Strategies To Modulate Heritable Epigenetic Defects in Cellular Machinery: Lessons from Nature

Abstract
:1. Introduction
2. Epigenetic Inheritance and Heritable Disorders
2.1. A Brief History of Epigenetic Inheritance and Their Influence on Cellular Homeostasis in Animal Models
2.2. Nature versus Nurture
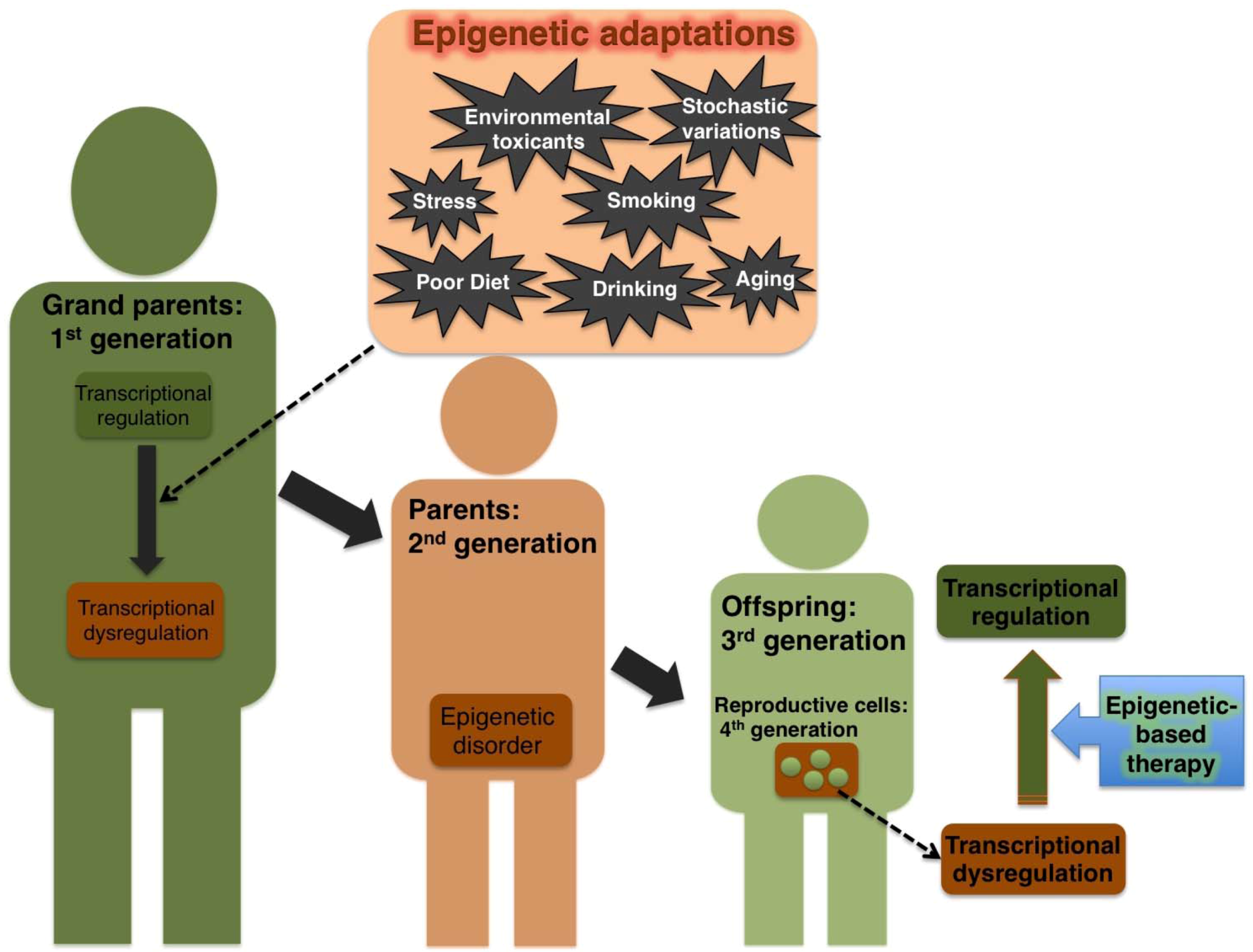
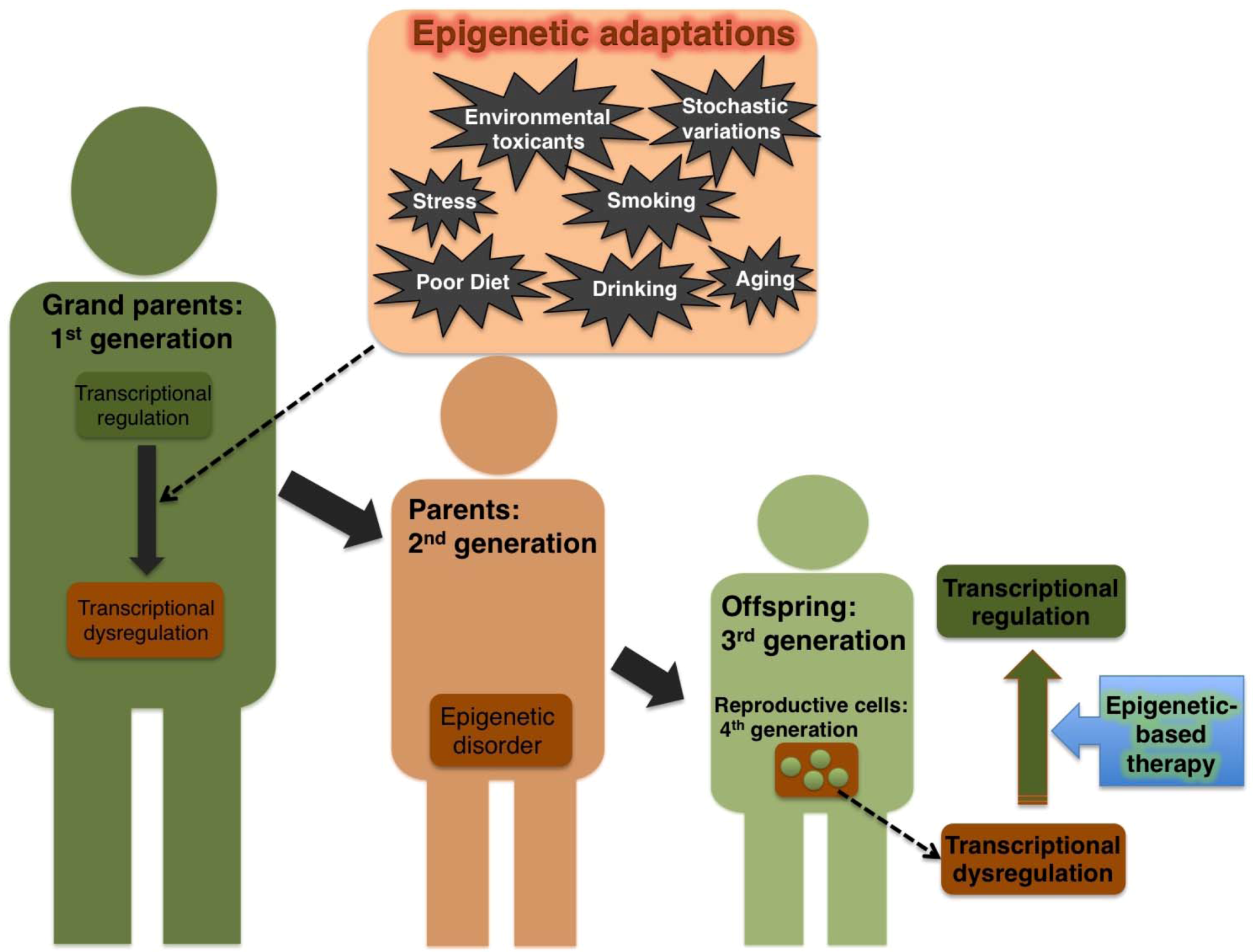
2.3. Effect of Environmental Cues on Transgenerational Epigenetic Damage in Humans
3. Epigenetic-Switch Based Future Medicine
3.1. Co-Ordinated Histone Modifications
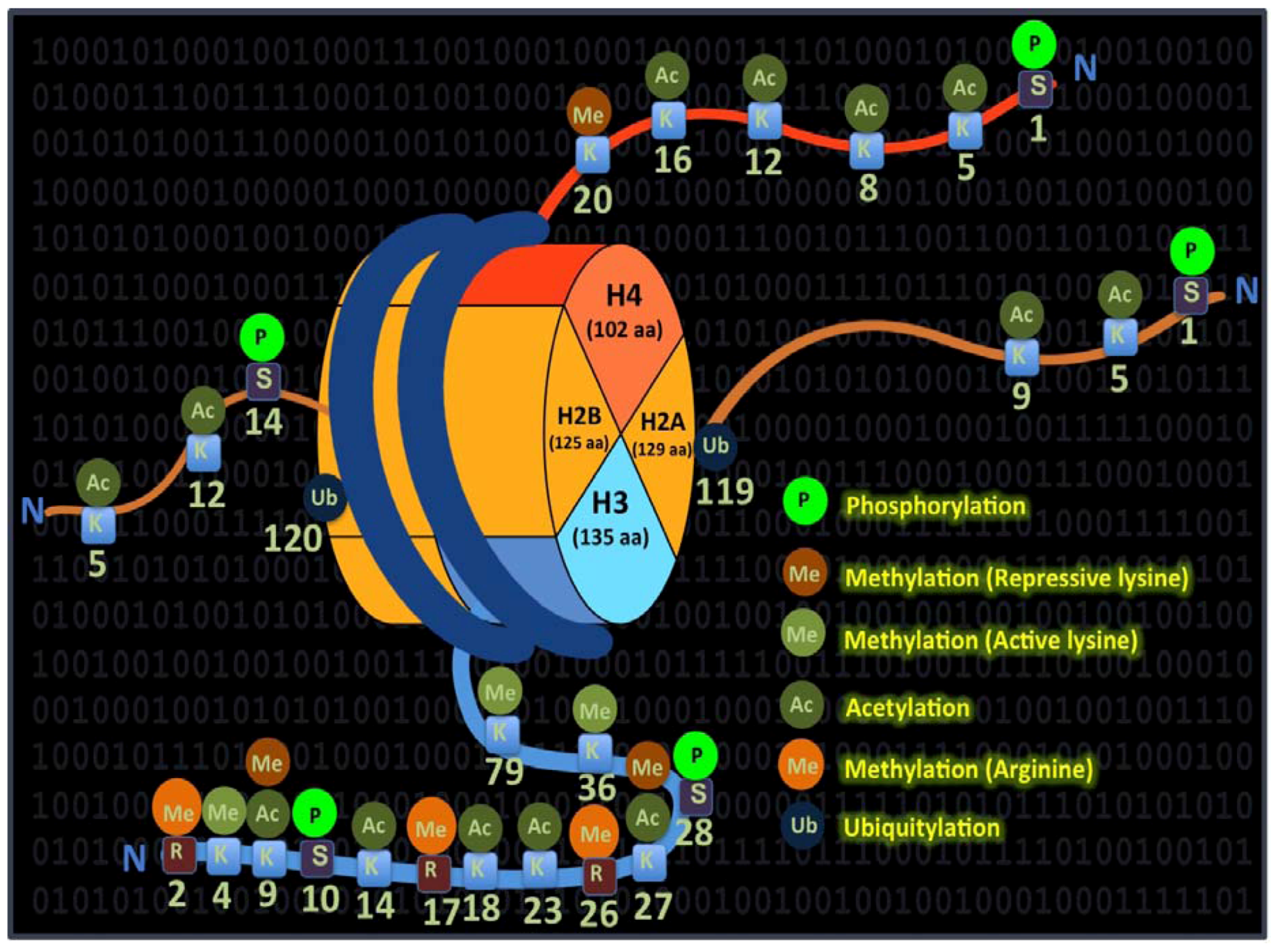
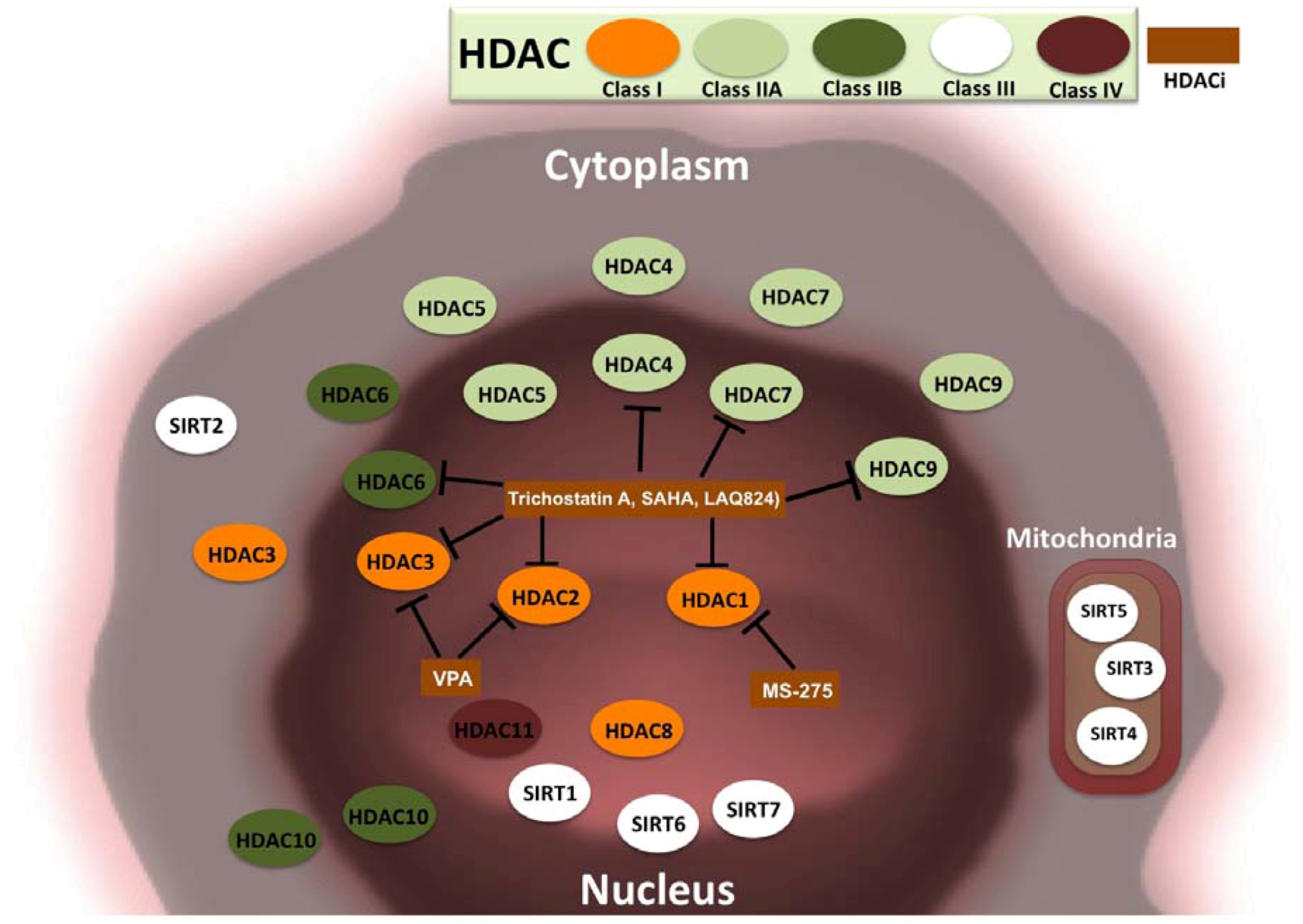
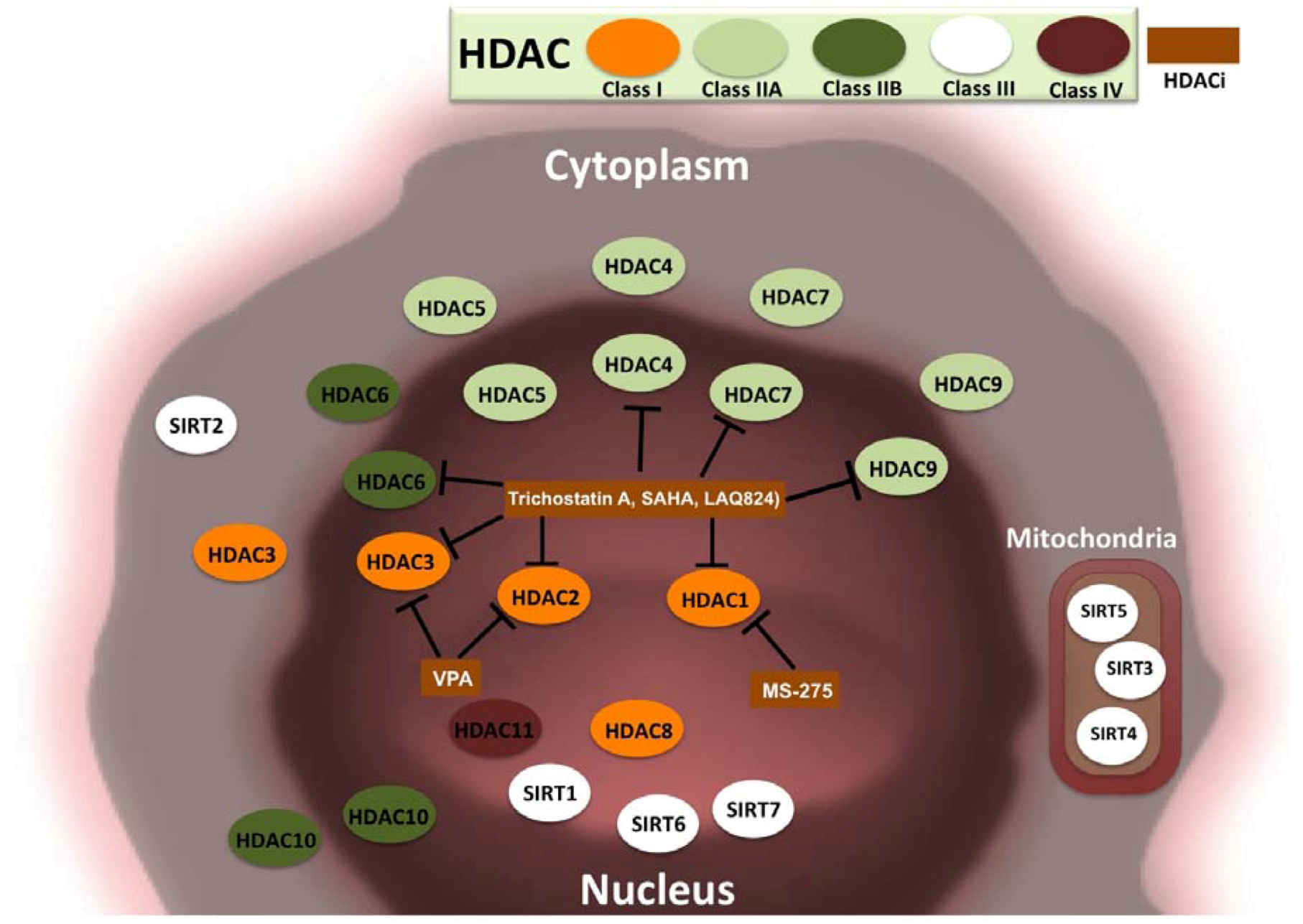
3.2. HDACs and Cancer
3.3. HDAC Inhibitors and Cancer Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors and compound type | Cancer type | Clinical limitations | References |
|---|---|---|---|
| Short chain fatty acids | |||
| Sodium butyrate | Leukemia, myeloma and breast cancer | Combination therapy is toxic | [62] |
