Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury

Abstract
:1. Introduction
1.1. Significance of Traumatic Brain Injury
1.2. Regulation of Intracellular Calcium
2. Voltage Gated Calcium Channels
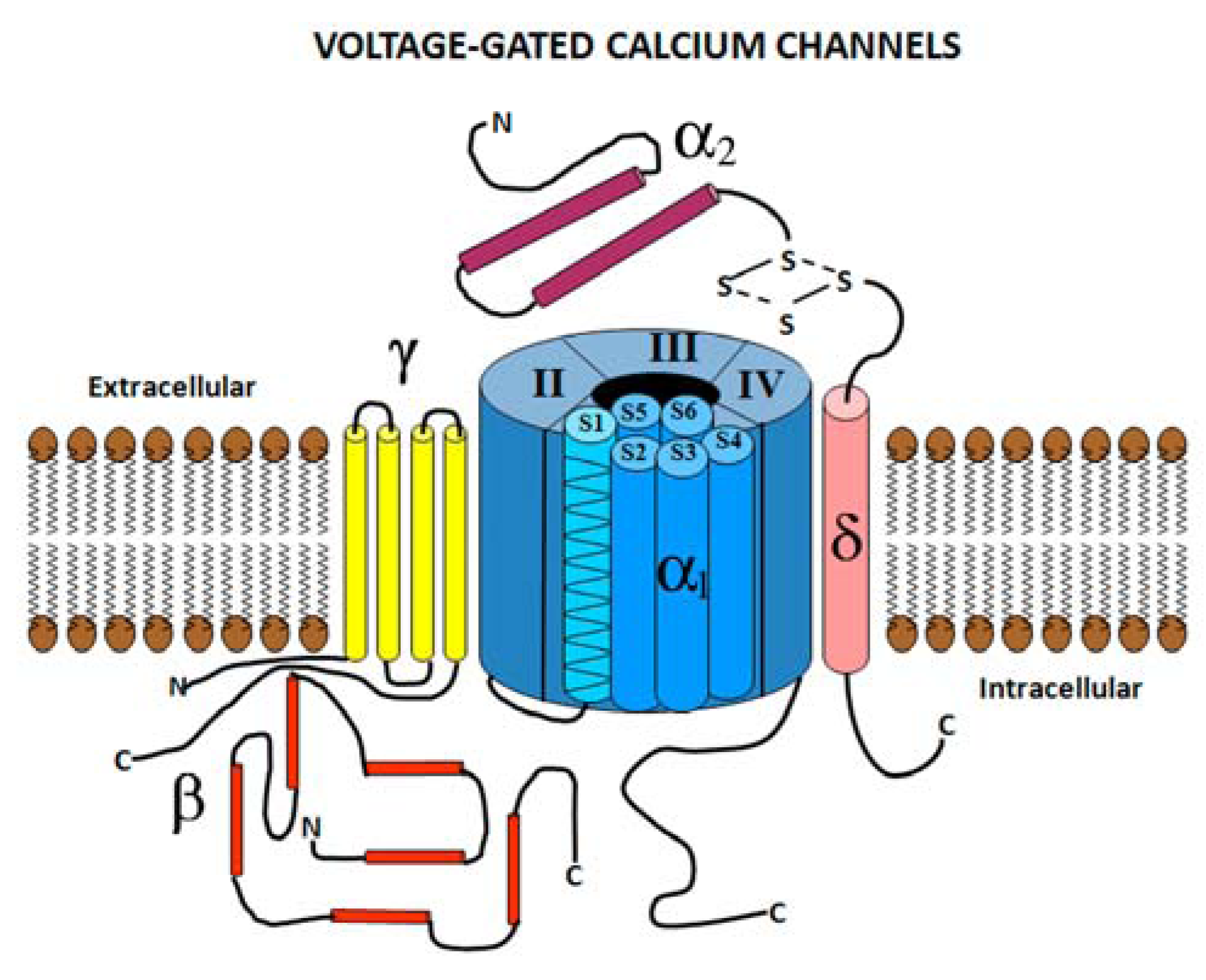
2.1. Voltage Gated Calcium Channel Structure
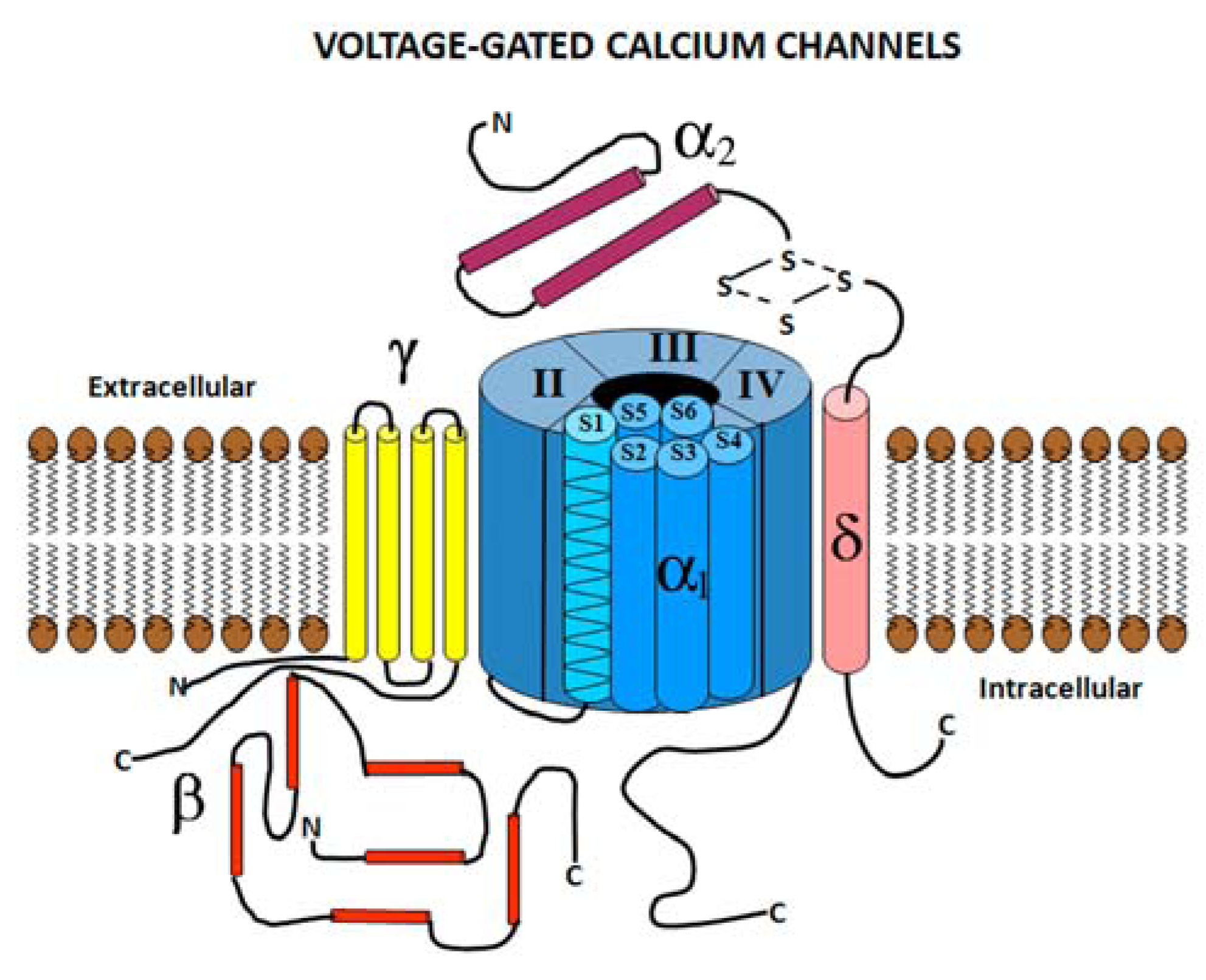
2.2. Voltage Gated Calcium Channel Subtypes

{kind=link}
{kind=link}
| Channel Type | L Cav1.1-1.4 | N Cav2.2 | P Cav2.1 | Q Cav2.1 | R Cav2.3 | T Cav3.1-3.3 | Ref. |
|---|---|---|---|---|---|---|---|
| Conductance (pS) | 25 | 11 to 20 | 9 to 20 | 15 to 16 | 15 to 20 | 8 | [40] |
| Selectivity (Ca2+>Ba2+) | 2:1 | 2:1 | 2:1 | ND | 1.3:1 | 1:1 | [40] |
| Activation Potential (mV) | −10 to −50 | −20 | −50 | −50 | −25 to −40 | −70 | [40] |
| Inactivation Kinetics (msec) | 150–2,000 | 100–200 | 500–1,000 | 500–1,000 | 50–100 | 10–70 | [40] |
| Calcium Blockers (IC50) | |||||||
| ω-conotoxin MVIIA | None | 78 nM–1 µM | None | None | None | None | [40,41,42] |
| ω-conotoxin GVIA | None | 28 nM–2 µM | None | None | None | None | [40,41,42] |
| ω-Agatoxin AgaIVA | None | None | 15 nM | 50 nM–1 µM | 50 nm | None | [41,43] |
| ω-conotoxin MVIIC | None | 18 nM | 18 nM | 50 nM–1 µM | None | None | [41,44] |
| ω-Agatoxin AgaIIIA | 1 nm | 1 nm | IC50 N/A | IC50 N/A | None | None | [45] |
| SNX-482 | None | None | 30–750 nm | 30–750 nm | 15–30 nM | None | [41,46,47] |
| Nimodipine | 0.135–2.6 µM | None | None | None | None | 5–11 µM | [48,49,50,51] |
| Nifedipine | 100 nM | None | None | None | None | 39 µM | [50] |
| Efonidipine | 10 µM | None | None | None | None | 1.3–13 µM | [51,52,53] |
| Amplodipine | 3–5 µM | None | None | None | None | 4–13 µM | [51,54,55] |
| Nicardipine | 9–26 µM | None | 32–97 µM | 32–97 µM | None | 5–13 µM | [55] |
| Verapamil | 0.6–1 µM | None | None | None | None | 20–30 µM | [49,56,57] |
| Diltiazem | 3–33 µM | None | None | None | None | 30 µM | [49,57] |
| Mibefradil | 1.7–21 µM | None | 208 µM | 208 µM | None | 0.5–11 µM | [51,58,59] |
2.3. Voltage Gated Calcium Channel Distribution in the Nervous System
2.4. Characterizing Voltage Gated Calcium Channels Based on Pharmacology
3. Experimental Evidence that Blockade of VGCC’s Can be Neuroprotective
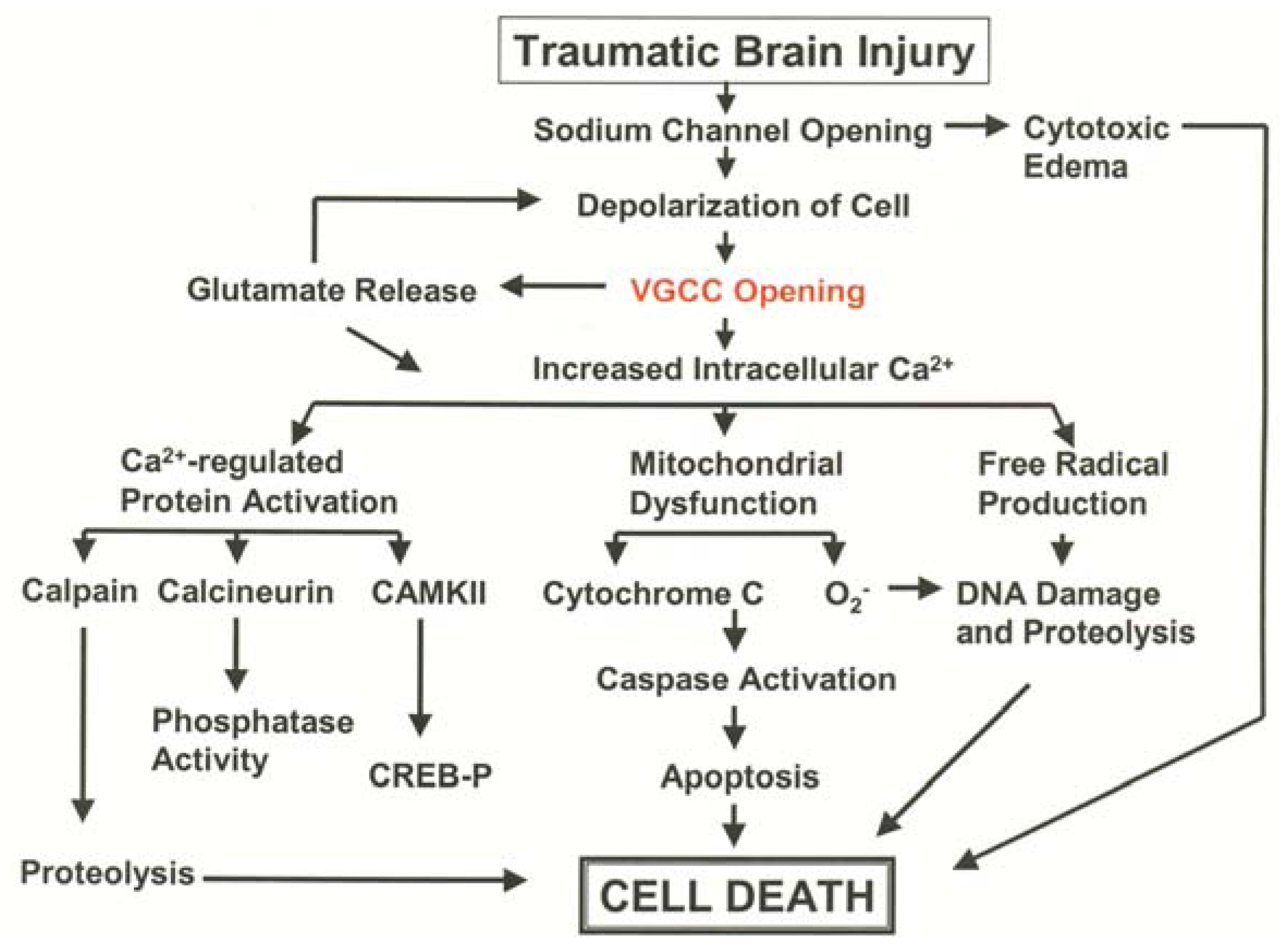
3.1. Pathological Calcium Accumulation Following TBI
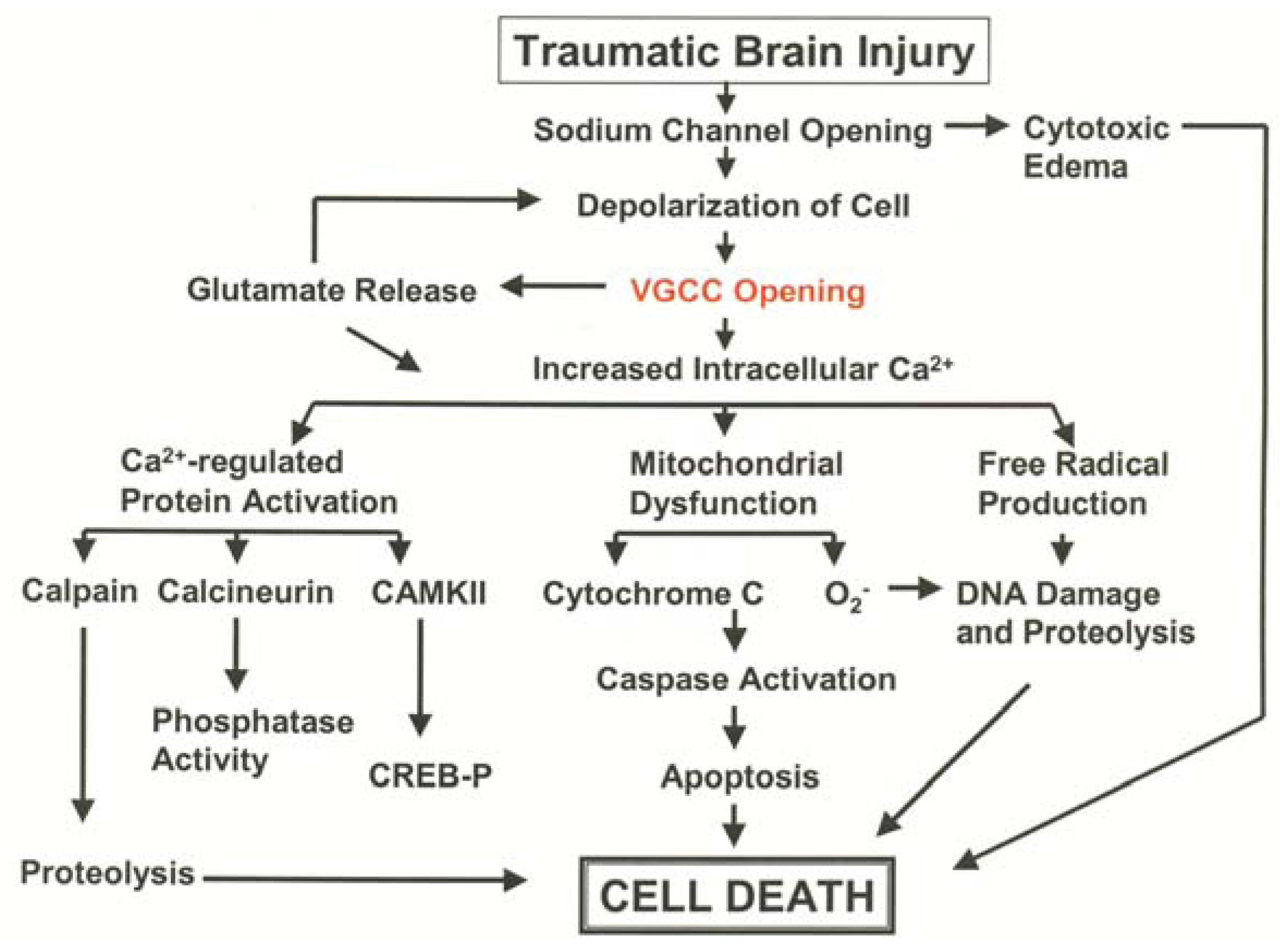
3.2. L-Type VGCC Antagonists to Treat Traumatic Brain Injury
3.3. N-Type VGCC Antagonists to Treat Traumatic Brain Injury
4. Conclusions
4.1. Lessons to be Learned from Ziconotide Development for Chronic Pain
4.2. Summary
Conflict of interest
References
- Faul, M.; Xu, L.; Wald, M.M.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010. [Google Scholar]
- Frost, R.B.; Farrer, T.J.; Primosch, M.; Hedges, D.W. Prevalence of traumatic brain injury in the general adult population: A meta-analysis. Neuroepidemiology 2012, 40, 154–159. [Google Scholar]
- Brooks, D.N.; Aughton, M.E. Cognitive recovery during the first year after severe blunt head injury. Int. Rehabil. Med. 1979, 1, 166–172. [Google Scholar]
- Brooks, N.; McKinlay, W.; Symington, C.; Beattie, A.; Campsie, L. Return to work within the first seven years of severe head injury. Brain Inj. 1987, 1, 5–19. [Google Scholar] [CrossRef]
- Benedictus, M.R.; Spikman, J.M.; van der Naalt, J. Cognitive and behavioral impairment in traumatic brain injury related to outcome and return to work. Arch. Phys. Med. Rehabil. 2010, 91, 1436–1441. [Google Scholar] [CrossRef]
- Bootes, K.; Chapparo, C. Difficulties with multitasking on return to work after TBI: A critical case study. Work 2010, 36, 207–216. [Google Scholar]
- Gilworth, G.; Eyres, S.; Carey, A.; Bhakta, B.B.; Tennant, A. Working with a brain injury: Personal experiences of returning to work following a mild or moderate brain injury. J. Rehabil. Med. 2008, 40, 334–339. [Google Scholar] [CrossRef]
- Shigaki, C.L.; Johnstone, B.; Schopp, L.H. Financial and vocational outcomes 2 years after traumatic brain injury. Disabil. Rehabil. 2009, 31, 484–489. [Google Scholar] [CrossRef]
- Lewin, J.; Sumners, D. Anorexia due to brain injury. Brain Inj. 1992, 6, 199–201. [Google Scholar] [CrossRef]
- Bullock, M.R.; Lyeth, B.G.; Muizelaar, J.P. Current status of neuroprotection trials for traumatic brain injury: Lessons from animal models and clinical studies. Neurosurgery 1999, 45, 207–217; discussion 217–220. [Google Scholar]
- Petersen, O.H.; Michalak, M.; Verkhratsky, A. Calcium signalling: Past, present and future. Cell Calcium 2005, 38, 161–169. [Google Scholar] [CrossRef]
- Carafoli, E. Intracellular calcium homeostasis. Annu. Rev. Biochem. 1987, 56, 395–433. [Google Scholar] [CrossRef]
- Miller, R.J. The control of neuronal Ca2+ homeostasis. Prog. Neurobiol. 1991, 37, 255–285. [Google Scholar] [CrossRef]
- Racay, P.; Kaplan, P.; Lehotsky, J. Control of Ca2+ homeostasis in neuronal cells. Gen. Physiol. Biophys. 1996, 15, 193–210. [Google Scholar]
- Racay, P.; Lehotsky, J. Intracellular and molecular aspects of Ca(2+)-mediated signal transduction in neuronal cells. Gen. Physiol. Biophys. 1996, 15, 273–289. [Google Scholar]
- Weber, J.T. Calcium homeostasis following traumatic neuronal injury. Curr. Neurovasc. Res. 2004, 1, 151–171. [Google Scholar] [CrossRef]
- Schanne, F.A.; Kane, A.B.; Young, E.E.; Farber, J.L. Calcium dependence of toxic cell death: A final common pathway. Science 1979, 206, 700–702. [Google Scholar]
- Berridge, M.J.; Irvine, R.F. Inositol phosphates and cell signalling. Nature 1989, 341, 197–205. [Google Scholar] [CrossRef]
- Carafoli, E.; Longoni, S. The plasma membrane in the control of the signaling function of calcium. Soc. Gen. Physiol. Ser. 1987, 42, 21–29. [Google Scholar]
