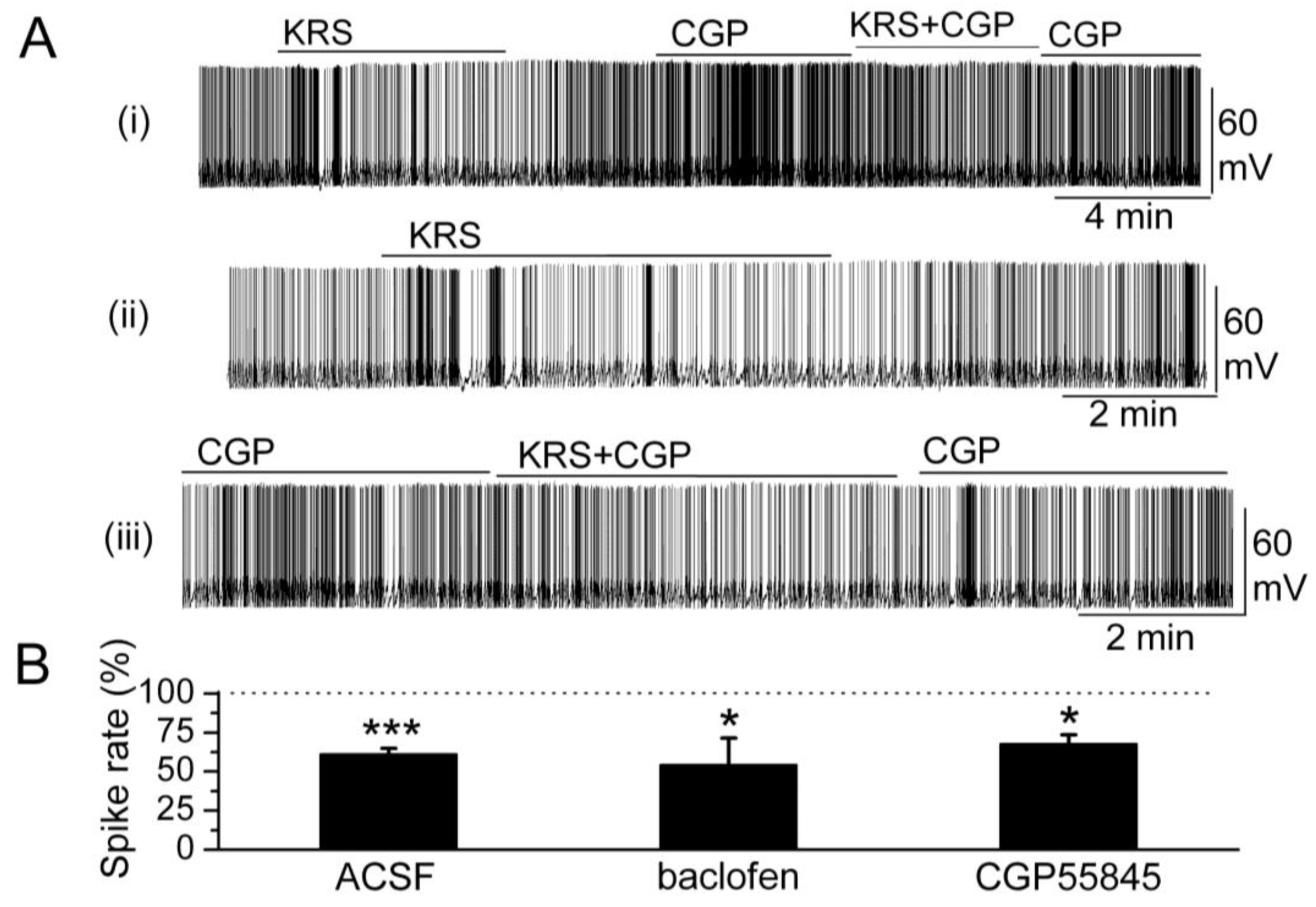
In the main olfactory bulb, GABAB receptors (GABABRs) are found mainly in the glomerular layer [63,64]. GABAB receptors belong to the class of metabotropic trans-membrane receptors and are linked via G-proteins to potassium channels [65]. Activation of GABAB receptors can stimulate the opening of K+ channels to hyperpolarize the membrane potential, reduce neuronal excitability and, subsequently, prevent release of neurotransmitter. Activation of GABAB receptors is known to reduce mitral cell excitability [66,67]. KRS-5Me-4-OCF3-evoked reduction of mitral cell firing and membrane hyperpolarization persists in the presence of GABABR antagonist CGP55845 (10 μM) (Figure 8). The GABAB receptor agonist (R)-baclofen (50 μM) alone evokes a strong decrease in firing and hyperpolarizes the membrane potential of mitral cells. In the presence of GABABR agonist baclofen, KRS-5Me-4-OCF3 further reduces mitral cell firing and hyperpolarizes the membrane potential indicating that the inhibitory effect of KRS-5Me-4-OCF3 persists irrespective of GABAB receptor activation or blockade [18].
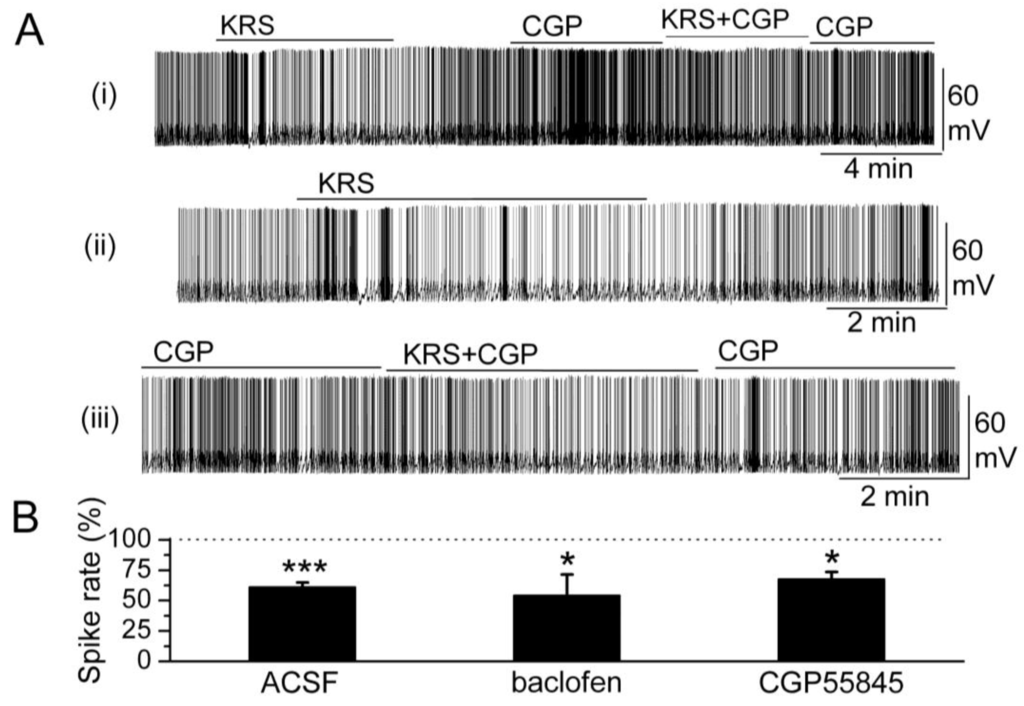
Figure 8.
GABAB receptors are not involved in KRS-5Me-4-OCF3-evoked inhibition. (A) Original recording from a mitral cell during KRS-5Me-4-OCF3 application in ACSF condition and in the presence of GABABR antagonist CGP55845 (10 μM). The upper trace (i) illustrates the effect of KRS-5Me-4-OCF3 on spiking of a mitral cell and the effect of adding CGP55845. The upper trace is shown at an extended time scale in the middle (ii) and lower trace (iii). (B) The normalized results showed the persistence of KRS-5Me-4-OCF3-evoked inhibitory effects on spiking of mitral cells in the presence of the GABAB receptor antagonist CGP55845 (10 µM) and the GABAB receptor agonist (R)-baclofen (50 µM) (* p < 0.05, *** p < 0.001). The data for the effect of KRS-5Me-4-OCF3 on spiking of mitral cells were normalized with respect to ACSF, or CGP88545 or baclofen alone. From [18].
