Are AMPA Receptor Positive Allosteric Modulators Potential Pharmacotherapeutics for Addiction?

{kind=link}
{kind=link}
Abstract
:1. Overview of Drug Addiction: The Need for Better Pharmacotherapeutics
2. “Addiction” versus “Substance Dependence”
2.1. “Bottom-up”: Subcortical Neuroplasticity Drives the Development of Habitual Drug-Seeking Behavior
2.2. “Top-down”: Repeated Drug-Induced Insults to Prefrontal Cortices Impairs Executive Functioning
3. Glutamatergic Mechanisms in Memory Formation: A Brief Overview
4. Extinction/Exposure Strategies: Rescuing Behavioral Regulation
5. Facilitating ILC-NAcs Glutamate Signaling
6. AMPA PAMs and Addiction: Preclinical Studies
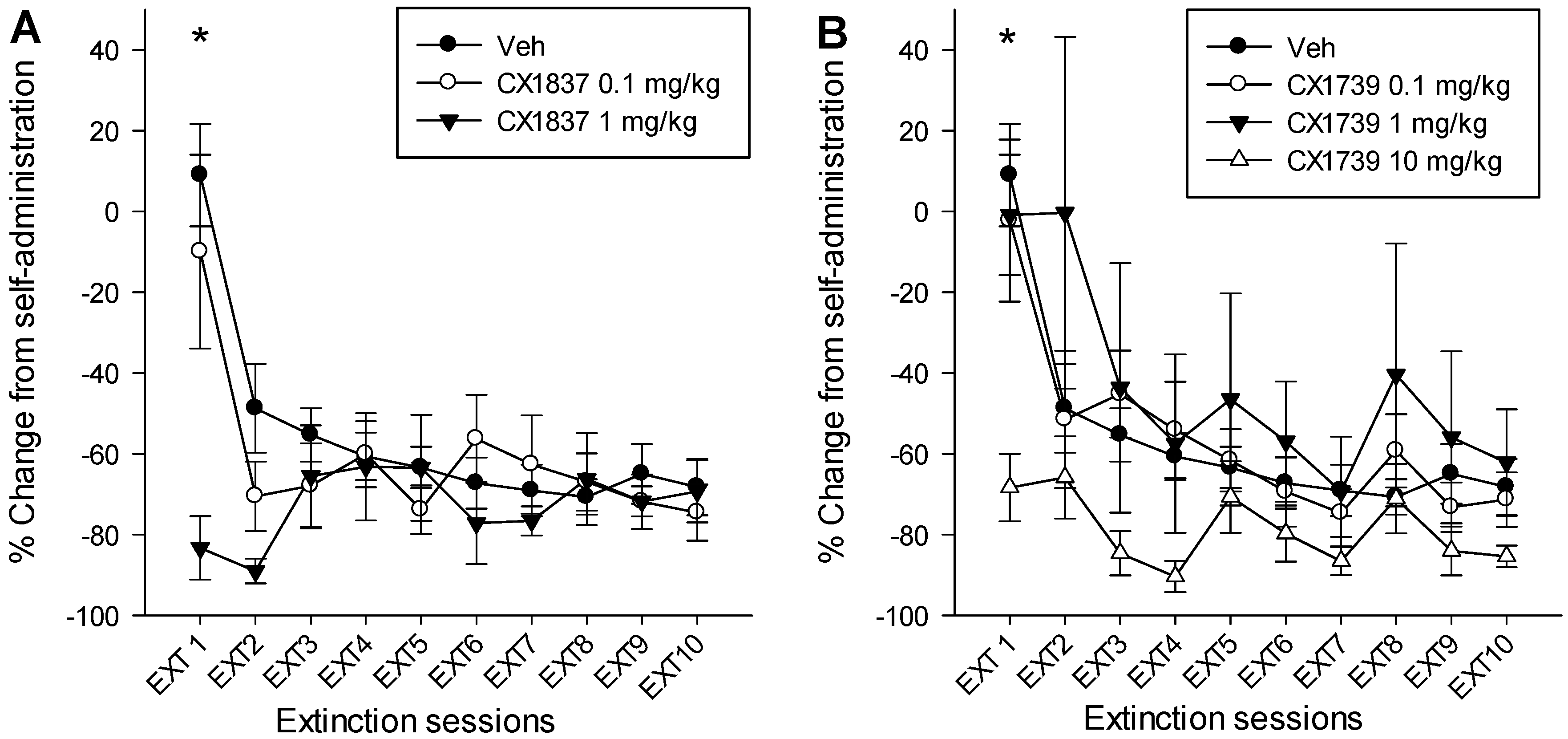
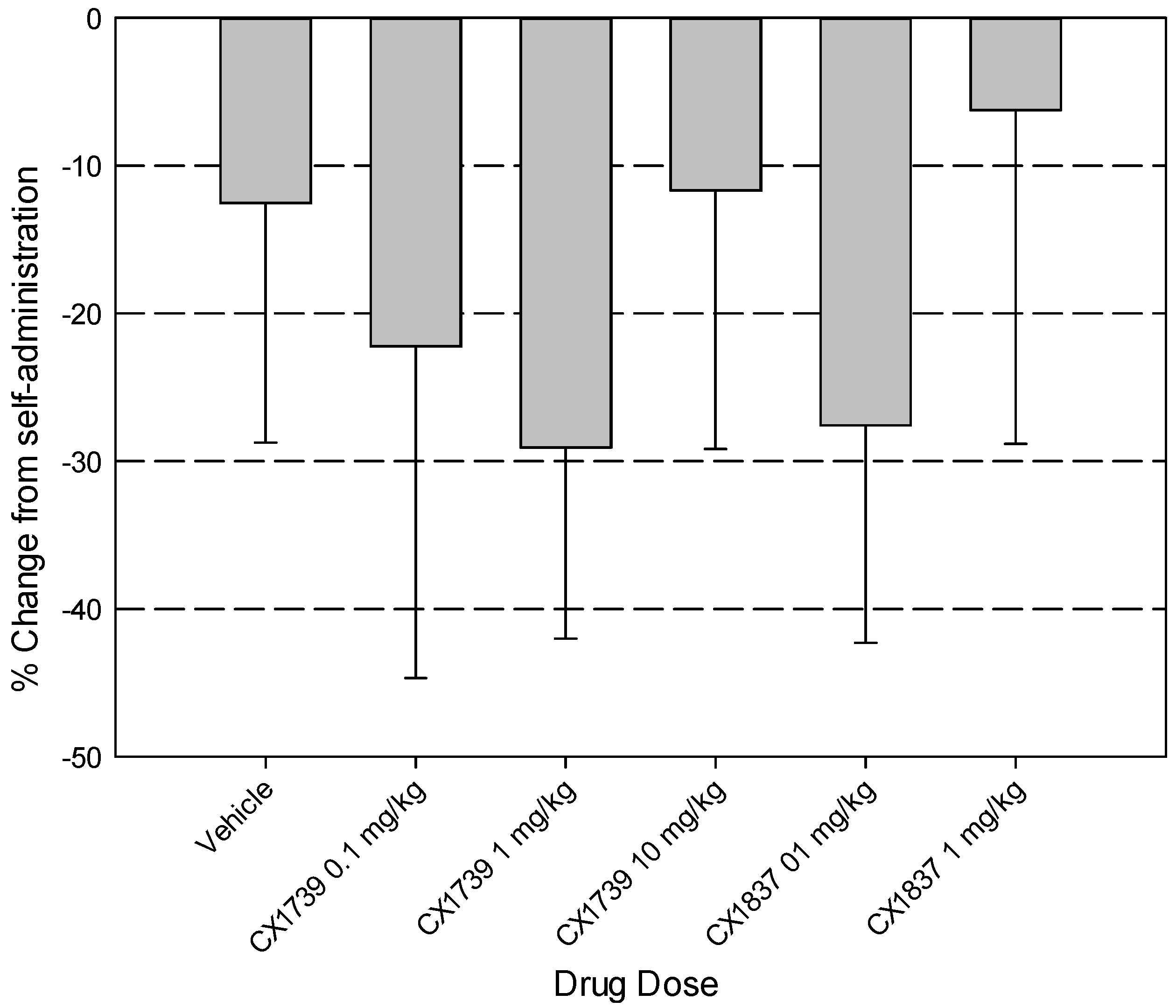
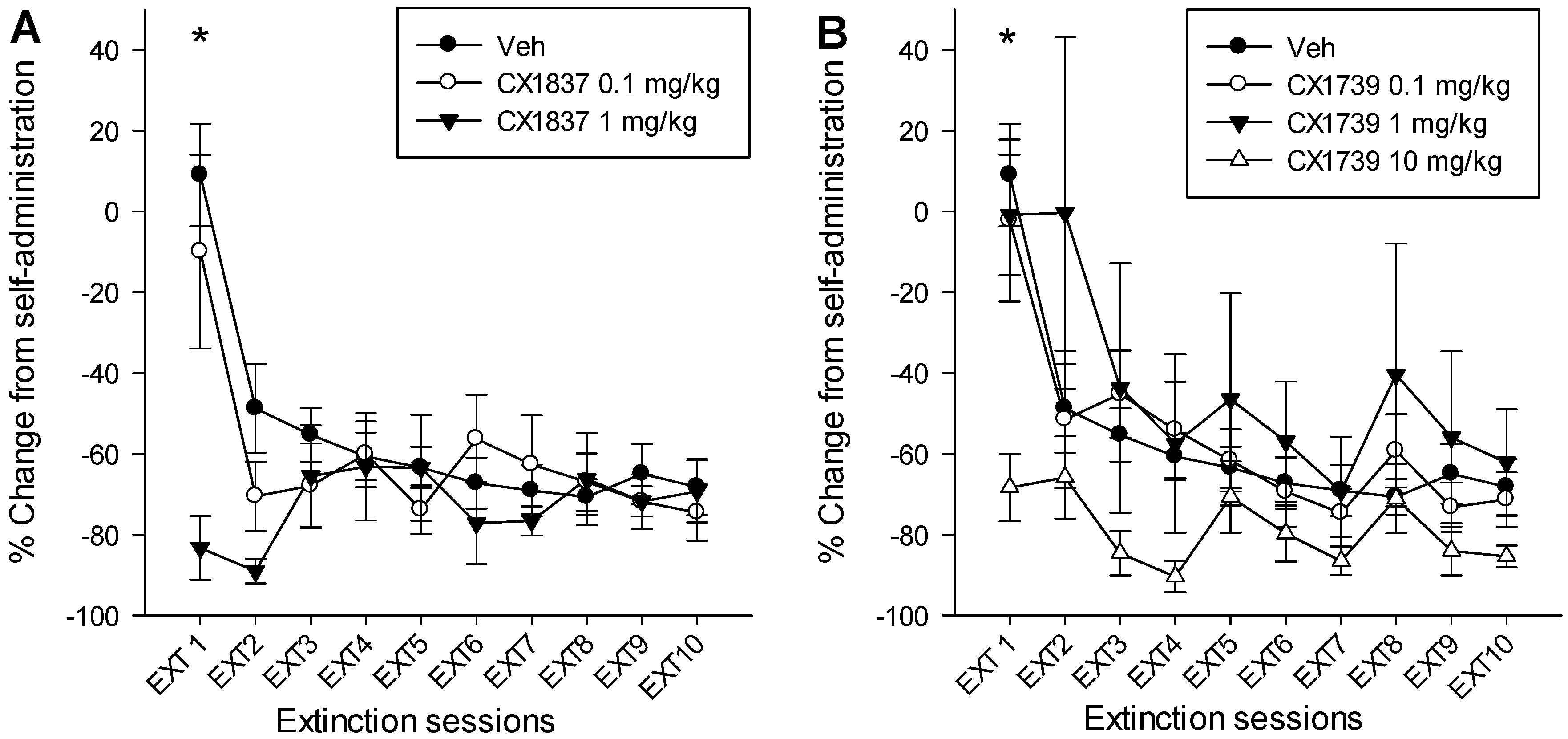
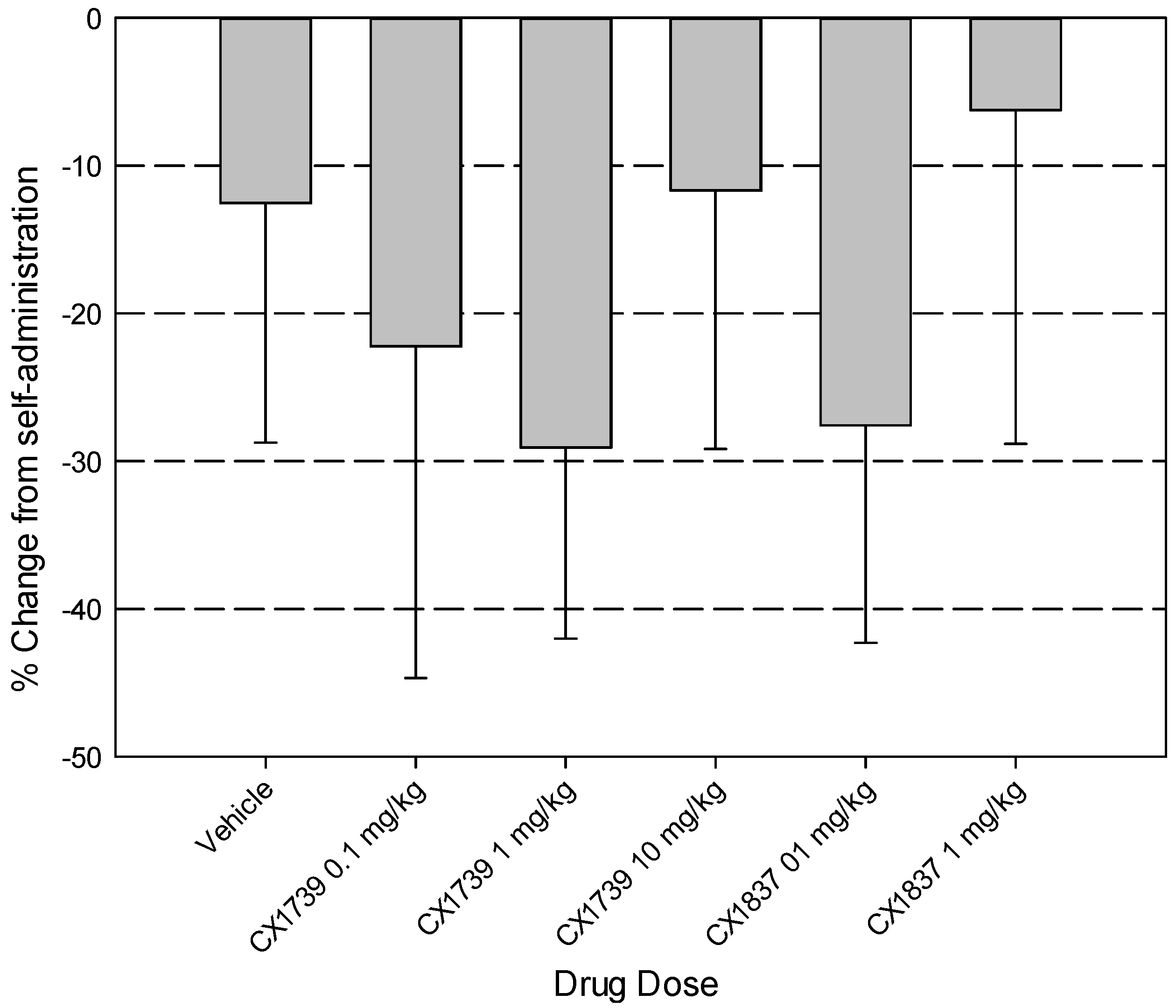
7. Discussion
8. Conclusions
Conflicts of Interest
Acknowledgments
References
- Koob, G.F.; Sanna, P.P.; Bloom, F.E. Neuroscience of addiction. Neuron 1998, 21, 467–476. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Volkow, N.; Seamans, J. Unmanageable motivation in addiction: A pathology in prefrontal-accumbens glutamate transmission. Neuron 2005, 45, 647–650. [Google Scholar] [CrossRef]
- Ericson, N. Substance Abuse : The Nation’s Number One Health Problem. Available online: https://www.ncjrs.gov/pdffiles1/ojjdp/fs200117.pdf (accessed on 28 October 2013).
- Harwood, H.; Bouchery, E. The Economic Costs of Drug Abuse in the United States, 1992-2002. 2004. Available online: https://www.ncjrs.gov/App/abstractdb/AbstractDBDetails.aspx?id=207303 (accessed on 28 October 2013).
- Fiscal Year 2008 Budget Request | National Institute on Drug Abuse. Available online: http://www.drugabuse.gov/about-nida/legislative-activities/testimony-to-congress/2007/03/fiscal-year-2008-budget-request (accessed on 28 October 2013).
- Substance Abuse and Mental Health Services Administration; US Department of Health and Human Services. Results from the 2011 National Survey on Drug Use and Health: Summary of National Findings. Available online: http://www.samhsa.gov/data/nsduh/2k11results/nsduhresults2011.htm (accessed on 28 October 2013).