- Campbell, K.P.; Knudson, C.M.; Imagawa, T.; Leung, A.T.; Sutko, J.L.; Kahl, S.D.; Raab, C.R.; Madson, L. Identification and characterization of the high affinity [3h]ryanodine receptor of the junctional sarcoplasmic reticulum Ca2+ release channel. J. Biol. Chem. 1987, 262, 6460–6463. [Google Scholar]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001; Chapter xviii; pp. 814, 818. [Google Scholar]
- Catterall, W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995, 64, 493–531. [Google Scholar] [CrossRef]
- Dunlap, K.; Luebke, J.I.; Turner, T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995, 18, 89–98. [Google Scholar] [CrossRef]
- Miljanich, G.P.; Ramachandran, J. Antagonists of neuronal calcium channels: Structure, function, and therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 707–734. [Google Scholar] [CrossRef]
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M.E. Calcium channel diversity and neurotransmitter release: The omega-conotoxins and omega-agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867. [Google Scholar] [CrossRef]
- Tsien, R.W.; Nilius, B. Cardiac calcium currents at the level of single channels. Experientia 1987, 43, 1169–1172. [Google Scholar] [CrossRef]
- Cain, S.M.; Snutch, T.P. Voltage-gated calcium channels and disease. BioFactors 2011, 37, 197–205. [Google Scholar] [CrossRef]
- Dolphin, A.C. Calcium channel auxiliary alpha2delta and beta subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 2012, 13, 542–555. [Google Scholar] [CrossRef]
- Kim, H.L.; Kim, H.; Lee, P.; King, R.G.; Chin, H. Rat brain expresses an alternatively spliced form of the dihydropyridine-sensitive L-type calcium channel alpha 2 subunit. Proc. Natl. Acad. Sci. USA 1992, 89, 3251–3255. [Google Scholar] [CrossRef]
- Perez-Reyes, E.; Wei, X.Y.; Castellano, A.; Birnbaumer, L. Molecular diversity of L-type calcium channels. Evidence for alternative splicing of the transcripts of three non-allelic genes. J. Biol. Chem. 1990, 265, 20430–20436. [Google Scholar]
- Allen, S.E.; Darnell, R.B.; Lipscombe, D. The neuronal splicing factor nova controls alternative splicing in N-type and P-type Cav2 calcium channels. Channels 2010, 4, 483–489. [Google Scholar]
- Williams, M.E.; Marubio, L.M.; Deal, C.R.; Hans, M.; Brust, P.F.; Philipson, L.H.; Miller, R.J.; Johnson, E.C.; Harpold, M.M.; Ellis, S.B. Structure and functional characterization of neuronal alpha 1e calcium channel subtypes. J. Biol. Chem. 1994, 269, 22347–22357. [Google Scholar]
- Senatore, A.; Spafford, J.D. Gene transcription and splicing of T-type channels are evolutionarily-conserved strategies for regulating channel expression and gating. PLoS One 2012, 7, e37409. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Liao, P.; Wang, J.J.; Yu de, J.; Soong, T.W. Alternative splicing modulates diltiazem sensitivity of cardiac and vascular smooth muscle Ca(v)1.2 calcium channels. Br. J. Pharmacol. 2010, 160, 1631–1640. [Google Scholar] [CrossRef]
- Zuccotti, A.; Clementi, S.; Reinbothe, T.; Torrente, A.; Vandael, D.H.; Pirone, A. Structural and functional differences between l-type calcium channels: Crucial issues for future selective targeting. Trends Pharmacol. Sci. 2011, 32, 366–375. [Google Scholar] [CrossRef]
- Helton, T.D.; Kojetin, D.J.; Cavanagh, J.; Horne, W.A. Alternative splicing of a beta4 subunit proline-rich motif regulates voltage-dependent gating and toxin block of Cav2.1 Ca2+ channels. J. Neurosci. 2002, 22, 9331–9339. [Google Scholar]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harbor Perspectives in Biology 2011, 3, a003947. [Google Scholar] [CrossRef]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef]
- Sochivko, D.; Pereverzev, A.; Smyth, N.; Gissel, C.; Schneider, T.; Beck, H. The Ca(v)2.3 Ca(2+) channel subunit contributes to R-type Ca(2+) currents in murine hippocampal and neocortical neurones. J. Physiol. 2002, 542, 699–710. [Google Scholar] [CrossRef]
- Sharman, J.L.; Benson, H.E.; Pawson, A.J.; Lukito, V.; Mpamhanga, C.P.; Bombail, V.; Davenport, A.P.; Peters, J.A.; Spedding, M.; Harmar, A.J.; et al. IUPHAR-DB: Updated database content and new features. Nucl. Acids Res. 2013, 41, 1083–1088. [Google Scholar] [CrossRef]
- Pringos, E.; Vignes, M.; Martinez, J.; Rolland, V. Peptide neurotoxins that affect voltage-gated calcium channels: A close-up on omega-agatoxins. Toxins 2011, 3, 17–42. [Google Scholar] [CrossRef]
- Bowman, D.; Alexander, S.; Lodge, D. Pharmacological characterisation of the calcium channels coupled to the plateau phase of kcl-induced intracellular free Ca2+ elevation in chicken and rat synaptosomes. Neuropharmacology 1993, 32, 1195–1202. [Google Scholar] [CrossRef]
- Stephens, G.J.; Page, K.M.; Burley, J.R.; Berrow, N.S.; Dolphin, A.C. Functional expression of rat brain cloned alpha1e calcium channels in cos-7 cells. Pflugers Arch. 1997, 433, 523–532. [Google Scholar] [CrossRef]
- McDonough, S.I.; Swartz, K.J.; Mintz, I.M.; Boland, L.M.; Bean, B.P. Inhibition of calcium channels in rat central and peripheral neurons by omega-conotoxin mviic. J. Neurosci. 1996, 16, 2612–2623. [Google Scholar]
- Ertel, E.A.; Warren, V.A.; Adams, M.E.; Griffin, P.R.; Cohen, C.J.; Smith, M.M. Type iii omega-agatoxins: A family of probes for similar binding sites on L- and N-type calcium channels. Biochemistry 1994, 33, 5098–5108. [Google Scholar] [CrossRef]
- Arroyo, G.; Aldea, M.; Fuentealba, J.; Albillos, A.; Garcia, A.G. Snx482 selectively blocks P/Q Ca2+ channels and delays the inactivation of Na+ channels of chromaffin cells. Eur. J. Pharmacol. 2003, 475, 11–18. [Google Scholar] [CrossRef]
- Lu, Z.J.; Pereverzev, A.; Liu, H.L.; Weiergraber, M.; Henry, M.; Krieger, A.; Smyth, N.; Hescheler, J.; Schneider, T. Arrhythmia in isolated prenatal hearts after ablation of the Cav2.3 (alpha1e) subunit of voltage-gated Ca2+ channels. Cell. Phys. Biochem. 2004, 14, 11–22. [Google Scholar]