Figure 8.
GABAB receptors are not involved in KRS-5Me-4-OCF3-evoked inhibition. (A) Original recording from a mitral cell during KRS-5Me-4-OCF3 application in ACSF condition and in the presence of GABABR antagonist CGP55845 (10 μM). The upper trace (i) illustrates the effect of KRS-5Me-4-OCF3 on spiking of a mitral cell and the effect of adding CGP55845. The upper trace is shown at an extended time scale in the middle (ii) and lower trace (iii). (B) The normalized results showed the persistence of KRS-5Me-4-OCF3-evoked inhibitory effects on spiking of mitral cells in the presence of the GABAB receptor antagonist CGP55845 (10 µM) and the GABAB receptor agonist (R)-baclofen (50 µM) (* p < 0.05, *** p < 0.001). The data for the effect of KRS-5Me-4-OCF3 on spiking of mitral cells were normalized with respect to ACSF, or CGP88545 or baclofen alone. From [18].
7. Blockade of GABAA Receptors Reverses Enaminone-Induced Inhibition of Mitral Cells
Activation of GABAA receptors on mitral cells suppresses their excitability and activity by decreasing the firing rate and hyperpolarizing the membrane potential [68,69]. GABA directly inhibits mitral cells; an observation that corresponds with the finding that GABA receptors are abundant in mitral cells [69,70]. In rodents, the GABAA receptor has been found to form pentameric complex of unknown stoichiometry with subunits of four classes (alpha 1 to alpha 6, beta 1 to beta 3, gamma 1 to gamma 3 and delta). Specifically, mitral cells express the alpha 1, beta 1, beta 2, beta 3, and gamma 2 mRNAs strongly and the alpha 3 mRNA weakly [69]. In the presence of GABA, KRS-5Me-4-OCF3 reduces the firing of mitral cells even further [18]. This enhancement of the KRS-5Me-4-OCF3-evoked inhibition of mitral cells in the presence of increased extracellular GABA levels (50 µM) indicates that KRS-5Me-4-OCF3-evoked inhibition is mediated by direct activation of GABA receptors rather than by increased GABA levels (Figure 9). An increase in endogenous GABA level can result from increased GABA release or from reduced GABA re-absorption and/or degradation. Increased GABA levels or blockade of GABAA receptors can result in over excitation of neurons and epileptiform activity.
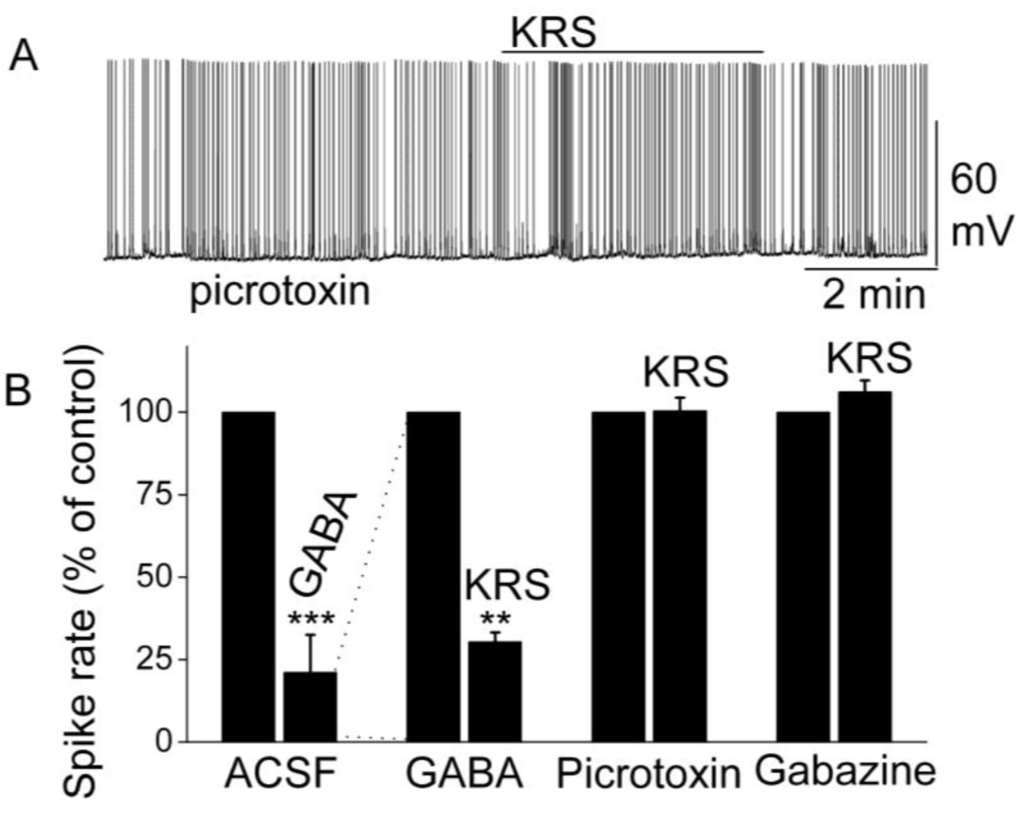
Figure 9.
KRS-5Me-4-OCF3-evoked inhibition of mitral cell spiking activity is blocked by GABAA receptor antagonists. (A) Original recording obtained from a mitral cell showed that picrotoxin (20 µM) blocks 20 µM KRS-5Me-4-OCF3-evoked suppression of neuronal firing. (B) The normalized and averaged results showed KRS-5Me-4-OCF3-evoked inhibitory effects on spiking of mitral cells in the presence of GABAA receptor agonist GABA (50 µM), and antagonist picrotoxin (20 µM) and gabazine (5 µM) (** p < 0.01, *** p < 0.001). The data for the effect of GABA or KRS-5Me-4-OCF3 on spiking of MCs are normalized with respect to control condition. From [18].
Figure 9.
KRS-5Me-4-OCF3-evoked inhibition of mitral cell spiking activity is blocked by GABAA receptor antagonists. (A) Original recording obtained from a mitral cell showed that picrotoxin (20 µM) blocks 20 µM KRS-5Me-4-OCF3-evoked suppression of neuronal firing. (B) The normalized and averaged results showed KRS-5Me-4-OCF3-evoked inhibitory effects on spiking of mitral cells in the presence of GABAA receptor agonist GABA (50 µM), and antagonist picrotoxin (20 µM) and gabazine (5 µM) (** p < 0.01, *** p < 0.001). The data for the effect of GABA or KRS-5Me-4-OCF3 on spiking of MCs are normalized with respect to control condition. From [18].