- Sofuoglu, M.; DeVito, E.E.; Waters, A.J.; Carroll, K.M. Cognitive enhancement as a treatment for drug addictions. Neuropharmacology 2013, 64, 452–463. [Google Scholar] [CrossRef]
- Sofuoglu, M. Cognitive enhancement as a pharmacotherapy target for stimulant addiction. Addiction 2010, 105, 38–48. [Google Scholar] [CrossRef]
- Hendershot, C.S.; Witkiewitz, K.; George, W.H.; Marlatt, G.A. Relapse prevention for addictive behaviors. Subst. Abuse Treat. Prev. Policy 2011, 6, 17. [Google Scholar] [CrossRef]
- Brandon, T.H.; Vidrine, J.I.; Litvin, E.B. Relapse and relapse prevention. Annu. Rev. Clin. Psychol. 2007, 3, 257–284. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV-TR, 4th ed.; American Psychiatric Publishing: Washington, WA, USA, 2000. [Google Scholar]
- Maddux, J.F.; Desmond, D.P. Addiction or dependence? Addiction 2000, 95, 661–665. [Google Scholar] [CrossRef]
- O’Brien, C.P.; Volkow, N.D.; Li, T.-K. What’s in a word? Addiction versus dependence in DSM-V. Am. J. Psychiatry 2006, 163, 2014. [Google Scholar]
- O’Brien, C. Addiction and dependence in DSM-V. Addiction 2011, 106, 866–867. [Google Scholar]
- Ahmed, S.H. Validation crisis in animal models of drug addiction: beyond non-disordered drug use toward drug addiction. Neurosci. Biobehav. Rev. 2010, 35, 172–184. [Google Scholar] [CrossRef]
- Ahmed, S.H. The science of making drug-addicted animals. Neuroscience 2012, 211, 107–125. [Google Scholar] [CrossRef]
- Kalivas, P.; Volkow, N.D.N. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol. Psychiatry 2011, 16, 974–986. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, WA, USA, 2013. [Google Scholar]
- Berridge, K.C.; Robinson, T.E. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 1998, 28, 309–369. [Google Scholar] [CrossRef]
- Feltenstein, M.W.; See, R.E. The neurocircuitry of addiction: An overview. Br. J. Pharmacol. 2008, 154, 261–274. [Google Scholar] [CrossRef]
- Hyman, S.E.; Malenka, R.C. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat. Rev. Neurosci. 2001, 2, 695–703. [Google Scholar] [CrossRef]
- Spanagel, R.; Weiss, F. The dopamine hypothesis of reward: past and current status. Trends Neurosci. 1999, 22, 521–527. [Google Scholar] [CrossRef]
- Graybiel, A.M. Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci. 2008, 31, 359–387. [Google Scholar] [CrossRef]
- Kalivas, P.W. Neurobiology of cocaine addiction: implications for new pharmacotherapy. Am. J. Addict. 2007, 16, 71–78. [Google Scholar] [CrossRef]
- Gass, J.T.; Olive, M.F. Glutamatergic substrates of drug addiction and alcoholism. Biochem. Pharmacol. 2008, 75, 218–265. [Google Scholar] [CrossRef]
- Cleva, R.; Gass, J. Neuroanatomical structures underlying the extinction of drug-seeking behavior. Open Addict. J. 2010, 3, 63–75. [Google Scholar] [CrossRef]
- Kalivas, P.W.; O’Brien, C. Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 2008, 33, 166–180. [Google Scholar] [CrossRef]
- Kalivas, P. Neurocircuitry of addiction. In Neuropsychopharmacology; Davis, K.L., Charney, D., Coyle, J.T., Nemeroff, C., Eds.; Lippincott, Williams, & Wilkins: Philadelphia, PA, USA, 2002; pp. 1357–1366. [Google Scholar]
- Goldstein, R.Z.; Volkow, N.D. Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 2011, 12, 652–669. [Google Scholar] [CrossRef]
- Kalivas, P.; Volkow, N. The neural basis of addiction: A pathology of motivation and choice. Am. J. Psychiatry 2005, 162, 1403–1413. [Google Scholar]
- Jentsch, J.D.; Taylor, J.R. Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology 1999, 146, 373–390. [Google Scholar] [CrossRef]
- Miller, E.K.; Cohen, J.D. An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 2001, 24, 167–202. [Google Scholar] [CrossRef]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef]
- Field, M.; Cox, W.M. Attentional bias in addictive behaviors: a review of its development, causes, and consequences. Drug Alcohol Depend. 2008, 97, 1–20. [Google Scholar] [CrossRef]