- Xu, W.; Lipscombe, D. Neuronal Ca(v)1.3alpha(1) l-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 2001, 21, 5944–5951. [Google Scholar]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union Of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharm. Rev. 2005, 57, 411–425. [Google Scholar]
- Stengel, W.; Jainz, M.; Andreas, K. Different potencies of dihydropyridine derivatives in blocking T-type but not L-type Ca2+ channels in neuroblastoma-glioma hybrid cells. Eur. J. Pharmacol. 1998, 342, 339–345. [Google Scholar] [CrossRef]
- Furukawa, T.; Nukada, T.; Namiki, Y.; Miyashita, Y.; Hatsuno, K.; Ueno, Y.; Yamakawa, T.; Isshiki, T. Five different profiles of dihydropyridines in blocking T-type Ca(2+) channel subtypes (Ca(v)3.1 (alpha(1g)), Ca(v)3.2 (alpha(1h)), and Ca(v)3.3 (alpha(1i))) expressed in Xenopus oocytes. Eur. J. Pharmacol. 2009, 613, 100–107. [Google Scholar]
- Horiba, M.; Muto, T.; Ueda, N.; Opthof, T.; Miwa, K.; Hojo, M.; Lee, J.K.; Kamiya, K.; Kodama, I.; Yasui, K. T-type Ca2+ channel blockers prevent cardiac cell hypertrophy through an inhibition of calcineurin-nfat3 activation as well as L-type Ca2+ channel blockers. Life Sci. 2008, 82, 554–560. [Google Scholar] [CrossRef]
- Masumiya, H.; Kase, J.; Tanaka, Y.; Tanaka, H.; Shigenobu, K. Frequency-dependent blockade of T-type Ca2+ current by efonidipine in cardiomyocytes. Life Sci. 2000, 68, 345–351. [Google Scholar] [CrossRef]
- Kwan, Y.W.; Bangalore, R.; Lakitsh, M.; Glossmann, H.; Kass, R.S. Inhibition of cardiac L-type calcium channels by quaternary amlodipine: Implications for pharmacokinetics and access to dihydropyridine binding site. J. Mol. Cell Cardiol. 1995, 27, 253–262. [Google Scholar] [CrossRef]
- Furukawa, T.; Yamakawa, T.; Midera, T.; Sagawa, T.; Mori, Y.; Nukada, T. Selectivities of dihydropyridine derivatives in blocking Ca(2+) channel subtypes expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 1999, 291, 464–473. [Google Scholar]
- Freeze, B.S.; McNulty, M.M.; Hanck, D.A. State-dependent verapamil block of the cloned human Ca(v)3.1 T-type Ca(2+) channel. Mol. Pharmacol. 2006, 70, 718–726. [Google Scholar]
- Kuga, T.; Sadoshima, J.; Tomoike, H.; Kanaide, H.; Akaike, N.; Nakamura, M. Actions of Ca2+ antagonists on two types of Ca2+ channels in rat aorta smooth muscle cells in primary culture. Circ. Res. 1990, 67, 469–480. [Google Scholar] [CrossRef]
- Xi, Q.; Angus, J.A. Evidence against an action of mibefradil at N-type voltage-operated calcium channels. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 364, 430–436. [Google Scholar] [CrossRef]
- Aczel, S.; Kurka, B.; Hering, S. Mechanism of voltage- and use-dependent block of class a Ca2+ channels by mibefradil. Br. J. Pharmacol. 1998, 125, 447–454. [Google Scholar] [CrossRef]
- Perez-Reyes, E.; Lory, P. Molecular biology of T-type calcium channels. CNS Neurol. Disord. Drug Targets 2006, 5, 605–609. [Google Scholar] [CrossRef]
- Perez-Reyes, E.; Van Deusen, A.L.; Vitko, I. Molecular pharmacology of human Cav3.2 T-type Ca2+ channels: Block by antihypertensives, antiarrhythmics, and their analogs. J. Pharmacol. Exp. Ther. 2009, 328, 621–627. [Google Scholar]
- McDonough, S.I.; Bean, B.P. Mibefradil inhibition of T-type calcium channels in cerebellar purkinje neurons. Mol. Pharmacol. 1998, 54, 1080–1087. [Google Scholar]
- Cain, S.M.; Snutch, T.P. T-type calcium channels in burst-firing, network synchrony, and epilepsy. Biochim. Biophys. Acta 2013, 1828, 1572–1578. [Google Scholar] [CrossRef]
- Takemura, M.; Kiyama, H.; Fukui, H.; Tohyama, M.; Wada, H. Autoradiographic visualization in rat brain of receptors for omega-conotoxin gvia, a newly discovered calcium antagonist. Brain Res. 1988, 451, 386–389. [Google Scholar] [CrossRef]
- Takemura, M.; Kiyama, H.; Fukui, H.; Tohyama, M.; Wada, H. Distribution of the omega-conotoxin receptor in rat brain. An autoradiographic mapping. Neuroscience 1989, 32, 405–416. [Google Scholar] [CrossRef]
- Timmermann, D.B.; Westenbroek, R.E.; Schousboe, A.; Catterall, W.A. Distribution of high-voltage-activated calcium channels in cultured gamma-aminobutyric acidergic neurons from mouse cerebral cortex. J. Neurosci. Res. 2002, 67, 48–61. [Google Scholar] [CrossRef]
- Westenbroek, R.E.; Sakurai, T.; Elliott, E.M.; Hell, J.W.; Starr, T.V.; Snutch, T.P.; Catterall, W.A. Immunochemical identification and subcellular distribution of the alpha 1a subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418. [Google Scholar]
- Nakanishi, S.; Fujii, A.; Kimura, T.; Sakakibara, S.; Mikoshiba, K. Spatial distribution of omega-agatoxin IVA binding sites in mouse brain slices. J. Neurosci. Res. 1995, 41, 532–539. [Google Scholar] [CrossRef]
- McCleskey, E.W.; Fox, A.P.; Feldman, D.H.; Cruz, L.J.; Olivera, B.M.; Tsien, R.W.; Yoshikami, D. Omega-conotoxin: Direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 4327–4331. [Google Scholar] [CrossRef]
- Rhee, J.S.; Ishibashi, H.; Akaike, N. Calcium channels in the gabaergic presynaptic nerve terminals projecting to meynert neurons of the rat. J. Neurochem. 1999, 72, 800–807. [Google Scholar] [CrossRef]
- Zaman, T.; Lee, K.; Park, C.; Paydar, A.; Choi, J.H.; Cheong, E.; Lee, C.J.; Shin, H.S. Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron 2011, 70, 95–108. [Google Scholar] [CrossRef]
- Muller, R.; Struck, H.; Ho, M.S.; Brockhaus-Dumke, A.; Klosterkotter, J.; Broich, K.; Hescheler, J.; Schneider, T.; Weiergraber, M. Atropine-sensitive hippocampal theta oscillations are mediated by Cav2.3 R-type Ca(2+) channels. Neuroscience 2012, 205, 125–139. [Google Scholar]