| Sodium Phenyl butyrate | Leukemias and myelodysplasia | High doses causes neurological toxicity | [63] |
| Sodium valproate | Leukemias, myelo dysplasia and cervical cancer | Neurological toxicity | [64] |
| Hydroxamic acids | |||
| Suberoylanilide hydroxamic acid | Leukemia, lymphoma and solid tumors | Dose limiting toxicity, dehydration, fatigue, diarrhoea and anorexia. | [65] |
| NVP-LAQ824 | Leukemia, lymphoma and solid tumors | Toxicity in bone marrow and cardiac cells. Fever, fatigue and nausea | [66] |
| PXD101 | Advanced solid tumors | Tiredness, fatigue and low-grade nausea | [67] |
| Others | |||
| MS-275 | Leukemia | Toxicity, nausea and vomiting | [68] |
3.4. Epigenetic Switches in Cellular Reprograming
| Inhibitors | HDAC activity | Uses | Comments | References |
|---|---|---|---|---|
| Aliphatic chain fatty acids | ||||
| Sodium butyrate (NaB) | Inhibit most HDACs except class III and HDACs 6 and 10 of class II | Cellular reprogramming, differentiation and self renewal | Dramatically enhances reprogramming efficiency in both mouse and human embryonic stem cells | [76,77] |
| Valproic acid (VPA) | Both Class I and II with higher potency against HDACs 2 and 3 | Cellular reprogramming, differentiation and self renewal | Self renewal of embryonic carcinoma cells, enhance reprogramming efficiency with fewer factors and induce neuronal differentiation | [75,78] |
| Hydroxamic acids | ||||
| Trichostatin A | Higher potency against HDACs 1,2,3,4, 6,7 and 9 and active against HDAC8. | Cellular reprogramming, differentiation and self renewal | Self renewal of mouse embryonic stem cells, embryonic carcinoma cells and neurosphere cells | [79,80,81] |
| Suberoylanilide hydroxamic acid | Higher potency against HDACs 1,2,3,4, 6,7 and 9 and active against HDAC8. | Cellular reprogramming and differentiation | Enhances reprogramming efficiency and Induces neuronal differentiation | [82] |
| Scriptaid | Class I and II with higher potency against HDACs 1, 3, 6 and 8. | Cellular reprogramming | Enhances reprogramming efficiency | [81] |
| Oxamflatin | Class I and II with higher potency against HDACs 1, 3, 6, 7 and 8. | Cellular reprogramming | Enhances somatic nucleus reprogramming | [80] |
| M344 | Class I and II with higher potency against HDACs 1,2 and 3 | Cellular differentiation | Induces neuronal differentiation. | [82] |
| M-Carboxycinnamic acid bishydroxamide (CBHA) | Class I and II with higher potency against HDACs 1 and 3 | Cellular reprogramming | Enhances reprogramming efficiency | [84] |
| Others | ||||
| Apicidin | Higher potency against HDACs 2 and 3 | Cellular reprogramming | Enhances reprogramming efficiency | [75] |
| Chlamydocin | Higher potency against Class I | Self renewal | Self renewal of hematopoietic stem cells | [83] |
| MS-275 | Higher potency against class I especially HDAC1 | Cellular reprogramming and differentiation | Induces neural differentiation | [75,82] |