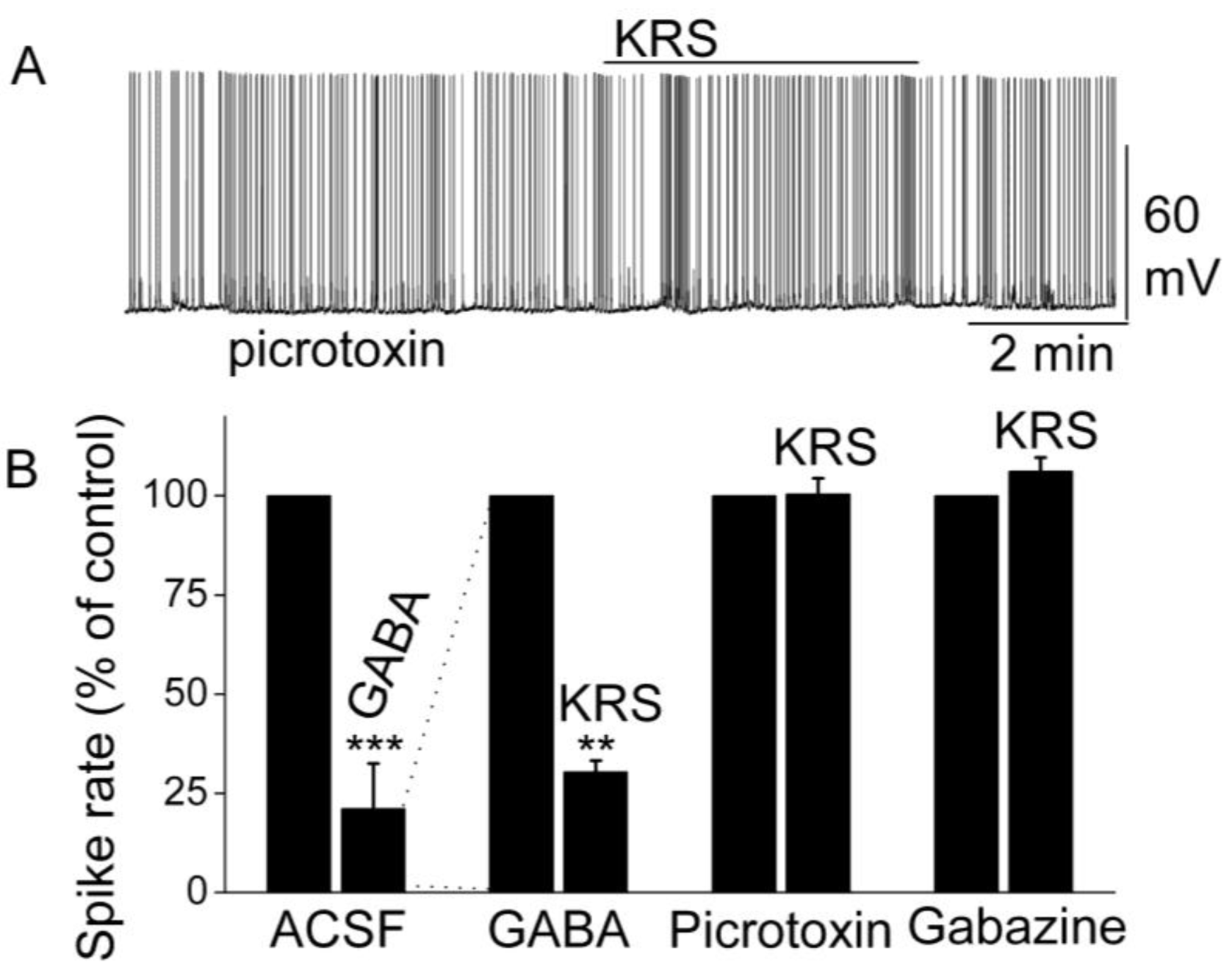
The inhibition of mitral cell activity by KRS-5Me-4-OCF3 can be eliminated by blocking GABAA receptors with receptor antagonists such as picrotoxin or gabazine (Figure 9). This result points to a KRS-5Me-4-OCF3 effect that is mediated either through enhancing extracellular GABA levels or a direct action on GABAA receptors. The latter option is supported by the fact that, in voltage clamp recordings from mitral cells, KRS-5Me-4-OCF3 produces an outward current (~15 pA), which is blocked in the presence of gabazine [18].
Furthermore, increasing extracellular GABA levels does not block the KRS-5ME-4-OCF3-induced inhibition of neuronal excitability. The fate of extracellular GABA can be described by two scenarios [71]; in one scenario, extracellular GABA is enzymatically degraded; and alternatively, after GABA is released from synaptic terminals, it may be removed from the extracellular space by GABA uptake back into axon terminals and/or into glial cells by plasma membrane transporters (GATs). Captured GABA is then degraded by the enzyme GABA transaminase (GABA-T). Vigabatrin is an irreversible and selective GABA-T inhibitor that results in extracellular accumulation of GABA in the synaptic cleft. It induces a reduction in mitral cell firing rate. In the presence of vigabatrin, application of KRS-5Me-4-OCF3 results in further reduction of the mitral cell-firing rate accompanied by hyperpolarization of the membrane potential (Figure 10A,C). The persistence of the KRS-5Me-4-OCF3 effect in the presence of vigabatrin indicates that enaminone-evoked neuronal inhibition is not mediated by GABA-T [18].
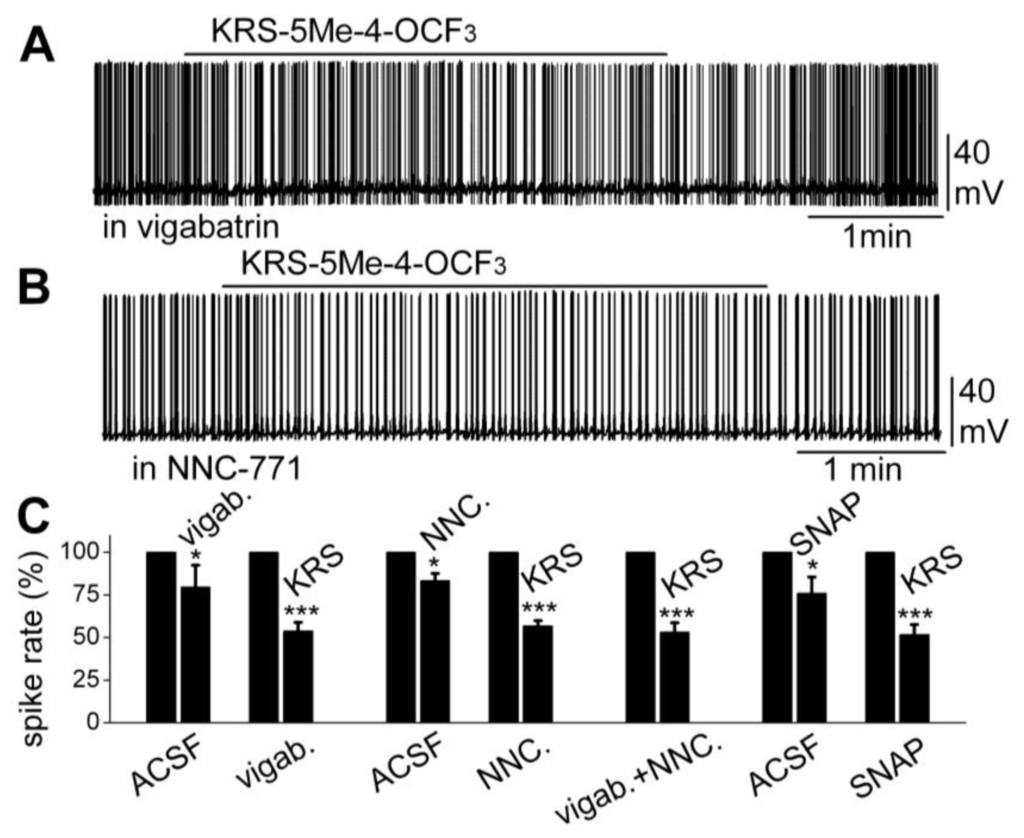
Figure 10.
Neither an inhibitor of GABA reuptake nor an inhibitor of GABA degradation enzyme GABA-T blocked the KRS-5Me-4-OCF3-evoked mitral cell inhibition. (A) Original recording from a mitral cell showing that spiking inhibition evoked by KRS-5Me-4-OCF3 persisted in the presence of vigabatrin (200 µM). (B) Original recording from a mitral cell showing that spiking inhibition evoked by KRS-5Me-4-OCF3 persisted in the presence of NNC-711 (10 µM). (C) Summary of results from normalized and averaged data (* p < 0.05, *** p < 0.001). SNAP (20 μM). From [18].
Figure 10.
Neither an inhibitor of GABA reuptake nor an inhibitor of GABA degradation enzyme GABA-T blocked the KRS-5Me-4-OCF3-evoked mitral cell inhibition. (A) Original recording from a mitral cell showing that spiking inhibition evoked by KRS-5Me-4-OCF3 persisted in the presence of vigabatrin (200 µM). (B) Original recording from a mitral cell showing that spiking inhibition evoked by KRS-5Me-4-OCF3 persisted in the presence of NNC-711 (10 µM). (C) Summary of results from normalized and averaged data (* p < 0.05, *** p < 0.001). SNAP (20 μM). From [18].