- Weiergraber, M.; Kamp, M.A.; Radhakrishnan, K.; Hescheler, J.; Schneider, T. The Ca(v)2.3 voltage-gated calcium channel in epileptogenesis—Shedding new light on an enigmatic channel. Neurosci. Biobehav. Rev. 2006, 30, 1122–1144. [Google Scholar] [CrossRef]
- Klugbauer, N.; Lacinova, L.; Marais, E.; Hobom, M.; Hofmann, F. Molecular diversity of the calcium channel alpha2delta subunit. J. Neurosci. 1999, 19, 684–691. [Google Scholar]
- Rettig, J.; Sheng, Z.H.; Kim, D.K.; Hodson, C.D.; Snutch, T.P.; Catterall, W.A. Isoform-specific interaction of the alpha1a subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and snap-25. Proc. Natl. Acad. Sci. USA 1996, 93, 7363–7368. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Yokoyama, C.T.; Catterall, W.A. Interaction of the synprint site of N-type Ca2+channels with the c2b domain of synaptotagmin I. Proc. Natl. Acad. Sci. USA 1997, 94, 5405–5410. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Steinhauser, C. Ion channels in glial cells. Brain Res. Brain Res. Rev. 2000, 32, 380–412. [Google Scholar] [CrossRef]
- Moreno Davila, H. Molecular and functional diversity of voltage-gated calcium channels. Ann. N. Y. Acad. Sci. 1999, 868, 102–117. [Google Scholar] [CrossRef]
- Masumiya, H.; Shijuku, T.; Tanaka, H.; Shigenobu, K. Inhibition of myocardial L- and T-type Ca2+ currents by efonidipine: Possible mechanism for its chronotropic effect. Eur. J. Pharmacol. 1998, 349, 351–357. [Google Scholar] [CrossRef]
- Galetin, T.; Tevoufouet, E.E.; Sandmeyer, J.; Matthes, J.; Nguemo, F.; Hescheler, J.; Weiergraber, M.; Schneider, T. Pharmacoresistant Ca(v) 2.3 (E-type/R-type) voltage-gated calcium channels influence heart rate dynamics and may contribute to cardiac impulse conduction. Cell Biochem. Funct. 2012. [Google Scholar] [CrossRef]
- Olivera, B.M.; Gray, W.R.; Zeikus, R.; McIntosh, J.M.; Varga, J.; Rivier, J.; de Santos, V.; Cruz, L.J. Peptide neurotoxins from fish-hunting cone snails. Science 1985, 230, 1338–1343. [Google Scholar]
- Adams, D.J.; Berecki, G. Mechanisms of conotoxin inhibition of N-type (Ca2.2) calcium channels. Biochim. Biophys. Acta 2013, in press. [Google Scholar]
- D'Ascenzo, M.; Vairano, M.; Andreassi, C.; Navarra, P.; Azzena, G.B.; Grassi, C. Electrophysiological and molecular evidence of L-(Cav1), N- (Cav2.2), and R- (Cav2.3) type Ca2+ channels in rat cortical astrocytes. Glia 2004, 45, 354–363. [Google Scholar]
- Newcomb, R.; Szoke, B.; Palma, A.; Wang, G.; Chen, X.; Hopkins, W.; Cong, R.; Miller, J.; Urge, L.; Tarczy-Hornoch, K.; et al. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula hysterocrates gigas. Biochemistry 1998, 37, 15353–15362. [Google Scholar] [CrossRef]
- Wang, G.; Dayanithi, G.; Newcomb, R.; Lemos, J.R. An R-type Ca(2+) current in neurohypophysial terminals preferentially regulates oxytocin secretion. J. Neurosci. 1999, 19, 9235–9241. [Google Scholar]
- Furukawa, T.; Nukada, T.; Miura, R.; Ooga, K.; Honda, M.; Watanabe, S.; Koganesawa, S.; Isshiki, T. Differential blocking action of dihydropyridine Ca2+ antagonists on a T-type Ca2+ channel (alpha1g) expressed in Xenopus oocytes. J. Cardiovasc. Pharmacol. 2005, 45, 241–246. [Google Scholar] [CrossRef]
- Clozel, J.P.; Ertel, E.A.; Ertel, S.I. Discovery and main pharmacological properties of mibefradil (RO 40-5967), the first selective T-type calcium channel blocker. J. Hypertens. Suppl. 1997, 15, S17–S25. [Google Scholar] [CrossRef]
- Bernal, J.; Lee, J.H.; Cribbs, L.L.; Perez-Reyes, E. Full reversal of Pb++ block of L-type Ca++ channels requires treatment with heavy metal antidotes. J. Pharmacol. Exp. Ther. 1997, 282, 172–180. [Google Scholar]
- Lee, J.H.; Gomora, J.C.; Cribbs, L.L.; Perez-Reyes, E. Nickel block of three cloned T-type calcium channels: Low concentrations selectively block alpha1h. Biophys. J. 1999, 77, 3034–3042. [Google Scholar] [CrossRef]
- Nelson, M.T.; Woo, J.; Kang, H.W.; Vitko, I.; Barrett, P.Q.; Perez-Reyes, E.; Lee, J.H.; Shin, H.S.; Todorovic, S.M. Reducing agents sensitize c-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J. Neurosci. 2007, 27, 8250–8260. [Google Scholar] [CrossRef]
- Shcheglovitov, A.; Vitko, I.; Lazarenko, R.M.; Orestes, P.; Todorovic, S.M.; Perez-Reyes, E. Molecular and biophysical basis of glutamate and trace metal modulation of voltage-gated Ca(v)2.3 calcium channels. J. Gen. Physiol. 2012, 139, 219–234. [Google Scholar] [CrossRef]
- Siesjo, B.K. Calcium and cell death. Magnesium 1989, 8, 223–237. [Google Scholar]
- Siesjo, B.K.; Agardh, C.D.; Bengtsson, F. Free radicals and brain damage. Cerebrovasc. Brain Metab. Rev. 1989, 1, 165–211. [Google Scholar]
- Siesjo, B.K.; Bengtsson, F.; Grampp, W.; Theander, S. Calcium, excitotoxins, and neuronal death in the brain. Ann. N. Y. Acad. Sci. 1989, 568, 234–251. [Google Scholar] [CrossRef]
- Young, W. Role of calcium in central nervous system injuries. J Neurotrauma 1992, 9 (Suppl 1), S9–S25. [Google Scholar]
- Nilsson, P.; Laursen, H.; Hillered, L.; Hansen, A.J. Calcium movements in traumatic brain injury: The role of glutamate receptor-operated ion channels. J Cereb. Blood. Flow. Metab. 1996, 16, 262–270. [Google Scholar]
- Cater, H.L.; Gitterman, D.; Davis, S.M.; Benham, C.D.; Morrison, B., 3rd; Sundstrom, L.E. Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: Role of glutamate receptors and voltage-dependent calcium channels. J. Neurochem. 2007, 101, 434–447. [Google Scholar]
- Floyd, C.L.; Gorin, F.A.; Lyeth, B.G. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia 2005, 51, 35–46. [Google Scholar] [CrossRef]
- Floyd, C.L.; Rzigalinski, B.A.; Weber, J.T.; Sitterding, H.A.; Willoughby, K.A.; Ellis, E.F. Traumatic injury of cultured astrocytes alters inositol (1,4,5)-trisphosphate-mediated signaling. Glia 2001, 33, 12–23. [Google Scholar] [CrossRef]