- Kalivas, P.W. Addiction as a pathology in prefrontal cortical regulation of corticostriatal habit circuitry. Neurotox. Res. 2008, 14, 185–189. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Schmidt, W.J. Glutamatergic mechanisms in addiction. Mol. Psychiatry 2003, 8, 373–382. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Lalumiere, R.T.; Knackstedt, L.; Shen, H. Glutamate transmission in addiction. Neuropharmacology 2009, 56, 169–173. [Google Scholar] [CrossRef]
- Gass, J.T.; Olive, M.F.F. Positive allosteric modulation of mGluR5 receptors facilitates extinction of a cocaine contextual memory. Biol. Psychiatry 2009, 65, 717–720. [Google Scholar] [CrossRef]
- Peters, J.; Kalivas, P.W.; Quirk, G.J. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn. Mem. 2009, 16, 279–288. [Google Scholar] [CrossRef]
- LaLumiere, R.T.; Smith, K.C.; Kalivas, P.W. Neural circuit competition in cocaine-seeking: Roles of the infralimbic cortex and nucleus accumbens shell. Eur. J. Neurosci. 2012, 35, 614–622. [Google Scholar] [CrossRef]
- Cleva, R.M.; Gass, J.T.; Widholm, J.J.; Olive, M.F. Glutamatergic targets for enhancing extinction learning in drug addiction. Curr. Neuropharmacol. 2010, 8, 394–408. [Google Scholar] [CrossRef]
- Olive, M.F.; Cleva, R.M.; Kalivas, P.W.; Malcolm, R.J. Glutamatergic medications for the treatment of drug and behavioral addictions. Pharmacol. Biochem. Behav. 2012, 100, 801–810. [Google Scholar] [CrossRef]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar]
- Nicoll, R.A.; Roche, K.W. Long-term potentiation: Peeling the onion. Neuropharmacology 2013, 74, 18–22. [Google Scholar] [CrossRef]
- Lamprecht, R.; LeDoux, J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Chang, P.K.-Y.; Verbich, D.; McKinney, R.A. AMPA receptors as drug targets in neurological disease—Advantages, caveats, and future outlook. Eur. J. Neurosci. 2012, 35, 1908–1916. [Google Scholar] [CrossRef]
- Lynch, G.; Gall, C.M. Ampakines and the threefold path to cognitive enhancement. Trends Neurosci. 2006, 29, 554–562. [Google Scholar] [CrossRef]
- Jog, M.S. Building neural representations of habits. Science 1999, 286, 1745–1749. [Google Scholar] [CrossRef]
- Everitt, B.J.; Robbins, T.W. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 2005, 8, 1481–1489. [Google Scholar] [CrossRef]
- Marlatt, G.A. Cue exposure and relapse prevention in the treatment of addictive behaviors. Addict. Behav. 1990, 15, 395–399. [Google Scholar] [CrossRef]
- Conklin, C.A.; Tiffany, S.T. Applying extinction research and theory to cue-exposure addiction treatments. Addiction 2002, 97, 155–167. [Google Scholar] [CrossRef]
- Havermans, R.C.; Jansen, A.T.M. Increasing the efficacy of cue exposure treatment in preventing relapse of addictive behavior. Addict. Behav. 2003, 28, 989–994. [Google Scholar] [CrossRef]
- Epstein, D.H.; Preston, K.L.; Stewart, J.; Shaham, Y. Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology 2006, 189, 1–16. [Google Scholar] [CrossRef]
- Bouton, M.E. Context and behavioral processes in extinction. Learn. Mem. 2004, 11, 485–494. [Google Scholar] [CrossRef]
- Bouton, M. Context, ambiguity, and unlearning: Sources of relapse after behavioral extinction. Biol. Psychiatry 2002, 52, 976–986. [Google Scholar] [CrossRef]
- Crombag, H.S.; Bossert, J.M.; Koya, E.; Shaham, Y. Context-induced relapse to drug seeking: A review. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3233–3243. [Google Scholar] [CrossRef]
- Rescorla, R. A Spontaneous recovery. Learn. Mem. 2004, 11, 501–509. [Google Scholar] [CrossRef]
- Taylor, J.R.; Olausson, P.; Quinn, J.J.; Torregrossa, M.M. Targeting extinction and reconsolidation mechanisms to combat the impact of drug cues on addiction. Neuropharmacology 2009, 56, 186–195. [Google Scholar] [CrossRef]
- Sutton, M.A.; Schmidt, E.F.; Choi, K.-H.; Schad, C.A.; Whisler, K.; Simmons, D.; Karaian, D.A.; Monteggia, L.M.; Neve, R.L.; Self, D.W. Extinction-induced upregulation in AMPA receptors reduces cocaine- seeking behaviour. Nature 2003, 421, 70–75. [Google Scholar] [CrossRef]
- Fuchs, R.A.; Branham, R.K.; See, R.E. Different neural substrates mediate cocaine seeking after abstinence versus extinction training: A critical role for the dorsolateral caudate-putamen. J. Neurosci. 2006, 26, 3584–3588. [Google Scholar] [CrossRef]