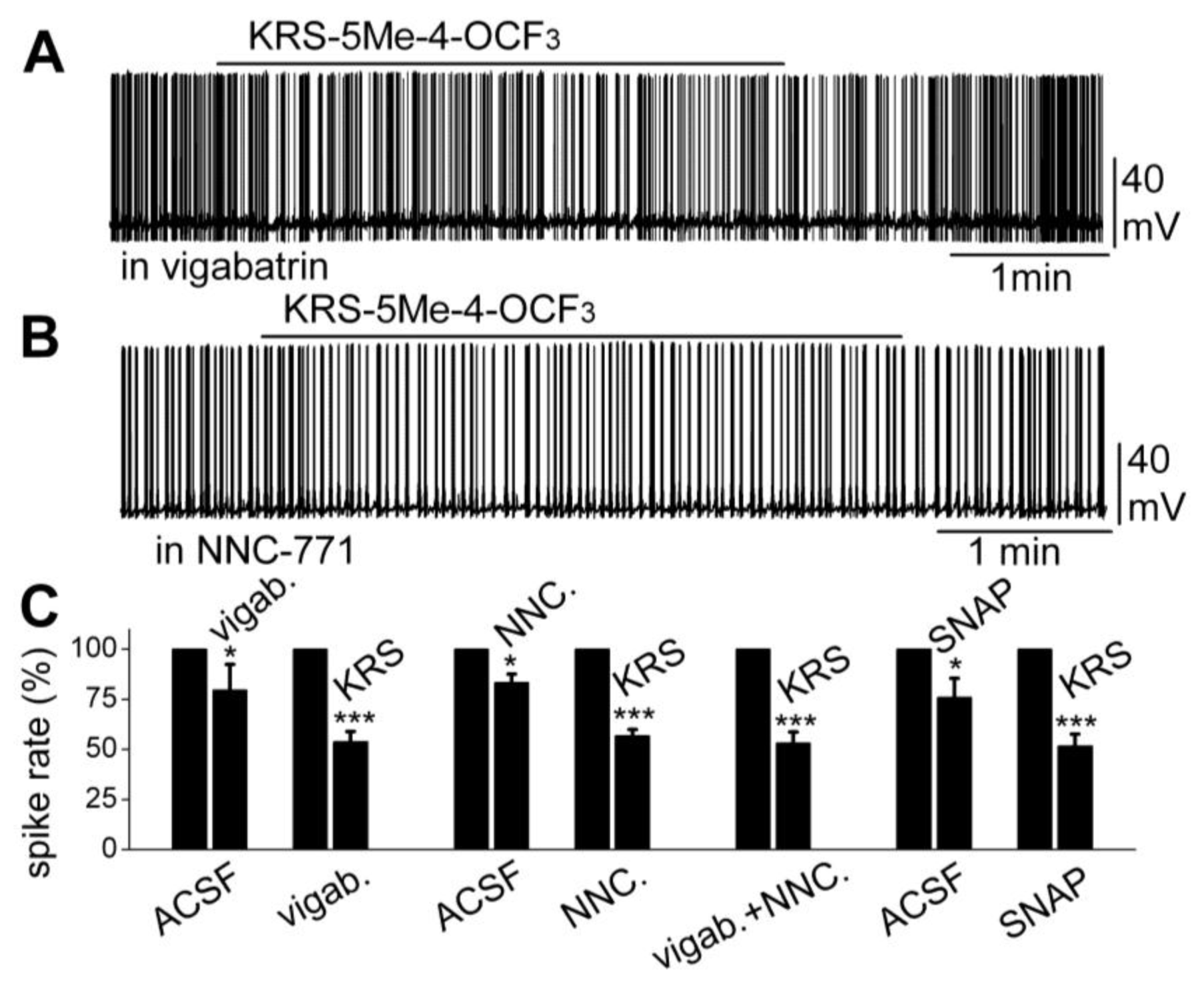
The next questions to answer were if enaminones interact with GATs to regulate GABA re-uptake and if GATs modulate KRS-5Me-4-OCF3-evoked neuronal inhibition. NNC-711 is a potent and selective inhibitor of GABA uptake by GAT-1 and (S)-SNAP 5114 is a selective inhibitor by GAT-2 and GAT-3. Bath application of either NNC-711 or (S)-SNAP 5114 reduces mitral cell spiking and modulates the membrane potential. In the presence of either GABA-uptake blocker, KRS-5Me-4-OCF3 further reduces the firing rate and hyperpolarizes mitral cells (Figure 10B,C). Even in the presence of both the GABA reuptake inhibitor NNC-711 and the GABA-T inhibitor vigabatrin, the inhibitory effect of KRS-5Me-4-OCF3 remains fully intact (Figure 10C). Neither the GABA reuptake transporters, GATs, nor GABA transaminase, GABA-T, interfere with KRS-5Me-4-OCF3-evoked neuronal inhibition, indicating that an extracellular increase of GABA does not contribute to the pharmacological effects of KRS-5ME-4-OCF3 [18].
Both GABAA and GABAB receptors are present in the main olfactory bulb but participate in the regulation of mitral cell excitability in distinct ways. Whereas GABAA receptors directly regulate the excitability of mitral cells, GABAB receptors indirectly regulate of mitral cells via presynaptic inhibition of olfactory receptor neuron terminals [14,50]. Anti-epileptic drugs such as benzodiazepines are known to interact with GABAA receptors [53], which confirms the observation that KRS-5Me-4-OCF3 does not interact with GABAB receptors but rather interacts directly with GABAA receptors to decrease neuronal activity of mitral cells.
The chemical structure of enaminones is critical in determining their mode of action in terms of neuronal inhibition and anticonvulsant effects. E139 (Figure 5) suppresses excitatory synaptic transmission by enhancing extracellular GABA levels [6] and blocking TTX-sensitive sodium channels and, thereby, directly inhibiting postsynaptic neuronal excitability [16]. Other enaminones with chemical moieties different from KRS-5Me-4-OCF3 interact with GABAA receptors [72,73,74]. The comparative molecular field analysis (CoMFA) and comparative molecular similarity (CoMSIA) techniques provide additional enaminone derivatives that most likely target GABA receptors. These techniques generate models to define the specific structural and electrostatic features essential for enhanced binding of enaminones to the putative GABA receptor [75].