- LaPlaca, M.C.; Thibault, L.E. An in vitro traumatic injury model to examine the response of neurons to a hydrodynamically-induced deformation. Ann. Biomed. Eng. 1997, 25, 665–677. [Google Scholar] [CrossRef]
- Rzigalinski, B.A.; Weber, J.T.; Willoughby, K.A.; Ellis, E.F. Intracellular free calcium dynamics in stretch-injured astrocytes. J. Neurochem. 1998, 70, 2377–2385. [Google Scholar]
- Shahlaie, K.; Lyeth, B.G.; Gurkoff, G.G.; Muizelaar, J.P.; Berman, R.F. Neuroprotective effects of selective N-type VGCC blockade on stretch-injury-induced calcium dynamics in cortical neurons. J. Neurotrauma 2010, 27, 175–187. [Google Scholar] [CrossRef]
- Staal, J.A.; Dickson, T.C.; Gasperini, R.; Liu, Y.; Foa, L.; Vickers, J.C. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 2010, 112, 1147–1155. [Google Scholar] [CrossRef]
- Weber, J.T.; Rzigalinski, B.A.; Ellis, E.F. Traumatic injury of cortical neurons causes changes in intracellular calcium stores and capacitative calcium influx. J. Biol. Chem. 2001, 276, 1800–1807. [Google Scholar] [CrossRef]
- Weber, J.T.; Rzigalinski, B.A.; Willoughby, K.A.; Moore, S.F.; Ellis, E.F. Alterations in calcium-mediated signal transduction after traumatic injury of cortical neurons. Cell Calcium 1999, 26, 289–299. [Google Scholar] [CrossRef]
- Wolf, J.A.; Stys, P.K.; Lusardi, T.; Meaney, D.; Smith, D.H. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 2001, 21, 1923–1930. [Google Scholar]
- Young, W.; Koreh, I. Potassium and calcium changes in injured spinal cords. Brain Res. 1986, 365, 42–53. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Weber, J.T.; Liang, S.; Willoughby, K.A.; Sitterding, H.A.; Rzigalinski, B.A.; Ellis, E.F. Nmda receptor activation contributes to a portion of the decreased mitochondrial membrane potential and elevated intracellular free calcium in strain-injured neurons. J. Neurotrauma 2002, 19, 1619–1629. [Google Scholar] [CrossRef]
- Geddes, D.M.; LaPlaca, M.C.; Cargill, R.S., 2nd. Susceptibility of hippocampal neurons to mechanically induced injury. Exp. Neurol. 2003, 184, 420–427. [Google Scholar] [CrossRef]
- Geddes-Klein, D.M.; Serbest, G.; Mesfin, M.N.; Cohen, A.S.; Meaney, D.F. Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J. Neurochem. 2006, 97, 462–474. [Google Scholar] [CrossRef]
- LaPlaca, M.C.; Thibault, L.E. Dynamic mechanical deformation of neurons triggers an acute calcium response and cell injury involving the n-methyl-d-aspartate glutamate receptor. J. Neurosci. Res. 1998, 52, 220–229. [Google Scholar] [CrossRef]
- Shahlaie, K.; Gurkoff, G.G.; Lyeth, B.G.; Muizelaar, J.P.; Berman, R.F. Neuroprotective effects of SNX-185 in an in vitro model of TBI with a second insult. Restor. Neurol. Neurosci. 2013, 31, 141–153. [Google Scholar]
- Chen, T.; Willoughby, K.A.; Ellis, E.F. Group I metabotropic receptor antagonism blocks depletion of calcium stores and reduces potentiated capacitative calcium entry in strain-injured neurons and astrocytes. J. Neurotrauma 2004, 21, 271–281. [Google Scholar] [CrossRef]
- Buki, A.; Okonkwo, D.O.; Wang, K.K.; Povlishock, J.T. Cytochrome C release and caspase activation in traumatic axonal injury. J. Neurosci. 2000, 20, 2825–2834. [Google Scholar]
- Conti, A.C.; Raghupathi, R.; Trojanowski, J.Q.; McIntosh, T.K. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J. Neurosci. 1998, 18, 5663–5672. [Google Scholar]
- Eldadah, B.A.; Faden, A.I. Caspase pathways, neuronal apoptosis, and CNS injury. J. Neurotrauma 2000, 17, 811–829. [Google Scholar] [CrossRef]
- Keane, R.W.; Kraydieh, S.; Lotocki, G.; Alonso, O.F.; Aldana, P.; Dietrich, W.D. Apoptotic and antiapoptotic mechanisms after traumatic brain injury. J. Cereb. Blood. Flow. Metab. 2001, 21, 1189–1198. [Google Scholar]
- Newcomb, J.K.; Zhao, X.; Pike, B.R.; Hayes, R.L. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp. Neurol. 1999, 158, 76–88. [Google Scholar] [CrossRef]
- Raghupathi, R.; Graham, D.I.; McIntosh, T.K. Apoptosis after traumatic brain injury. J. Neurotrauma 2000, 17, 927–938. [Google Scholar] [CrossRef]
- Robertson, C.L.; Puskar, A.; Hoffman, G.E.; Murphy, A.Z.; Saraswati, M.; Fiskum, G. Physiologic progesterone reduces mitochondrial dysfunction and hippocampal cell loss after traumatic brain injury in female rats. Exp. Neurol. 2006, 197, 235–243. [Google Scholar] [CrossRef]
- Robertson, C.L.; Saraswati, M.; Fiskum, G. Mitochondrial dysfunction early after traumatic brain injury in immature rats. J. Neurochem. 2007, 101, 1248–1257. [Google Scholar] [CrossRef]
- Singh, I.N.; Sullivan, P.G.; Deng, Y.; Mbye, L.H.; Hall, E.D. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: Implications for neuroprotective therapy. J. Cereb. Blood. Flow. Metab. 2006, 26, 1407–1418. [Google Scholar] [CrossRef]
- White, R.J.; Reynolds, I.J. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J. Physiol. 1997, 498 (Pt 1), 31–47. [Google Scholar]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 1997, 14, 23–34. [Google Scholar] [CrossRef]
- Mustafa, A.G.; Singh, I.N.; Wang, J.; Carrico, K.M.; Hall, E.D. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J. Neurochem 2010, 114, 271–280. [Google Scholar]
- Braughler, J.M.; Duncan, L.A.; Goodman, T. Calcium enhances in vitro free radical-induced damage to brain synaptosomes, mitochondria, and cultured spinal cord neurons. J. Neurochem 1985, 45, 1288–1293. [Google Scholar] [CrossRef]
- Braughler, J.M.; Hall, E.D. Involvement of lipid peroxidation in CNS injury. J. Neurotrauma 1992, 9 (Suppl 1), S1–S7. [Google Scholar]
- Odland, R.M.; Sutton, R.L. Hyperosmosis of cerebral injury. Neurological Res. 1999, 21, 500–508. [Google Scholar]
- Tavalin, S.J.; Ellis, E.F.; Satin, L.S. Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J. Neurophysiol. 1995, 74, 2767–2773. [Google Scholar]