- Di Ciano, P.; Robbins, T.W.; Everitt, B.J. Differential effects of nucleus accumbens core, shell, or dorsal striatal inactivations on the persistence, reacquisition, or reinstatement of responding for a drug-paired conditioned reinforcer. Neuropsychopharmacology 2008, 33, 1413–1425. [Google Scholar] [CrossRef]
- Peters, J.; LaLumiere, R.T.; Kalivas, P.W. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J. Neurosci. 2008, 28, 6046–6053. [Google Scholar] [CrossRef]
- LaLumiere, R.T.; Kalivas, P.W. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 2008, 28, 3170–3177. [Google Scholar] [CrossRef]
- Knackstedt, L.A.; Moussawi, K.; LaLumiere, R.T.; Schwendt, M.; Klugmann, M.; Kalivas, P.W. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J. Neurosci. 2010, 30, 7984–7992. [Google Scholar]
- Childress, A.; Mozley, P. Limbic activation during cue-induced cocaine craving. Am. J. Psychiatry 1999, 156, 1–15. [Google Scholar]
- Ongür, D.; Price, J.L. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb. Cortex 2000, 10, 206–219. [Google Scholar] [CrossRef]
- McFarland, K.; Kalivas, P.W. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 2001, 21, 8655–8663. [Google Scholar]
- Kalivas, P.W.; McFarland, K. Brain circuitry and the reinstatement of cocaine-seeking behavior. Psychopharmacology 2003, 168, 44–56. [Google Scholar] [CrossRef]
- McFarland, K.; Lapish, C.C.; Kalivas, P.W. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci. 2003, 23, 3531–7353. [Google Scholar]
- LaLumiere, R.T.; Niehoff, K.E.; Kalivas, P.W. The infralimbic cortex regulates the consolidation of extinction after cocaine self-administration. Learn. Mem. 2010, 17, 168–175. [Google Scholar] [CrossRef]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef]
- Ghasemzadeh, M.B.; Vasudevan, P.; Mueller, C.; Seubert, C.; Mantsch, J.R. Region specific alterations in glutamate receptor expression and subcellular distribution following extinction of cocaine self-administration. Brain Res. 2009, 1267, 89–102. [Google Scholar]
- Bachtell, R.K.; Choi, K.-H.; Simmons, D.L.; Falcon, E.; Monteggia, L.M.; Neve, R.L.; Self, D.W. Role of GluR1 expression in nucleus accumbens neurons in cocaine sensitization and cocaine-seeking behavior. Eur. J. Neurosci. 2008, 27, 2229–2240. [Google Scholar] [CrossRef]
- Lynch, G. Memory enhancement: the search for mechanism-based drugs. Nat. Neurosci. 2002, 5, S1035–S1038. [Google Scholar] [CrossRef]
- Lynch, G. Glutamate-based therapeutic approaches: ampakines. Curr. Opin. Pharmacol. 2006, 6, 82–88. [Google Scholar] [CrossRef]
- Arai, A.C.; Kessler, M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr. Drug Targets 2007, 8, 583–602. [Google Scholar] [CrossRef]
- Lynch, G.; Palmer, L.C.; Gall, C.M. The likelihood of cognitive enhancement. Pharmacol. Biochem. Behav. 2011, 99, 116–129. [Google Scholar]
- Swanson, G. Targeting AMPA and kainate receptors in neurological disease: therapies on the horizon? Neuropsychopharmacology 2009, 34, 249–250. [Google Scholar] [CrossRef]
- Black, M.D. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology 2005, 179, 154–163. [Google Scholar] [CrossRef]
- Marenco, S.; Weinberger, D.R. Therapeutic potential of positive AMPA receptor modulators in the treatment of neuropsychiatric disorders. CNS Drugs 2006, 20, 173–185. [Google Scholar] [CrossRef]
- Jin, R.; Clark, S.; Weeks, A.M.; Dudman, J.T.; Gouaux, E.; Partin, K.M. Mechanism of positive allosteric modulators acting on AMPA receptors. J. Neurosci. 2005, 25, 9027–9036. [Google Scholar] [CrossRef]
- ONeill, M.; Bleakman, D. AMPA receptor potentiators for the treatment of CNS disorders. CNS Neurol. Disord. 2004, 3, 181–194. [Google Scholar]
- Christopoulos, A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef]
- Olney, J.W. Excitatory transmitter neurotoxicity. Neurobiol. Aging 1994, 15, 259–260. [Google Scholar] [CrossRef]
- Staubli, U.; Rogers, G.; Lynch, G. Facilitation of glutamate receptors enhances memory. Proc. Natl. Acad. Sci. USA 1994, 91, 777–781. [Google Scholar] [CrossRef]