8. A Substituted Anilino Enaminone Exhibits Characteristics of a Positive Allosteric Modulator of GABAA Receptors
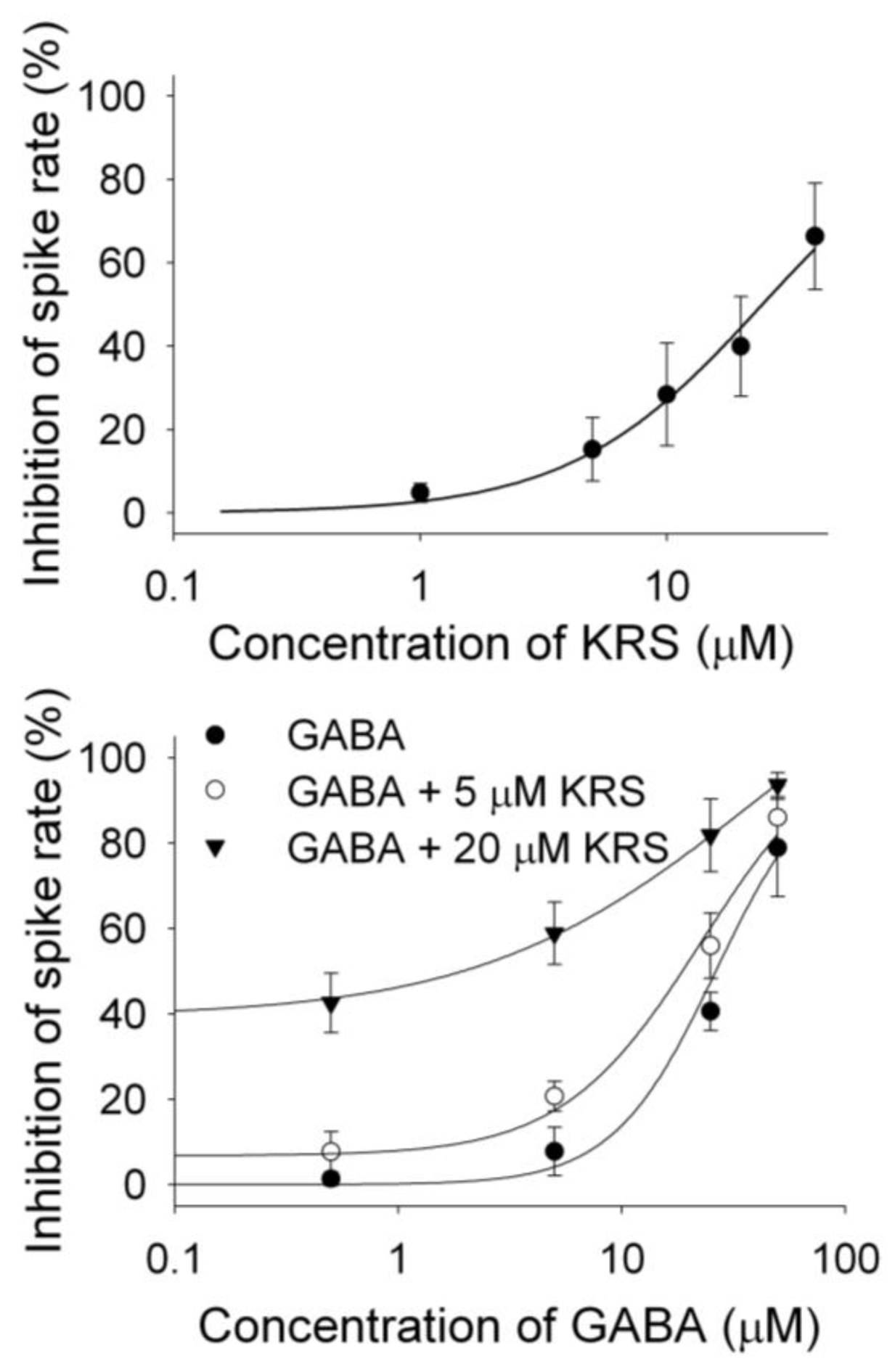
KRS-5Me-4-OCF3 enhances the inhibitory effect of GABA on mitral cells (Figure 9B), suggesting that KRS-5Me-4-OCF3 acts as a positive allosteric modulator of GABAA receptors. GABAA receptors are the main target for positive allosteric modulators such as benzodiazepines [76,77,78]. A positive allosteric modulator facilitates GABAA receptor function by binding at a site distinct from the GABA binding site and by increasing GABA affinity for the GABAA receptor. This can be tested by examining the concentration-response relationships of KRS-5Me-4-OCF3 and GABA in the absence and presence of KRS-5Me-4-OCF3 (0 μM, 5 μM, 20 μM) (Figure 11) [18]. The stoichiometry of drug and receptor interaction is 1:1 based on a fitted Hill coefficient (n) value of 1.11. The inhibitory effect of KRS-5Me-4-OCF3 is concentration-dependent with an EC50 of 24.5 μM. GABA-evoked inhibition is concentration-dependent (Figure 11B). The concentration-response curves are left shifted by KRS-5Me-4-OCF3, which suggests that KRS-5Me-4-OCF3 mostly likely binds at non-GABA binding sites on the GABA receptor [18]. The EC50 of GABA fitted by the Hill equation is 28.8 μM for GABA only, 19.9 μM for GABA plus 5 μM KRS-5Me-4-OCF3, and 10.5 μM for GABA plus 20 μM KRS-5Me-4-OCF3. KRS-5Me-4-OCF3 enhances the affinity of GABA for GABA binding sites suggesting an action of KRS-5Me-4-OCF3 as a positive allosteric modulator of the GABAA receptor. The in vivo data, which show an anticonvulsant effect of KRS-5Me-4-OCF3, correspond well with the suggested cellular mechanism of its action. The corresponding in vitro and in vivo results also indicate that recording in mitral cells is an appropriate method to elucidate the bioactivity of enaminones.
9. KRS-5Me-4-OCF3 Binds at the Benzodiazepine Site
GABA is the major inhibitory neurotransmitter in the brain. The GABAA receptor is a ligand-gated ion channel that binds GABA but it possesses distinct binding sites for GABA, benzodiazepines, barbiturates, ethanol [79], inhaled anesthetics, and neuroactive steroids. Compounds such as benzodiazepines, neuroactive steroids, and barbiturates act as allosteric modulators of GABAA receptors and have also been identified as useful anxiolytics, anticonvulsants, anesthetics, and sedative-hypnotics. Positive allosteric modulators increase the affinity of GABA for the binding site.
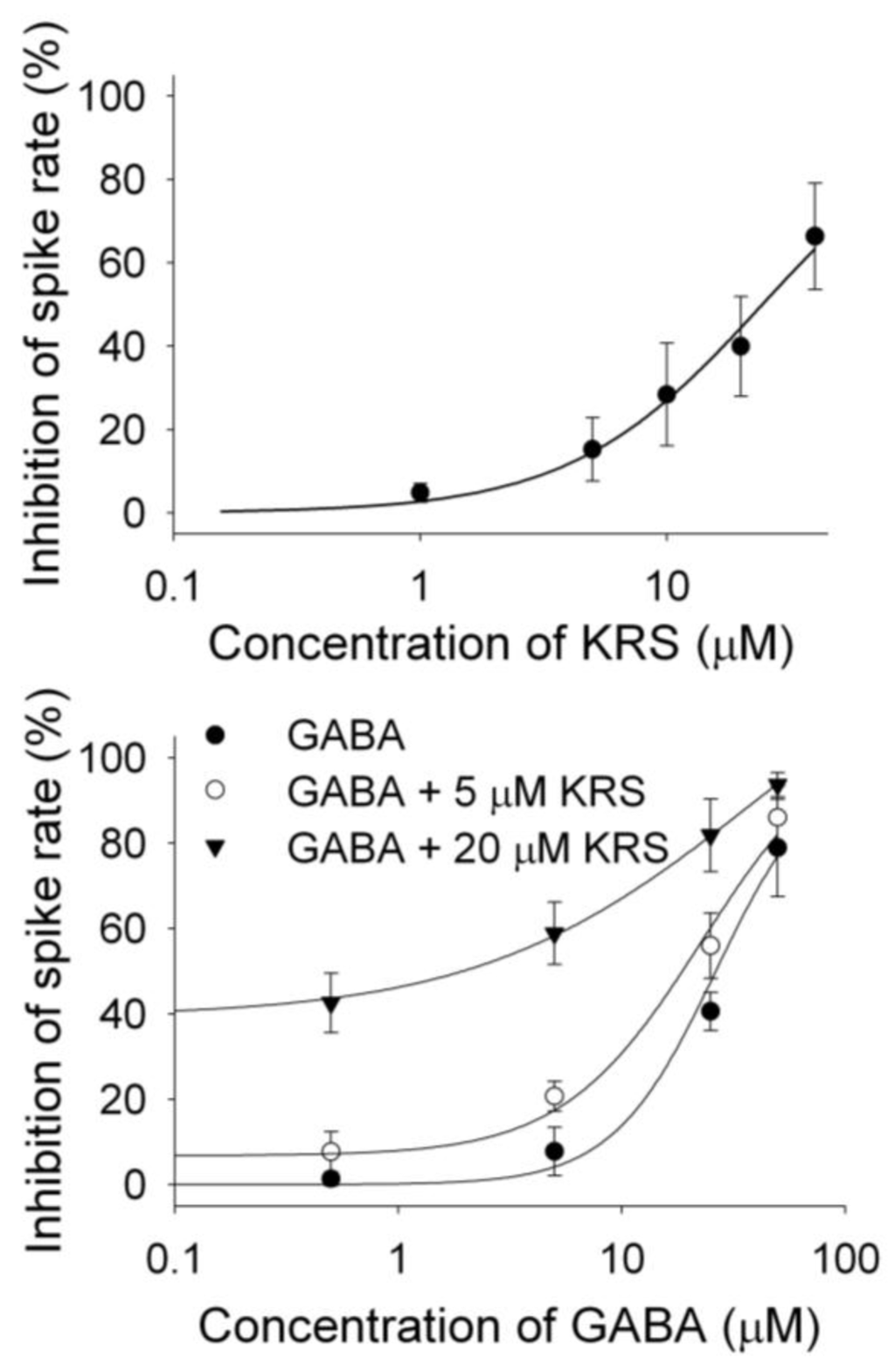
Figure 11.