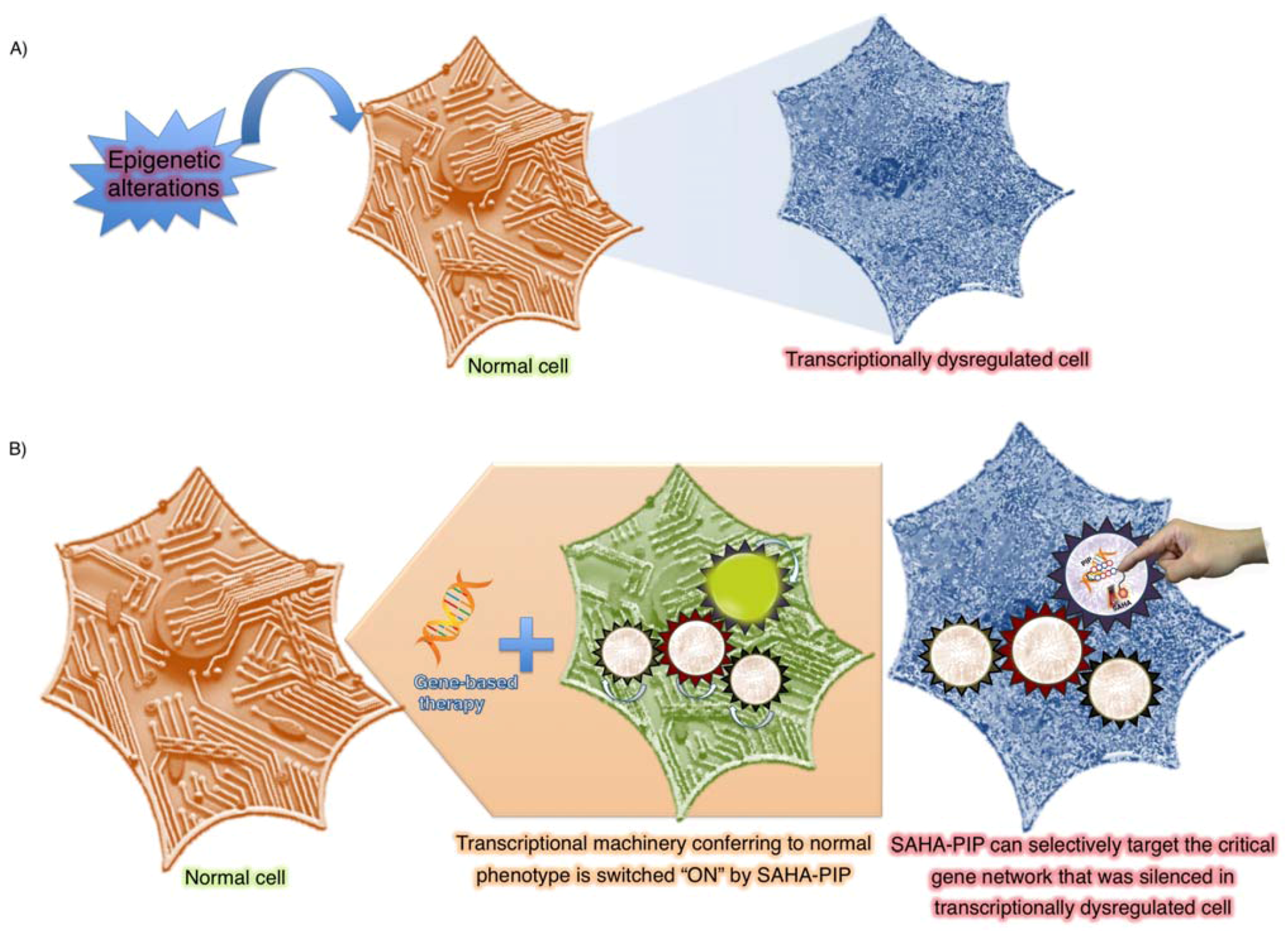
4. Tailor-Made Epigenetic-Based Switches
4.1. Need for Selectivity
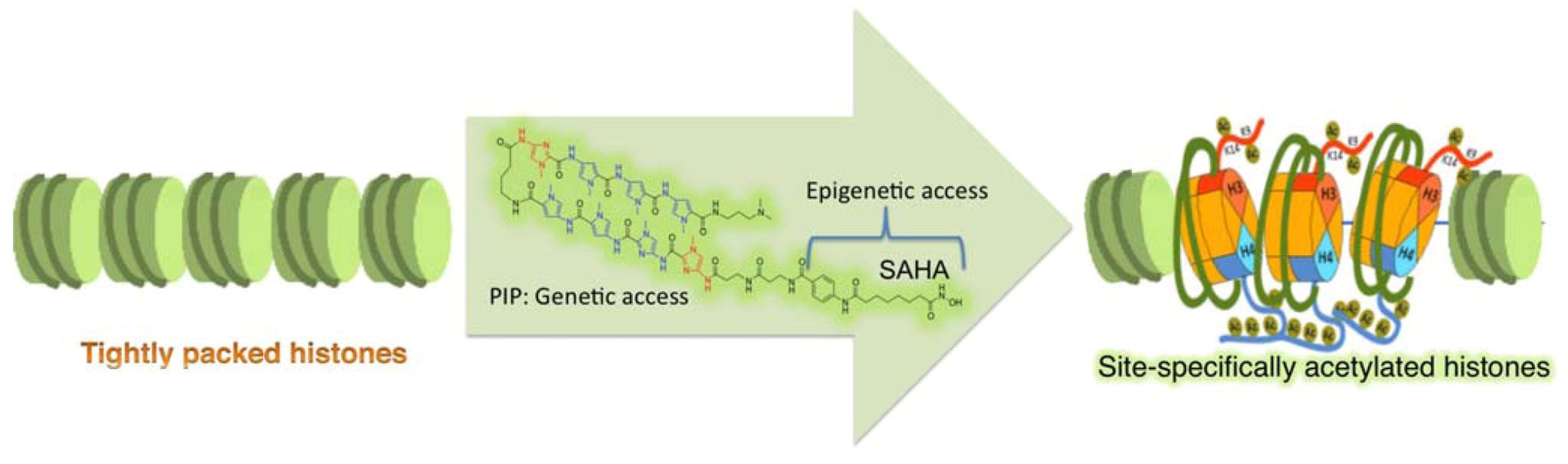
4.2. Programmable Genetic Switches To Reset Epigenome
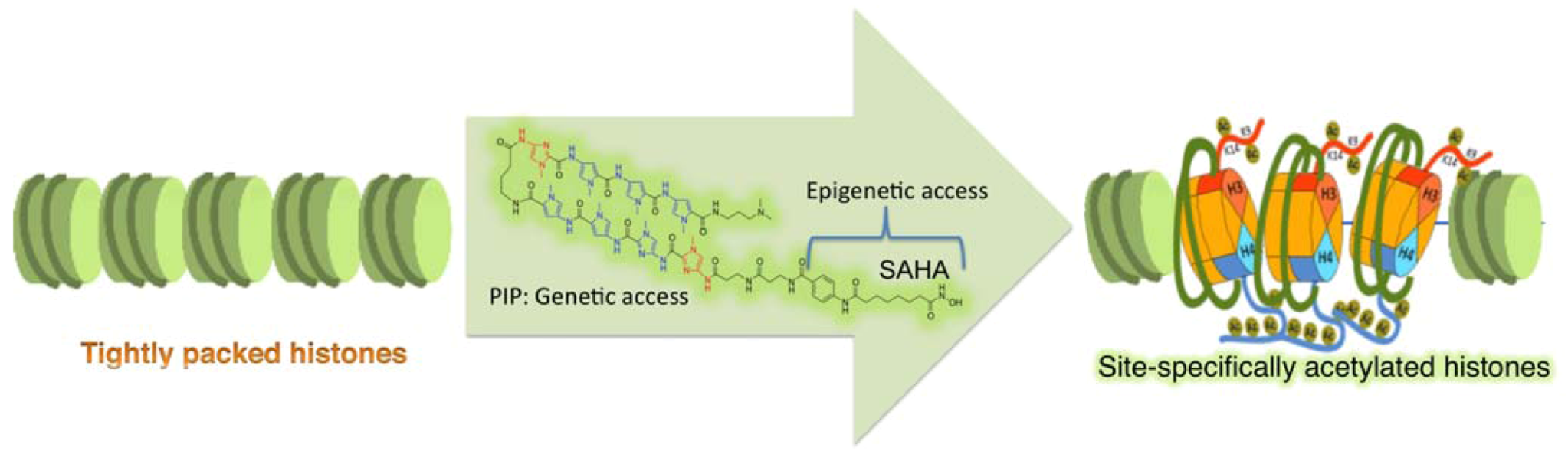
5. Future Strategies To Tinker with Epigenetic Inheritance
5.1. Heritable Histone Modifications as Effective Therapeutic Targets
5.2. Dynamic Histone Modifications – Can It Be Therapeutically Targeted?
5.3. Prospects of Tailor-Made Epigenetic Switches
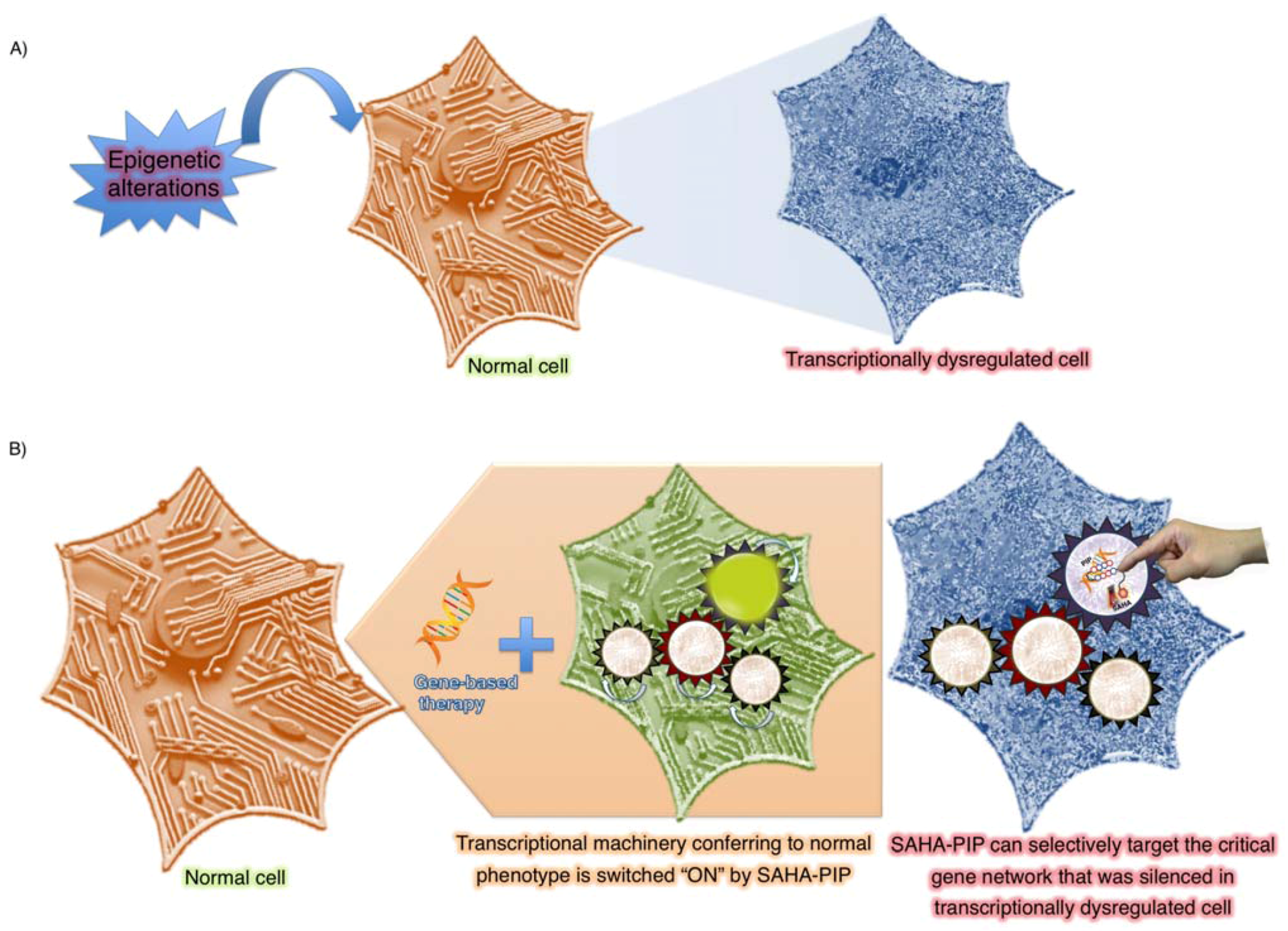
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Gordon, J.E. The New Science of Strong Materials, or Why You Don’t Fall through the Floor, 2nd ed; Pelican-Penguin: London, UK, 1976; pp. 17–33. [Google Scholar]
- Sapp, J. Genesis: the Evolution of Biology; Oxford University Press: Oxford, U.K, 2003; pp. 5–10. [Google Scholar]
- Lamb, M.J.; Jablonka, E. Evolution in Four Dimensions: Genetic, Epigenetic, Behavioral, and Symbolic Variation in the History of Life; MIT Press: Cambridge, MA, USA, 2005; pp. 5–34. [Google Scholar]
- Bhushan, B. Biomimetics-lessons from nature: an overview. Phil. Trans. R. Soc. A. 2009, 367, 1445–1486. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar]
- Frances, M.B.; Mukund, T.; Satyajit, M. Evolutionary cell biology: Lessons from diversity. Nat. Cell Biol. 2012, 14, 651. [Google Scholar] [CrossRef]
- Muneoka, K.; Allan, C.H.; Yang, X.; Lee, J.; Han, M. Mammalian regeneration and regenerative medicine. Birth Defects Research. Part C. Embryo Today 2008, 84, 265–280. [Google Scholar]
- Riggs, A.D.; Russo, V.E.A.; Martienssen, R.A. Epigenetic mechanisms of gene regulation; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1996; pp. 231–267. [Google Scholar]
- Baltimore, D. Our genome unveiled. Nature 2001, 409, 814–816. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar]
- Stein, G.S.; Zaidi, S.K.; Stein, J.L.; Lian, J.B.; van Wijnen, A.J.; Montecino, M.; Young, D.W.; Javed, A.; Pratap, J.; Choi, J.Y.; et al. Transcription-factor-mediated epigenetic control of cell fate and lineage commitment. Biochem. Cell Biol. 2009, 87, 1–6. [Google Scholar] [CrossRef]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar]
- Jablonka, E.; Raz, G. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef]
- Garber, K. Breaking the silence: the rise of epigenetic therapy. J. Natl. Cancer Inst. 2002, 94, 874–875. [Google Scholar] [CrossRef]
- Pfeifer, A.; Verma, I.M. Gene therapy: promises and problems. Annu. Rev. Genomics Hum. Genet. 2001, 2, 177–211. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Chin, L.; Anderson, J.N.; Futreal, P.A. Cancer genomics: from discovery science to personalized medicine. Nat. Med. 2011, 17, 297–303. [Google Scholar]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar]
- Waddington, C.H. The epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar]
- Rando, O.J.; Verstrepen, K.J. Timescales of genetic and epigenetic inheritance. Cell 2007, 128, 655–668. [Google Scholar] [CrossRef]
- Jablonka, E.; Lamb, M.J. Epigenetic Inheritance and Evolution: The Lamarckian Dimension; Oxford University Press: Oxford, U.K, 1995; pp. 133–160. [Google Scholar]
- Canli, T. Genomic psychology: an emerging paradigm. EMBO Rep. 2007, 8, S30–S34. [Google Scholar] [CrossRef]
- Huxley, J. Epigenetics. Nature 1956, 177, 807. [Google Scholar]
- Hattman, S. DNA methylation of T-even bacteriophages and of their nonglucosylated mutants: Its role in P1-directed restriction. Virology 1970, 42, 359–367. [Google Scholar] [CrossRef]
- Waterland, R.A.; Jirtle, R.L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef]
- Richards, E.J. Inherited epigenetic variation-revisiting soft inheritance. Nature Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef]