- Mattson, M. Excitotoxic and excitoprotective mechanisms. Neuromol. Med. 2003, 3, 65–94. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef]
- Shaffer, C.L.; Hurst, R.S.; Scialis, R.J.; Osgood, S.M.; Bryce, D.K.; Hoffmann, W.E.; Lazzaro, J.T.; Hanks, A.N.; Lotarski, S.; Weber, M.L.; et al. Positive allosteric modulation of AMPA receptors from efficacy to toxicity: The interspecies exposure-response continuum of the novel potentiator PF-4778574. J. Pharmacol. Exp. Ther. 2013, 347, 212–224. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Nishikawa, K.; Aoki, S.; Wada, K. A desensitization-selective potentiator of AMPA-type glutamate receptors. Br. J. Pharmacol. 2002, 136, 1033–1041. [Google Scholar] [CrossRef]
- Bramham, C.R.; Messaoudi, E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog. Neurobiol. 2005, 76, 99–125. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Overman, J.J.; Zhong, S.; Mueller, R.; Lynch, G.; Carmichael, S.T. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J. Neurosci. 2011, 31, 3766–3775. [Google Scholar] [CrossRef]
- Silverman, J.L.; Oliver, C.F.; Karras, M.N.; Gastrell, P.T.; Crawley, J.N. AMPAKINE enhancement of social interaction in the BTBR mouse model of autism. Neuropharmacology 2013, 64, 268–282. [Google Scholar] [CrossRef]
- Bowers, M.S.; Chen, B.T.; Bonci, A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron 2010, 67, 11–24. [Google Scholar] [CrossRef]
- Ghitza, U.E.; Zhai, H.; Wu, P.; Airavaara, M.; Shaham, Y.; Lu, L. Role of BDNF and GDNF in drug reward and relapse: A review. Neurosci. Biobehav. Rev. 2010, 35, 157–171. [Google Scholar] [CrossRef]
- Willcocks, A.L.; McNally, G.P. The role of medial prefrontal cortex in extinction and reinstatement of alcohol-seeking in rats. Eur. J. Neurosci. 2013, 37, 259–268. [Google Scholar] [CrossRef]
- Xue, Y.-X.; Luo, Y.-X.; Wu, P.; Shi, H.-S.; Xue, L.-F.; Chen, C.; Zhu, W.-L.; Ding, Z.-B.; Bao, Y.-P.; Shi, J.; et al. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science 2012, 336, 241–245. [Google Scholar] [CrossRef]
- Zayra Millan, E.; Milligan-Saville, J.; McNally, G.P. Memory retrieval, extinction, and reinstatement of alcohol seeking. Neurobiol. Learn. Mem. 2013, 101, 26–32. [Google Scholar] [CrossRef]
- Sacktor, T. How does PKMζ maintain long-term memory? Nat. Rev. Neurosci. 2010, 12, 9–15. [Google Scholar] [CrossRef]
- Volk, L.J.; Bachman, J.L.; Johnson, R.; Yu, Y.; Huganir, R.L. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 2013, 493, 420–423. [Google Scholar] [CrossRef]
- Lee, A.M.; Kanter, B.R.; Wang, D.; Lim, J.P.; Zou, M.E.; Qiu, C.; McMahon, T.; Dadgar, J.; Fischbach-Weiss, S.C.; Messing, R.O. Prkcz null mice show normal learning and memory. Nature 2013, 493, 416–419. [Google Scholar] [CrossRef]
- Lauterborn, J.C.; Pineda, E.; Chen, L.Y.; Ramirez, E.A.; Lynch, G.; Gall, C.M. Ampakines cause sustained increases in brain-derived neurotrophic factor signaling at excitatory synapses without changes in AMPA receptor subunit expression. Neuroscience 2009, 159, 283–295. [Google Scholar] [CrossRef]
- Lynch, G. Memory and the brain: unexpected chemistries and a new pharmacology. Neurobiol. Learn. Mem. 1998, 70, 82–100. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watterson, L.R.; Olive, M.F. Are AMPA Receptor Positive Allosteric Modulators Potential Pharmacotherapeutics for Addiction? Pharmaceuticals 2014, 7, 29-45. https://doi.org/10.3390/ph7010029
Watterson LR, Olive MF. Are AMPA Receptor Positive Allosteric Modulators Potential Pharmacotherapeutics for Addiction? Pharmaceuticals. 2014; 7(1):29-45. https://doi.org/10.3390/ph7010029
Chicago/Turabian StyleWatterson, Lucas R., and M. Foster Olive. 2014. "Are AMPA Receptor Positive Allosteric Modulators Potential Pharmacotherapeutics for Addiction?" Pharmaceuticals 7, no. 1: 29-45. https://doi.org/10.3390/ph7010029
APA StyleWatterson, L. R., & Olive, M. F. (2014). Are AMPA Receptor Positive Allosteric Modulators Potential Pharmacotherapeutics for Addiction? Pharmaceuticals, 7(1), 29-45. https://doi.org/10.3390/ph7010029