The concentration–response curves of KRS-5Me-4-OCF3–evoked inhibition and of GABA in the presence of KRS-5Me-4-OCF3. (A) The KRS-5Me-4-OCF3–evoked change in spiking rate was normalized to the control condition, and then averaged. Each point was the mean value ° SEM of 4 to 7 cells. The line is fit for the data to the Hill equation: y = Y0 + Axn/(Kdn + xn), where y is the inhibition of spiking rate, Y0 is minimal inhibition, A is maximal inhibition, Kd is the apparent dissociation constant for agents, and n is the Hill coefficient. Kd and n were estimated using a Marquadt nonlinear least-squares routine. (B) Shift of the concentration–response curves of GABA in the presence of KRS-5Me-4-OCF3 at different concentrations (0, 5, 20 μM). The lines are fits for the data to the above Hill equation. From [18].
Figure 11.
The concentration–response curves of KRS-5Me-4-OCF3–evoked inhibition and of GABA in the presence of KRS-5Me-4-OCF3. (A) The KRS-5Me-4-OCF3–evoked change in spiking rate was normalized to the control condition, and then averaged. Each point was the mean value ° SEM of 4 to 7 cells. The line is fit for the data to the Hill equation: y = Y0 + Axn/(Kdn + xn), where y is the inhibition of spiking rate, Y0 is minimal inhibition, A is maximal inhibition, Kd is the apparent dissociation constant for agents, and n is the Hill coefficient. Kd and n were estimated using a Marquadt nonlinear least-squares routine. (B) Shift of the concentration–response curves of GABA in the presence of KRS-5Me-4-OCF3 at different concentrations (0, 5, 20 μM). The lines are fits for the data to the above Hill equation. From [18].
In vivo data shows potent anticonvulsant effects of enaminones in chemically-induced epilepsy in animal models but with fewer side-effects [11,12]. The in vivo and in vitro results suggest that KRS-5Me-4-OCF3 binds to benzodiazepine sites on GABAA receptors to exert its effect. Indeed, flumazenil, a benzodiazepine site antagonist, slightly increases mitral cell firing. However, in the presence of flumazenil, KRS-5Me-4-OCF3 fails to inhibit firing or change the membrane potential of mitral cells [18]. This establishes that the enhancement of GABA by KRS-5Me-4-OCF3 is mediated at the classical benzodiazepine site.
The enaminone KRS-5Me-4-OCF3 acts as a novel positive allosteric modulator to decrease neuronal activity via direct regulation of GABAA receptors, which suggests that KRS-5Me-4-OCF3 could be a potential medication as anxiolytic, anticonvulsant, anesthetic, and sedative-hypnotic. A number of enaminone compounds provide protection against glutamate-mediated excitatory synaptic transmission by modulating GABAergic transmission [6,13]. We hypothesize the existence of an essential pharmacophore within the enaminone structure that possibly interacts with the GABA receptor. The exact site and structural requirements for optimal binding are unknown. Nevertheless, we hypothesize, because of the molecular similarities between the enaminone analogs discussed here, that they share a common binding pocket on the GABA receptor, which explains the probability of eliciting similar biological responses.
Enaminones with different chemical structure can use different mechanisms to inhibit neuronal activity. Anilino enaminone E139 suppresses excitatory synaptic transmission through increasing GABA levels [6]. Chemically, E139 differs in its structure from KRS-5Me-4-OCF3 in the C1 position of the cyclohexenone moiety. E139 has a methyl ester substitution of the cyclohexenone moiety, whereas the three compounds described here are non-esters and para- or meta-substituted in the aromatic moiety. Enaminone compounds such as KRS-5Me-4-OCF3 act as a positive allosteric modulator of GABAA receptors. We postulate that the para-substitution of an aniline enaminone is a critical factor for the interaction with target proteins. Benzylamino enaminones with a similar structure to anilino enaminones use a distinctly different cellular mechanism for anti-convulsion and to suppress glutamate-mediated excitatory synaptic transmission as shown for benzylamino enaminone compound 30 [17]. Structurally diverse enaminones with different site substitutions may form different pharmacophores in order to interact with specific protein targets. A better understanding of the structure-response relationship may pave the way to rational drug design for potential anticonvulsant and anxiolytic compounds.
Acknowledgments
We would like to gratefully acknowledge Matthew Ennis and Philip M. Heyward, and Kenneth R. Scott for his pioneering work. T.H. is supported by grants from NIH [MD007597] and NSF [IOS-1355034].
Conflicts of Interest
The authors declare no conflict of interest.
Author Contributions
Thomas Heinbockel, Ze-Jun Wang, and Patrice Jackson-Ayotunde wrote the manuscript. All authors approved the final version.
References
- Greenhill, J.V. Enaminones. Chem. Soc. Rev. 1977, 6, 277–294. [Google Scholar] [CrossRef]
- Edafiogho, I.O.; Qaddoumi, M.G.; Ananthalakshmi, K.V.; Phillips, O.A.; Kombian, S.B. Synthesis, neuronal activity and mechanisms of action of halogenated enaminones. Eur. J. Med. Chem. 2014, 76, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Edafiogho, I.O.; Hinko, C.N.; Chang, H.; Moore, J.A.; Mulzac, D.; Nicholson, J.M.; Scott, K.R. Synthesis and anticonvulsant activity of enaminones. J. Med. Chem. 1992, 35, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Edafiogho, I.O.; Kombian, S.B.; Ananthalakshmi, K.V.; Salama, N.N.; Eddington, N.D.; Wilson, T.L.; Alexander, M.S.; Jackson, P.L.; Hanson, C.D.; Scott, K.R. Enaminones: Exploring additional therapeutic activities. J. Pharm. Sci. 2007, 96, 2509–2531. [Google Scholar] [CrossRef] [PubMed]
- Edafiogho, I.O.; Phillips, O.A.; Udo, E.E.; Samuel, S.; Rethish, B. Synthesis, antibacterial and anticonvulsant evaluations of some cyclic enaminones. Eur. J. Med. Chem. 2009, 44, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Kombian, S.B.; Edafiogho, I.O.; Ananthalakshmi, K.V. Anticonvulsant enaminones depress excitatory synaptic transmission in the rat brain by enhancing extracellular GABA levels. Br. J. Pharmacol. 2005, 145, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, M.E.; Edafiogho, I.O.; Scott, K.R. LC/MS determination of the enaminones E139, DM5 and DM27 in rat serum. J. Pharm. Biomed. Anal. 2002, 30, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.E.; Nicholson, J.M.; Butcher, R.; Stables, J.P.; Edafiogho, I.O.; Goodwin, A.M.; Henson, M.C.; Smith, C.A.; Scott, K.R. Synthesis, characterization and anticonvulsant activity of enaminones. Part 6: Synthesis of substituted vinylic benzamides as potential anticonvulsants. Bioorg. Med. Chem. 1999, 7, 2415–2425. [Google Scholar] [CrossRef] [PubMed]
- Stables, J.P.; Kupferberg, H.J. The NIH Anticonvulsant Drug Development (ADD) Program: preclinical anticonvulsant screening project. In Molecular and Cellular Targets for Anti-epileptic Drugs; Avanzini, G., Regesta, G., Tanganelli, P., Avoli, M., Eds.; John Libbey & Company Ltd: Londen, UK, 1997; pp. 191–198. [Google Scholar]
- Eddington, N.D.; Cox, D.S.; Roberts, R.R.; Stables, J.P.; Powell, C.B.; Scott, K.R. Enaminones-versatile therapeutic pharmacophores. Further advances. Curr. Med. Chem. 2000, 7, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Eddington, N.D.; Cox, D.S.; Khurana, M.; Salama, N.N.; Stables, J.P.; Harrison, S.J.; Negussie, A.; Taylor, R.S.; Tran, U.Q.; Moore, J.A.; Barrow, J.C.; et al. Synthesis and anticonvulsant activity of enaminones. Part 7. Synthesis and anticonvulsant evaluation of ethyl 4-[(substituted phenyl)amino]-6-methyl-2-oxocyclohex-3-ene-1-carboxylates and their corresponding 5-methylcyclohex-2-enone derivatives. Eur. J. Med. Chem. 2003, 38, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Mulzac, D.; Scott, K.R. Profile of anticonvulsant activity and minimal toxicity of methyl 4-[(p-chlorophenyl)amino]-6-methyl-2-oxo-cyclohex-3-en-1-oate and some prototype antiepileptic drugs in mice and rats. Epilepsia 1993, 34, 1141–1146. [Google Scholar] [CrossRef]