- Arai, J.; Li, S.; Hartley, D.M.; Feig, L.A. Transgenerational rescue of a genetic defect in long-term potentiation and memory formation by juvenile enrichment. J. Neuroscience 2009, 29, 1496–1502. [Google Scholar] [CrossRef]
- Sollars, V.; Lu, X.; Xiao, L.; Wang, X.; Garfinkel, M.D.; Ruden, D.M. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat. Genet. 2003, 33, 70–74. [Google Scholar]
- Jablonka, E.; Lamb, M.J. Soft inheritance: challenging the modern synthesis. Genet. Mol. Biol. 2008, 31, 389–395. [Google Scholar] [CrossRef]
- Bernstein, E.; Allis, C.D. RNA meets chromatin. Genes Dev. 2005, 19, 1635–1655. [Google Scholar] [CrossRef]
- Henikoff, S.; Smith, M. Histone variants and epigenetics. Allis, D.C., Jenuwein, T., Reinberg, D., Caparros, M.L., Eds.; Epigenetics. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2007; pp. 249–264. [Google Scholar]
- Jiang, Y.H.; Bressler, J.; Beaudet, A.L. Epigenetics and human disease. Annu. Rev. Genomics Hum. Genet. 2004, 5, 479–510. [Google Scholar] [CrossRef]
- Pembrey, M.E.; Bygren, L.O.; Kaati, G.; Edvinsson, S.; Northstone, K.; Sjostrom, M.; Golding, J. Sex-specific, male-line transgenerational responses in humans. Eur. J. Hum. Genet. 2006, 14, 159–166. [Google Scholar]
- Lumey, L.H.; Stein, A.D. Offspring birth weights after maternal intrauterine undernutrition: a comparison within sibships. Am. J. Epidemiol. 1997, 146, 810–819. [Google Scholar] [CrossRef]
- Stein, A.D.; Lumey, L.H. The relationship between maternal and offspring birth weights after maternal prenatal famine exposure: the Dutch Famine Birth Cohort Study. Hum. Biol. 2000, 72, 641–654. [Google Scholar]
- Kaati, G.; Bygren, L.O.; Pembrey, M.; Sjostrom, M. Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. 2007, 15, 784–790. [Google Scholar] [CrossRef]
- Galler, J.R.; Seelig, C. Home-orienting behavior in rat pups: the effect of 2 and 3 generations of rehabilitation following intergenerational malnutrition. Dev. Psychobiol. 1981, 14, 541–548. [Google Scholar] [CrossRef]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef]
- Champagne, F.A. Epigenetic mechanisms and the transgenerational effects of maternal care. Front Neuroendocrinol. 2008, 29, 386–397. [Google Scholar] [CrossRef]
- Weaver, I.C. Epigenetic programming by maternal behavior and pharmacological intervention. Nature versus nurture: let’s call the whole thing off. Epigenetics 2007, 2, 22–28. [Google Scholar] [CrossRef]
- Lamb, M.J. Epigenetic inheritance and aging. Rev. Clin. Gerontol. 1994, 4, 97–105. [Google Scholar] [CrossRef]
- Marmorstein, R.; Trievel, R.C. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim. Biophys. Acta 2009, 1789, 58–68. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Polo, S.E.; Almouzni, G. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 2005, 220, 1–9. [Google Scholar] [CrossRef]
- Vidanes, G.M.; Bonilla, C.Y.; Toczyski, D.P. Complicated tails: histone modifications and the DNA damage response. Cell 2005, 121, 973–976. [Google Scholar] [CrossRef]
- Weinreich, M.; Palacios DeBeer, M.A.; Fox, C.A. The activities of eukaryotic replication origins in chromatin. Biochim. Biophys. Acta 2004, 1677, 142–157. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, F.; Hashimshony, T.; Keshet, I.; Cedar, H. Establishment of transcriptional competence in early and late S phase. Nature 2002, 420, 198–202. [Google Scholar]
- Santos-Rosa, H.; Caldas, C. Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer 2005, 41, 2381–2402. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar]
- Minucci, S.; Nervi, C.; Lo Coco, F.; Pelicci, P.G. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene 2001, 20, 3110–3115. [Google Scholar] [CrossRef]