- Geddes-Klein, D.M.; Schiffman, K.B.; Meaney, D.F. Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. J. Neurotrauma 2006, 23, 193–204. [Google Scholar] [CrossRef]
- Lusardi, T.A.; Wolf, J.A.; Putt, M.E.; Smith, D.H.; Meaney, D.F. Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J. Neurotrauma 2004, 21, 61–72. [Google Scholar] [CrossRef]
- Rzigalinski, B.A.; Liang, S.; McKinney, J.S.; Willoughby, K.A.; Ellis, E.F. Effect of Ca2+ on in vitro astrocyte injury. J. Neurochem. 1997, 68, 289–296. [Google Scholar]
- Goforth, P.B.; Ellis, E.F.; Satin, L.S. Mechanical injury modulates AMPA receptor kinetics via an NMDA receptor-dependent pathway. J. Neurotrauma 2004, 21, 719–732. [Google Scholar] [CrossRef]
- Goforth, P.B.; Ellis, E.F.; Satin, L.S. Enhancement of AMPA-mediated current after traumatic injury in cortical neurons. J. Neurosci 1999, 19, 7367–7374. [Google Scholar]
- Goforth, P.B.; Ren, J.; Schwartz, B.S.; Satin, L.S. Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. J. Neurophys. 2011, 105, 2350–2363. [Google Scholar] [CrossRef]
- Spaethling, J.; Le, L.; Meaney, D.F. NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiol. Dis. 2012, 46, 646–654. [Google Scholar] [CrossRef]
- Spaethling, J.M.; Klein, D.M.; Singh, P.; Meaney, D.F. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 2008, 25, 1207–1216. [Google Scholar] [CrossRef]
- Gurkoff, G.G.; Shahlaie, K.; Lyeth, B.G. In vitro mechanical strain trauma alters neuronal calcium responses: Implications for posttraumatic epilepsy. Epilepsia 2012, 53 (Suppl 1), 53–60. [Google Scholar]
- DeWitt, D.S.; Jenkins, L.W.; Prough, D.S. Enhanced vulnerability to secondary ischemic insults after experimental traumatic brain injury. New. Horiz. 1995, 3, 376–383. [Google Scholar]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 1998, 89, 507–518. [Google Scholar] [CrossRef]
- Katayama, Y.; Kawamata, T.; Tamura, T.; Hovda, D.A.; Becker, D.P.; Tsubokawa, T. Calcium-dependent glutamate release concomitant with massive potassium flux during cerebral ischemia in vivo. Brain Res. 1991, 558, 136–140. [Google Scholar] [CrossRef]
- Kochanek, P.M. Ischemic and traumatic brain injury: Pathobiology and cellular mechanisms. Crit. Care Med. 1993, 21, S333–S335. [Google Scholar] [CrossRef]
- Engel, D.C.; Slemmer, J.E.; Vlug, A.S.; Maas, A.I.; Weber, J.T. Combined effects of mechanical and ischemic injury to cortical cells: Secondary ischemia increases damage and decreases effects of neuroprotective agents. Neuropharmacology 2005, 49, 985–995. [Google Scholar]
- Stokes, B.T.; Fox, P.; Hollinden, G. Extracellular calcium activity in the injured spinal cord. Exp. Neurol. 1983, 80, 561–572. [Google Scholar] [CrossRef]
- Nilsson, P.; Hillered, L.; Olsson, Y.; Sheardown, M.J.; Hansen, A.J. Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J. Cereb. Blood Flow Metab. 1993, 13, 183–192. [Google Scholar] [CrossRef]
- Dienel, G.A. Regional accumulation of calcium in postischemic rat brain. J. Neurochem. 1984, 43, 913–925. [Google Scholar] [CrossRef]
- Fineman, I.; Hovda, D.A.; Smith, M.; Yoshino, A.; Becker, D.P. Concussive brain injury is associated with a prolonged accumulation of calcium: A 45Ca autoradiographic study. Brain Res. 1993, 624, 94–102. [Google Scholar] [CrossRef]
- Osteen, C.L.; Moore, A.H.; Prins, M.L.; Hovda, D.A. Age-dependency of 45calcium accumulation following lateral fluid percussion: Acute and delayed patterns. J. Neurotrauma 2001, 18, 141–162. [Google Scholar] [CrossRef]
- Deshpande, L.S.; Sun, D.A.; Sombati, S.; Baranova, A.; Wilson, M.S.; Attkisson, E.; Hamm, R.J.; DeLorenzo, R.J. Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci. Lett. 2008, 441, 115–119. [Google Scholar] [CrossRef]
- Sun, D.A.; Deshpande, L.S.; Sombati, S.; Baranova, A.; Wilson, M.S.; Hamm, R.J.; DeLorenzo, R.J. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur. J. Neurosci. 2008, 27, 1659–1672. [Google Scholar] [CrossRef]
- Hell, J.W.; Westenbroek, R.E.; Warner, C.; Ahlijanian, M.K.; Prystay, W.; Gilbert, M.M.; Snutch, T.P.; Catterall, W.A. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell. Biol. 1993, 123, 949–962. [Google Scholar] [CrossRef]
- Hoffmeister, F.; Benz, U.; Heise, A.; Krause, H.P.; Neuser, V. Behavioral effects of nimodipine in animals. Arzneimittel Forschung 1982, 32, 347–360. [Google Scholar]
- Lazarewicz, J.W.; Pluta, R.; Puka, M.; Salinska, E. Diverse mechanisms of neuronal protection by nimodipine in experimental rabbit brain ischemia. Stroke 1990, 21, IV108–110. [Google Scholar]
- Symon, L.; Harris, R.J.; Branston, N.M. Calcium ions and calcium antagonists in ischaemia. Acta Neurochir. (Wien) 1982, 63, 267–275. [Google Scholar]
- Babu, C.S.; Ramanathan, M. Post-ischemic administration of nimodipine following focal cerebral ischemic-reperfusion injury in rats alleviated excitotoxicity, neurobehavioural alterations and partially the bioenergetics. Int. J. Dev. Neurosci. 2011, 29, 93–105. [Google Scholar] [CrossRef]
- Steen, P.A.; Gisvold, S.E.; Milde, J.H.; Newberg, L.A.; Scheithauer, B.W.; Lanier, W.L.; Michenfelder, J.D. Nimodipine improves outcome when given after complete cerebral ischemia in primates. Anesthesiology 1985, 62, 406–414. [Google Scholar] [CrossRef]
- Gaab, M.R.; Haubitz, I.; Brawanski, A.; Korn, A.; Czech, T. Acute effects of nimodipine on the cerebral blood flow and intracranial pressure. Neurochirurgia 1985, 28 (Suppl 1), 93–99. [Google Scholar]
- Gelmers, H.J. The effects of nimodipine on the clinical course of patients with acute ischemic stroke. Acta Neurol. Scand. 1984, 69, 232–239. [Google Scholar] [CrossRef]
- Paci, A.; Ottaviano, P.; Trenta, A.; Iannone, G.; de Santis, L.; Lancia, G.; Moschini, E.; Carosi, M.; Amigoni, S.; Caresia, L. Nimodipine in acute ischemic stroke: A double-blind controlled study. Acta Neurol. Scand. 1989, 80, 282–286. [Google Scholar] [CrossRef]