- Ananthalakshmi, K.V.; Edafiogho, I.O.; Kombian, S.B. Anticonvulsant enaminone E139 suppresses epileptiform activity in rat hippocampal slices. Epilepsy Res. 2007, 76, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Ennis, M.; Hamilton, K.A.; Hayar, A. Neurochemistry of the main olfactory system. In Handbook of Neurochemistry and Molecular Neurobiology: Sensory Neurochemistry, 3rd ed.; Johnson, D.A., Ed.; Springer: Heidelberg, Germany, 2007; Volume 20, pp. 137–204. [Google Scholar]
- Heinbockel, T.; Heyward, P.M. Glutamate synapses in olfactory neural circuits. In Amino Acid Receptor Research; Paley, B.F., Warfield, T.E., Eds.; Nova Science Publishers: New York, NY, USA, 2008; Volume Chapter 16, pp. 379–414. [Google Scholar]
- Ananthalakshmi, K.V.; Edafiogho, I.O.; Kombian, S.B. Concentration-dependent effects of anticonvulsant enaminone methyl 4-(4'-bromophenyl)aminocyclohex-3-en-6-methyl-2-oxo-1-oate on neuronal excitability in vitro. Neuroscience 2006, 41, 345–356. [Google Scholar] [CrossRef]
- Edafiogho, I.O.; Ananthalakshmi, K.V.; Kombian, S.B. Anticonvulsant evaluation and mechanism of action of benzylamino enaminones. Bioorg. Med. Chem. 2006, 14, 5266–5272. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Sun, L.; Jackson, P.L.; Scott, K.R.; Heinbockel, T. A substituted anilino enaminone acts as a novel positive allosteric modulator of GABAA receptors in the mouse brain. J. Pharmacol. Exp. Ther. 2011, 336, 916–924. [Google Scholar]
- Schiebler, T.H.; Schmidt, W.; Zilles, K. Anatomie, 8 ed.; Springer Verlag: Berlin, Germany, 1999. [Google Scholar]
- Swanson, L.W. Brain Maps: Structure of the Rat Brain. A Laboratory Guide with Printed and Electronic Templates for Data, Models and Schematics, 3rd ed.; Elsevier: Amsterdam, the Netherhand, 2004. [Google Scholar]
- Shipley, M.T.; Ennis, M. Functional organization of olfactory system. J. Neurobiol. 1996, 30, 123–176. [Google Scholar] [CrossRef] [PubMed]
- Klenoff, J.R.; Greer, C.A. Postnatal development of olfactory receptor cell axonal arbors. J. Comp. Neurol. 1998, 390, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Meisami, E.; Mikhail, L.; Baim, D.; Bhatnagar, K.P. Human olfactory bulb: Aging of glomeruli and mitral cells and a search for the accessory olfactory bulb. Ann. NY Acad. Sci. 1998, 855, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Aungst, J.L.; Heyward, P.M.; Puche, A.C.; Karnup, S.V.; Hayar, A.; Szabo, G.; Shipley, M.T. Centre-surround inhibition among olfactory bulb glomeruli. Nature 2003, 426, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Pinching, A.J.; Powell, T.P. The neuropil of the glomeruli of the olfactory bulb. J. Cell. Sci. 1971, 9, 347–377. [Google Scholar] [PubMed]
- Wachowiak, M.; Shipley, M.T. Coding and synaptic processing of sensory information in the glomerular layer of the olfactory bulb. Sem. Cell. Dev. Biol. 2006, 17, 411–423. [Google Scholar] [CrossRef]
- Hayar, A.; Karnup, S.; Ennis, M.; Shipley, M.T. External tufted cells: a major excitatory element that coordinates glomerular activity. J. Neurosci. 2004, 24, 6676–6685. [Google Scholar] [CrossRef] [PubMed]
- Kiyokage, E.; Pan, Y.Z.; Shao, Z.; Kobayashi, K.; Szabo, G.; Yanagawa, Y.; Obata, K.; Okano, H.; Toida, K.; Puche, A.C.; et al. Molecular identity of periglomerular and short axon cells. J. Neurosci. 2010, 30, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Aroniadou-Anderjaska, V.; Zhou, F.M.; Priest, C.A.; Ennis, M.; Shipley, M.T. Tonic and synaptically evoked presynaptic inhibition of sensory input to the rat olfactory bulb via GABA(B) heteroreceptors. J. Neurophysiol. 2000, 84, 1194–1203. [Google Scholar] [PubMed]
- Murphy, G.J.; Darcy, D.P.; Isaacson, J.S. Intraglomerular inhibition: Signaling mechanisms of an olfactory microcircuit. Nat. Neurosci. 2005, 8, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Hayar, A.; Shipley, M.T.; Ennis, M. Olfactory bulb external tufted cells are synchronized by multiple intraglomerular mechanisms. J. Neurosci. 2005, 25, 8197–8208. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.W.; Heinbockel, T.; Hamilton, K.A.; Hayar, A.; Ennis, M. Metabotropic glutamate receptors and dendrodendritic synapses in the main olfactory bulb. Ann. NY Acad. Sci. 2009, 1170, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Laaris, N.; Puche, A.; Ennis, M. Complementary postsynaptic activity patterns elicited in olfactory bulb by stimulation of mitral/tufted and centrifugal fiber inputs to granule cells. J. Neurophysiol. 2007, 97, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Jackowski, A.; Parnavelas, J.G.; Lieberman, A.R. The reciprocal synapse in the external plexiform layer of the mammalian olfactory bulb. Brain Res. 1978, 159, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Rall, W.; Shepherd, G.M.; Reese, T.S.; Brightman, M.W. Dendrodendritic synaptic pathway for inhibition in the olfactory bulb. Exp. Neurol. 1966, 14, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Frazier, L.L.; Brunjes, P.C. Unilateral odor deprivation: Early postnatal changes in olfactory bulb cell density and number. J. Comp. Neurol. 1988, 269, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Meisami, E. A proposed relationship between increases in the number of olfactory receptor neurons, convergence ratio and sensitivity in the developing rat. Brain Res. Dev. Brain Res. 1989, 46, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Heinbockel, T.; Ennis, M. Metabotropic glutamate receptors and neural processing in the olfactory system. In Neural Pathways Research; Pichler, F.L., Ed.; Nova Science Publishers: New York, NY, USA, 2008; Volume Chapter 1, pp. 1–30. [Google Scholar]
- Pressler, R.T.; Strowbridge, B.W. Blanes cells mediate persistent feedforward inhibition onto granule cells in the olfactory bulb. Neuron 2006, 49, 889–904. [Google Scholar] [CrossRef]
- Lledo, P.M.; Alonso, M.; Grubb, M.S. Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 2006, 7, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Whitman, M.C.; Greer, C.A. Synaptic integration of adult-generated olfactory bulb granule cells: Basal axodendrodendritic centrifugal input precedes apical dendrodendritic local circuits. J. Neurosci. 2007, 27, 9951–9961. [Google Scholar] [CrossRef] [PubMed]
- Jahr, C.E.; Nicoll, R.A. Noradrenergic modulation of dendrodendritic inhibition in the olfactory bulb. Nature 1982, 297, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Schoppa, N.E.; Urban, N.N. Dendritic processing within olfactory bulb circuits. Trends Neurosci. 2003, 26, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Heinbockel, T.; Laaris, N.; Ennis, M. Metabotropic glutamate receptors in the main olfactory bulb drive granule cell-mediated inhibition. J. Neurophysiol. 2007, 97, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Heinbockel, T.; Heyward, P.; Conquet, F.; Ennis, M. Regulation of main olfactory bulb mitral cell excitability by metabotropic glutamate receptor mGluR1. J. Neurophysiol. 2004, 92, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.W.; Hayar, A.; Ennis, M. Activation of group I metabotropic glutamate receptors on main olfactory bulb granule cells and periglomerular cells enhances synaptic inhibition of mitral cells. J. Neurosci. 2007, 27, 5654–5663. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, J.S.; Strowbridge, B.W. Olfactory reciprocal synapses: Dendritic signaling in the CNS. Neuron 1998, 20, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Balu, R.; Pressler, R.T.; Strowbridge, B.W. Multiple modes of synaptic excitation of olfactory bulb granule cells. J. Neurosci. 2007, 27, 5621–5632. [Google Scholar] [CrossRef] [PubMed]