- He, L.Z.; Tolentino, T.; Grayson, P.; Zhong, S.; Warrell, R.P., Jr.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; Pandolfi, P. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J. Clin. Invest. 2001, 108, 1321–1330. [Google Scholar] [Green Version]
- Bereshchenko, O.R.; Gu, W.; Dalla-Favera, R. Acetylation inactivates the transcriptional repressor BCL6. Nature Genet. 2002, 32, 606–613. [Google Scholar] [Green Version]
- Kumar, R.; Wang, R.A.; Bagheri-Yarmand, R. Emerging roles of MTA family members in human cancers. Semin. Oncol. 2003, 30, 30–37. [Google Scholar] [Green Version]
- Toh, Y.; Ohga, T.; Endo, K.; Adachi, E.; Kusumoto, H.; Haraguchi, M.; Okamura, T.; Nicolson, G.L. Expression of the metastasis-associated MTA1 protein and its relationship to deacetylation of the histone H4 in esophageal squamous cell carcinomas. Int. J. Cancer 2004, 110, 362–367. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genet. 2005, 37, 391–400. [Google Scholar]
- Florean, C.; Schnekenburger, M.; Grandjenette, C.; Dicato, M.; Diederich, M. Epigenomics of leukemia: from mechanisms to therapeutic applications. Epigenomics 2011, 3, 581–609. [Google Scholar] [CrossRef]
- Tai, K.Y.; Shiah, S.G.; Shieh, Y.S.; Kao, Y.R.; Chi, C.Y.; Huang, E.; Lee, H.S.; Chang, L.C.; Yang, P.C.; Wu, C.W. DNA methylation and histone modification regulate silencing of epithelial cell adhesion molecule for tumor invasion and progression. Oncogene 2007, 26, 3989–3997. [Google Scholar]
- Musolino, C.; Sant'antonio, E.; Penna, G.; Alonci, A.; Russo, S.; Granata, A. Epigenetic therapy in myelodysplastic syndromes. Eur. J. Haematol. 2010, 84, 463–473. [Google Scholar] [CrossRef]
- Minucci, P.G. Pelicci Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Butler, L.M.; Zhou, X.; Xu, W.S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar]
- Garcia-Manero, G.; Tambaro, F.P.; Bekele, N.B.; Yang, H.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Kadia, T.M.; Konopleva, M.Y.; Faderl, S.; et al. Phase II Trial of Vorinostat with Idarubicin and Cytarabine for patients with newly diagnosed Acute Myelogenous Leukemia or Myelodysplastic Syndrome. J. Clin. Oncol. 2012, 30, 2204–2210. [Google Scholar]
- Katherine, V.; Tom, C.K. Overview of the Classical Histone Deacetylase Enzymes and Histone Deacetylase Inhibitors. ISRN Cell Biol. 2012. Article ID 130360. [Google Scholar] [CrossRef]
- Gore, S.D. Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 963–970. [Google Scholar]
- Raffoux, E.; Chaibi, P.; Dombret, H.; Degos, L. Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia. Haematologica. 2005, 90, 986–988. [Google Scholar]
- Cuneo, K.C.; Fu, A.; Osusky, K.; Huamani, J.; Hallahan, D.E.; Geng, L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs 2007, 18, 793–800. [Google Scholar] [CrossRef]
- Qian, X.; LaRochelle, W.J.; Ara, G.; Wu, F.; Petersen, K.D.; Thougaard, A.; Sehested, M.; Lichenstein, H.S.; Jeffers, M. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol. Cancer. Ther. 2006, 8, 2086–2095. [Google Scholar]
- Ryan, Q.C. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J. Clin. Oncol. 2005, 23, 3912–3922. [Google Scholar] [CrossRef]
- Chen, L.; Kasai, T.; Li, Y.; Sugii, Y.; Jin, G.; Okada, M.; Vaidyanath, A.; Mizutani, A.; Satoh, A.; Kudoh, T.; et al. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS One 2012, 7, e33544. [Google Scholar]
- Poss, K.D. Advances in understanding tissue regenerative capacity and mechanisms in animals. Nature Rev. Genet. 2010, 11, 710–722. [Google Scholar] [CrossRef]