- Krieglstein, J.; Lippert, K.; Poch, G. Apparent independent action of nimodipine and glutamate antagonists to protect cultured neurons against glutamate-induced damage. Neuropharmacology 1996, 35, 1737–1742. [Google Scholar] [CrossRef]
- Maeda, T.; Lee, S.M.; Hovda, D.A. Restoration of cerebral vasoreactivity by an l-type calcium channel blocker following fluid percussion brain injury. J. Neurotrauma 2005, 22, 763–771. [Google Scholar] [CrossRef]
- Ercan, M.; Inci, S.; Kilinc, K.; Palaoglu, S.; Aypar, U. Nimodipine attenuates lipid peroxidation during the acute phase of head trauma in rats. Neurosurg. Rev. 2001, 24, 127–130. [Google Scholar] [CrossRef]
- Bailey, I.; Bell, A.; Gray, J.; Gullan, R.; Heiskanan, O.; Marks, P.V.; Marsh, H.; Mendelow, D.A.; Murray, G.; Ohman, J.; et al. A trial of the effect of nimodipine on outcome after head injury. Acta Neurochir. (Wien.) 1991, 110, 97–105. [Google Scholar]
- Teasdale, G.; Bailey, I.; Bell, A.; Gray, J.; Gullan, R.; Heiskanan, O.; Marks, P.V.; Marsh, H.; Mendelow, D.A.; Murray, G.; et al. A randomized trial of nimodipine in severe head injury: Hit I. British/finnish co-operative head injury trial group. J. Neurotrauma 1992, 9, S545–S550. [Google Scholar]
- Group, E.S. A multicenter trial of the efficacy of nimodipine on outcome after severe head injury. The european study group on nimodipine in severe head injury. J. Neurosurg. 1994, 80, 797–804. [Google Scholar] [CrossRef]
- Murray, G.D.; Teasdale, G.M.; Schmitz, H. Nimodipine in traumatic subarachnoid haemorrhage: A re-analysis of the hit I and hit II trials. Acta Neurochir. (Wien.) 1996, 138, 1163–1167. [Google Scholar]
- Vergouwen, M.D.; Vermeulen, M.; Roos, Y.B. Effect of nimodipine on outcome in patients with traumatic subarachnoid haemorrhage: A systematic review. Lancet Neurol. 2006, 5, 1029–1032. [Google Scholar] [CrossRef]
- Aslan, A.; Gurelik, M.; Cemek, M.; Buyukokuroglu, M.; Goksel, H.M.; Eser, O. Nimodipine can diminish oxidative stress in patients with severe head trauma. J. Neurosurg. Sci. 2012, 56, 247–253. [Google Scholar]
- Aslan, A.; Gurelik, M.; Cemek, M.; Goksel, H.M.; Buyukokuroglu, M.E. Nimodipine can improve cerebral metabolism and outcome in patients with severe head trauma. Pharm. Res. 2009, 59, 120–124. [Google Scholar] [CrossRef]
- Langham, J.; Goldfrad, C.; Teasdale, G.; Shaw, D.; Rowan, K. Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst. Rev. 2003, CD000565. [Google Scholar]
- Williams, M.E.; Brust, P.F.; Feldman, D.H.; Patthi, S.; Simerson, S.; Maroufi, A.; McCue, A.F.; Velicelebi, G.; Ellis, S.B.; Harpold, M.M. Structure and functional expression of an omega-conotoxin-sensitive human N-type calcium channel. Science 1992, 257, 389–395. [Google Scholar]
- Dubel, S.J.; Starr, T.V.; Hell, J.; Ahlijanian, M.K.; Enyeart, J.J.; Catterall, W.A.; Snutch, T.P. Molecular cloning of the alpha-1 subunit of an omega-conotoxin-sensitive calcium channel. Proc. Natl. Acad. Sci. USA 1992, 89, 5058–5062. [Google Scholar]
- Volsen, S.G.; Day, N.C.; McCormack, A.L.; Smith, W.; Craig, P.J.; Beattie, R.; Ince, P.G.; Shaw, P.J.; Ellis, S.B.; Gillespie, A.; et al. The expression of neuronal voltage-dependent calcium channels in human cerebellum. Brain Res. Mol. Brain Res. 1995, 34, 271–282. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Wagner, J.A.; Snyder, S.H.; Thayer, S.A.; Olivera, B.M.; Miller, R.J. Brain voltage-sensitive calcium channel subtypes differentiated by omega-conotoxin fraction gvia. Proc. Natl. Acad. Sci. USA 1986, 83, 8804–8807. [Google Scholar] [CrossRef]
- Hirning, L.D.; Fox, A.P.; McCleskey, E.W.; Olivera, B.M.; Thayer, S.A.; Miller, R.J.; Tsien, R.W. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science 1988, 239, 57–61. [Google Scholar]
- Bowersox, S.; Mandema, J.; Tarczy-Hornoch, K.; Miljanich, G.; Luther, R.R. Pharmacokinetics of SNX-111, a selective N-type calcium channel blocker, in rats and cynomolgus monkeys. Drug Metab. Dispos. 1997, 25, 379–383. [Google Scholar]
- Bowersox, S.S.; Gadbois, T.; Singh, T.; Pettus, M.; Wang, Y.X.; Luther, R.R. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J. Pharmacol. Exp. Ther. 1996, 279, 1243–1249. [Google Scholar]
- Bowersox, S.S.; Luther, R. Pharmacotherapeutic potential of omega-conotoxin MVIIA (SNX-111), an N-type neuronal calcium channel blocker found in the venom of conus magus. Toxicon 1998, 36, 1651–1658. [Google Scholar]
- Berman, R.F.; Verweij, B.H.; Muizelaar, J.P. Neurobehavioral protection by the neuronal calcium channel blocker ziconotide in a model of traumatic diffuse brain injury in rats. J. Neurosurg. 2000, 93, 821–828. [Google Scholar] [CrossRef]
- Colbourne, F.; Li, H.; Buchan, A.M.; Clemens, J.A. Continuing postischemic neuronal death in CA1: Influence of ischemia duration and cytoprotective doses of NBQX and SNX-111 in rats. Stroke 1999, 30, 662–668. [Google Scholar] [CrossRef]
- Perez-Pinzon, M.A.; Yenari, M.A.; Sun, G.H.; Kunis, D.M.; Steinberg, G.K. SNX-111, a novel, presynaptic N-type calcium channel antagonist, is neuroprotective against focal cerebral ischemia in rabbits. J. Neurol. Sci. 1997, 153, 25–31. [Google Scholar] [CrossRef]
- Valentino, K.; Newcomb, R.; Gadbois, T.; Singh, T.; Bowersox, S.; Bitner, S.; Justice, A.; Yamashiro, D.; Hoffman, B.B.; Ciaranello, R.; et al. A selective N-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. Proc. Natl. Acad. Sci. USA 1993, 90, 7894–7897. [Google Scholar] [CrossRef]
- Hovda, D.A.; Fu, K.; Badie, H.; Samii, A.; Pinanong, P.; Becker, D.P. Administration of an omega-conopeptide one hour following traumatic brain injury reduces 45calcium accumulation. Acta Neurochir. Suppl. (Wien.) 1994, 60, 521–523. [Google Scholar]