- Willhite, D.C.; Nguyen, K.T.; Masurkar, A.V.; Greer, C.A.; Shepherd, G.M.; Chen, W.R. Viral tracing identifies distributed columnar organization in the olfactory bulb. Proc. Natl. Acad. Sci. USA 2006, 103, 12592–12597. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G.M.; Chen, W.R.; Greer, C.A. Olfactory bulb. In The Synaptic Organization of the Brain; Shepherd, G.M., Ed.; Oxford University Press: Oxford, UK, 2004; pp. 165–216. [Google Scholar]
- Brodie, M.J.; Elder, A.T.; Kwan, P. Epilepsy in later life. Lancet Neurol. 2009, 8, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; van Emde Boas, W.; Blume, W.; Elger, C.; Genton, P.; Lee, P.; Engel, J. Epileptic seizures and epilepsy: Definitions proposed by the International League against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005, 46, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Rall, T.W.; Schleifer, L.S. Drugs effective in the therapy of the epilepsies. In The Pharmacological Basis of Therapeutics; Goodman, A.G., Rall, T.W., Nies, A.S., Taylor, P., Eds.; Pergamon Press: New York, NY, USA, 1990; pp. 436–462. [Google Scholar]
- Devlin, A.L.; Odell, M.; Charlton, J.L.; Koppel, S. Epilepsy and driving: Current status of research. Epilepsy Res. 2012, 102, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Salome, C.; Salome-Grosjean, E.; Park, K.; Marieux, P.; Swendiman, E.D.; Stables, J.; Kohn, H. Synthesis and anticonvulsant activities of (R)-N-(4'-substituted)benzyl 2-acetamido-3-methoxypropionamides. J. Med. Chem. 2010, 53, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Rho, J.M.; Sankar, R. The pharmacologic basis of antiepileptic drug action. Epilepsia 1999, 40, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Dingledine, R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J. Neurophysiol. 1988, 59, 259–276. [Google Scholar] [PubMed]
- McNamara, J.O. Drugs effective in the therapy of the epilepsies. In The Pharmacological Basis of Therapeutics; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Gilman, A.G., Eds.; McGraw–Hill: New York, NY, USA, 1996; pp. 461–486. [Google Scholar]
- Snell, L.D.; Claffey, D.J.; Ruth, J.A.; Valenzuela, C.F.; Cardoso, R.; Wang, Z.; Levinson, S.R.; Sather, W.A.; Williamson, A.V.; Ingersoll, N.C.; et al. Novel structure having antagonist actions at both the glycine site of the N-methyl-d-aspartate receptor and neuronal voltage-sensitive sodium channels: Biochemical, electrophysiological, and behavioral characterization. J. Pharmacol. Exp. Ther. 2000, 292, 215–227. [Google Scholar] [PubMed]
- Wang, Z.J.; Snell, L.D.; Tabakoff, B.; Levinson, S.R. Inhibition of neuronal Na+ channels by the novel antiepileptic compound DCUKA: Identification of the diphenylureido moiety as an inactivation modifier. Exp. Neurol. 2002, 178, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Friary, R.J.; Gilligan, J.M.; Szajewski, R.P.; Falci, K.J.; Franck, R.W. Heterocyclic synthesis via the intramolecular acylation of enamines derived from amino acids. J. Org. Chem. 1973, 38, 3487–3491. [Google Scholar] [CrossRef]
- Scott, K.R.; Edafiogho, I.O.; Richardson, E.L.; Farrar, V.A.; Moore, J.A.; Tietz, E.I.; Hinko, C.N.; Chang, H.; El-Assadi, A.; Nicholson, J.M. Synthesis and anticonvulsant activity of enaminones. 2. Further structure activity correlations. J. Med. Chem. 1993, 36, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Bowery, N.G.; Hudson, A.L.; Price, G.W. GABAA and GABAB receptor site distribution in the rat central nervous system. Neuroscience 1987, 20, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.C.; Albin, R.L.; Young, A.B.; Penney, J.B. Distribution and kinetics of GABAB binding sites in rat central nervous system: a quantitative autoradiographic study. Neuroscience 1990, 34, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, H.Z.; Ye, N.; Zhang, J.; Wang, J.J. Role of GABAB receptors in GABA and baclofen-induced inhibition of adult rat cerebellar interpositus nucleus neurons in vitro. Brain Res. Bull. 2005, 67, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Palouzier-Paulignan, B.; Duchamp-Viret, P.; Hardy, A.B.; Duchamp, A. GABA(B) receptor-mediated inhibition of mitral/tufted cell activity in the rat olfactory bulb: A whole-cell patch-clamp study in vitro. Neuroscience 2002, 111, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, J.S.; Vitten, H. GABA(B) receptors inhibit dendrodendritic transmission in the rat olfactory bulb. J. Neurosci. 2003, 23, 2032–2039. [Google Scholar] [PubMed]
- Panzanelli, P.; Perazzini, A.Z.; Fritschy, J.M.; Sassoè-Pognetto, M. Heterogeneity of gamma-aminobutyric acid type A receptors in mitral and tufted cells of the rat main olfactory bulb. J. Comp. Neurol. 2005, 484, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Laurie, D.J.; Seeburg, P.H.; Wisden, W. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J. Neurosci. 1992, 12, 1063–1076. [Google Scholar] [PubMed]
- Persohn, E.; Malherbe, P.; Richards, J.G. Comparative molecular neuroanatomy of cloned GABAA receptor subunits in the rat CNS. J. Comp. Neurol. 1992, 326, 193–216. [Google Scholar] [CrossRef] [PubMed]
- Errante, L.D.; Williamson, A.; Spencer, D.D.; Petroff, O.A. Gabapentin and vigabatrin increase GABA in the human neocortical slice. Epilepsy Res. 2002, 49, 203–210. [Google Scholar] [CrossRef]
- Hogenkamp, D.J.; Johnstone, T.B.; Huang, J.C.; Li, W.Y.; Tran, M.; Whittemore, E.R.; Bagnera, R.E.; Gee, K.W. Enaminone amides as novel orally active GABAA receptor modulators. J. Med. Chem. 2007, 50, 3369–3379. [Google Scholar] [CrossRef] [PubMed]
- Reitz, A.B.; Fitzpatrick, L.J.; Jordan, A.D.; Sanfilippo, P.J. 5-[(Heteroaryl)alkyl]-3-oxo-pyrido[1,2-a]benzimidazole-4-carboxamide derivatives useful in treating central nervous system disorders. US patent 5,922,731, 13 July 1999. [Google Scholar]
- Yohannes, D.; Maynard, G.; Yuan, J.; Xie, L.; Lee, K.; Ghosh, M.; Luke, G.; Liu, X.; Nagal, A.; Vincent, L.; et al. Heterocyclic compounds as ligands of the GABAA receptor. US patent 6,653,471, 25 November 2003. [Google Scholar]
- Jackson, P.L.; Scott, K.R.; Southerland, W.M.; Fang, Y.Y. Enaminones 8: CoMFA and CoMSIA studies on some anticonvulsant enaminones. Bioorg. Med. Chem. 2009, 17, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.L. The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology 2009, 56, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Möhler, H.; Fritschy, J.M.; Rudolph, U. A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 2002, 300, 2–8. [Google Scholar] [PubMed]
- Munro, G.; Lopez-Garcia, J.A.; Rivera-Arconada, I.; Erichsen, H.K.; Nielsen, E.Ø.; Larsen, J.S.; Ahring, P.K.; Mirza, N.R. Comparison of the novel subtype-selective GABAA receptor-positive allosteric modulator NS11394 [3'-[5-(1-hydroxy-1-methyl-ethyl)-benzoimidazol-1-yl]-biphenyl-2-carbonitrile] with diazepam, zolpidem, bretazenil, and gaboxadol in rat models of inflammatory and neuropathic pain. J. Pharmacol. Exp. Ther. 2008, 327, 969–981. [Google Scholar] [CrossRef]
- Santhakumar, V.; Wallner, M.; Otis, T.S. Ethanol acts directly on extrasynaptic subtypes of GABAA receptors to increase tonic inhibition. Alcohol 2007, 41, 211–221. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).