- Samii, A.; Badie, H.; Fu, K.; Luther, R.R.; Hovda, D.A. Effects of an N-type calcium channel antagonist (SNX-111; ziconotide) on calcium-45 accumulation following fluid-percussion injury. J. Neurotrauma 1999, 16, 879–892. [Google Scholar] [CrossRef]
- Verweij, B.H.; Muizelaar, J.P.; Vinas, F.C.; Peterson, P.L.; Xiong, Y.; Lee, C.P. Improvement in mitochondrial dysfunction as a new surrogate efficiency measure for preclinical trials: Dose-response and time-window profiles for administration of the calcium channel blocker ziconotide in experimental brain injury. J. Neurosurg. 2000, 93, 829–834. [Google Scholar]
- Verweij, B.H.; Muizelaar, J.P.; Vinas, F.C.; Peterson, P.L.; Xiong, Y.; Lee, C.P. Mitochondrial dysfunction after experimental and human brain injury and its possible reversal with a selective N-type calcium channel antagonist (SNX-111). Neurol. Res. 1997, 19, 334–339. [Google Scholar]
- Xiong, Y.; Peterson, P.L.; Verweij, B.H.; Vinas, F.C.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction after experimental traumatic brain injury: Combined efficacy of SNX-111 and U-101033E. J. Neurotrauma 1998, 15, 531–544. [Google Scholar] [CrossRef]
- Lee, L.L.; Galo, E.; Lyeth, B.G.; Muizelaar, J.P.; Berman, R.F. Neuroprotection in the rat lateral fluid percussion model of traumatic brain injury by SNX-185, an N-type voltage-gated calcium channel blocker. Exp. Neurol. 2004, 190, 70–78. [Google Scholar]
- Hinzman, J.M.; Thomas, T.C.; Quintero, J.E.; Gerhardt, G.A.; Lifshitz, J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. J. Neurotrauma 2012, 29, 1197–1208. [Google Scholar] [CrossRef]
- Pruneau, D.; Angus, J.A. Omega-conotoxin GVIA, the N-type calcium channel inhibitor, is sympatholytic but not vagolytic: Consequences for hemodynamics and autonomic reflexes in conscious rabbits. J. Cardiovasc. Pharmacol. 1990, 16, 675–680. [Google Scholar] [CrossRef]
- Wright, C.E.; Angus, J.A. Prolonged cardiovascular effects of the N-type Ca2+ channel antagonist omega-conotoxin GVIA in conscious rabbits. J. Cardiovasc. Pharmacol. 1997, 30, 392–399. [Google Scholar] [CrossRef]
- Malmberg, A.B.; Yaksh, T.L. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995, 60, 83–90. [Google Scholar] [CrossRef]
- Penn, R.D.; Paice, J.A. Adverse effects associated with the intrathecal administration of ziconotide. Pain 2000, 85, 291–296. [Google Scholar] [CrossRef]
- Atanassoff, P.G.; Hartmannsgruber, M.W.; Thrasher, J.; Wermeling, D.; Longton, W.; Gaeta, R.; Singh, T.; Mayo, M.; McGuire, D.; Luther, R.R. Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative pain. Reg. Anesth. Pain Med. 2000, 25, 274–278. [Google Scholar]
- Staats, P.S.; Yearwood, T.; Charapata, S.G.; Presley, R.W.; Wallace, M.S.; Byas-Smith, M.; Fisher, R.; Bryce, D.A.; Mangieri, E.A.; Luther, R.R.; et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or aids: A randomized controlled trial. JAMA 2004, 291, 63–70. [Google Scholar] [CrossRef]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Smith, H.S.; Deer, T.R. Safety and efficacy of intrathecal ziconotide in the management of severe chronic pain. Ther. Clin. Risk Manag. 2009, 5, 521–534. [Google Scholar] [CrossRef]
- Webster, L.R.; Fisher, R.; Charapata, S.; Wallace, M.S. Long-term intrathecal ziconotide for chronic pain: An open-label study. J. Pain Symptom Manage. 2009, 37, 363–372. [Google Scholar] [CrossRef]
- Wheaton, P.; Mathias, J.L.; Vink, R. Impact of pharmacological treatments on outcome in adult rodents after traumatic brain injury: A meta-analysis. J. Psychopharm. 2011, 25, 1581–1599. [Google Scholar] [CrossRef]
- Ishiguro, M.; Wellman, T.L.; Honda, A.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Emergence of a R-type Ca2+ channel (Cav 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ. Res. 2005, 96, 419–426. [Google Scholar]
- Wang, F.; Yin, Y.H.; Jia, F.; Jiang, J.Y. Antagonism of r-type calcium channels significantly improves cerebral blood flow after subarachnoid hemorrhage in rats. J. Neurotrauma 2010, 27, 1723–1732. [Google Scholar] [CrossRef]
- Hansen, P.B. Functional and pharmacological consequences of the distribution of voltage-gated calcium channels in the renal blood vessels. Acta Physiol. 2013, 207, 690–699. [Google Scholar] [CrossRef]
- Hansen, P.B.; Jensen, B.L.; Andreasen, D.; Friis, U.G.; Skott, O. Vascular smooth muscle cells express the alpha(1a) subunit of a P-/Q-type voltage-dependent Ca(2+)channel, and it is functionally important in renal afferent arterioles. Circ. Res. 2000, 87, 896–902. [Google Scholar] [CrossRef]
- Hansen, P.B.; Poulsen, C.B.; Walter, S.; Marcussen, N.; Cribbs, L.L.; Skott, O.; Jensen, B.L. Functional importance of L- and P/Q-type voltage-gated calcium channels in human renal vasculature. Hypertension 2011, 58, 464–470. [Google Scholar] [CrossRef]
- Canova, D.; Roatta, S.; Micieli, G.; Bosone, D. Cerebral oxygenation and haemodynamic effects induced by nimodipine in healthy subjects. Funct. Neurol. 2012, 27, 169–176. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gurkoff, G.; Shahlaie, K.; Lyeth, B.; Berman, R. Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury. Pharmaceuticals 2013, 6, 788-812. https://doi.org/10.3390/ph6070788
Gurkoff G, Shahlaie K, Lyeth B, Berman R. Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury. Pharmaceuticals. 2013; 6(7):788-812. https://doi.org/10.3390/ph6070788
Chicago/Turabian StyleGurkoff, Gene, Kiarash Shahlaie, Bruce Lyeth, and Robert Berman. 2013. "Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury" Pharmaceuticals 6, no. 7: 788-812. https://doi.org/10.3390/ph6070788
APA StyleGurkoff, G., Shahlaie, K., Lyeth, B., & Berman, R. (2013). Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury. Pharmaceuticals, 6(7), 788-812. https://doi.org/10.3390/ph6070788