- Brockes, J.P.; Kumar, A. Plasticity and reprogramming of differentiated cells in amphibian regeneration. Nature Rev. Mol. Cell Biol. 2002, 3, 566–574. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2007, 126, 663–676. [Google Scholar] [CrossRef]
- Christen, B.; Robles, V.; Raya, M.; Paramonov, I.; Belmonte, J.C. Regeneration and reprogramming compared. BMC Biol. 2010, 8, 5. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Plath, K. Epigenetic reprogramming and induced pluripotency. Development 2009, 136, 509–523. [Google Scholar] [CrossRef]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef]
- Mali, P.; Chou, B.K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate Greatly Enhances Derivation of Human Induced Pluripotent Stem Cells by Promoting Epigenetic Remodeling and the Expression of Pluripotency-Associated Genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef]
- Chen, H.P.; Denicola, M.; Qin, X. HDAC inhibition promotes cardiogenesis and the survival of embryonic stem cells through proteasome-dependent pathway. J. Cellular Biochem. 2011, 112, 3246–3255. [Google Scholar] [CrossRef]
- Balasubramaniyan, V.; Boddeke, E.; Bakels, R. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience 2006, 143, 939–951. [Google Scholar] [CrossRef]
- Brons, I.G.M.; Smithers, L.E.; Trotter, M.W.B. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar]
- Schneider, J.W.; Gao, Z.; Li, S. Small-molecule activation of neuronal cell fate. Nature Chem. Biol. 2008, 4, 408–410. [Google Scholar] [CrossRef]
- Haumaitre, C.; Lenoir, O.; Scharfmann, R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol. Cell. Biol. 2008, 28, 6373–6383. [Google Scholar] [CrossRef]
- Hao, Y.; Creson, T.; Zhang, L.; Li, P.; Du, F.; Yuan, P.; Gould, T.D.; Manji, H.K.; Chen, G. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J. Neurosci. 2004, 24, 6590–6599. [Google Scholar] [CrossRef]
- Rossler, R.; Boddeke, E.; Copray, S. Differentiation of non-mesencephalic neural stem cells towards dopaminergic neurons. Neuroscience 2010, 170, 417–428. [Google Scholar] [CrossRef]
- Dai, X.; Hao, J.; Hou, X.J. Somatic nucleus reprogramming is significantly improved by m-carboxycinnamic acid bishydroxamide, a histone deacetylase inhibitor. J. Biol. Chem. 2010, 285, 31002–31010. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O`Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar]
- Pandian, G.N.; Sugiyama, H. Programmable genetic switches to control transcriptional machinery of pluripotency. Biotechnol. J. 2012, 7, 798–809. [Google Scholar]
- Cloud, J. Why your DNA isn’t your destiny. Time Magazine 2010, 61, 175. [Google Scholar]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer cell 2012, 22, 9–20. [Google Scholar] [CrossRef]
- Kashiwazaki, G.; Bando, T.; Yoshidome, T.; Masui, S.; Takagaki, T.; Hashiya, K.; Pandian, G.N.; Yasuoka, J.; Akiyoshi, K.; Sugiyama, H. Synthesis and biological properties of highly sequence-specific-alkylating N-Methylpyrrole-N-Methylimidazole polyamide conjugates. J. Med. Chem. 2012, 55, 2057–2066. [Google Scholar]
- Shinohara, K.; Bando, T.; Sasaki, S.; Sakakibara, Y.; Minoshima, M.; Sugiyama, H. Antitumor activity of sequence-specific alkylating agents: Pyrrole-imidazole CBI conjugates with indole linker. Cancer Sci. 2006, 97, 219–225. [Google Scholar] [CrossRef]
- Ohtsuki, A.; Kimura, M.T.; Minoshima, M.; Suzuki, T.; Ikeda, M.; Bando, T.; Nagase, H.; Shinohara, K.; Sugiyama, H. Synthesis and properties of PI polyamide-SAHA conjugate. Tetrahedron Lett. 2009, 50, 7288–7292. [Google Scholar] [CrossRef]
- Pandian, G.N.; Shinohara, K.; Ohtsuki, A.; Nakano, Y.; Masafumi, M.; Bando, T.; Nagase, H.; Yamada, Y.; Watanabe, A.; Terada, N.; et al. Synthetic small molecules for epigenetic activation of pluripotent genes in mouse embryonic fibroblasts. ChemBioChem 2011, 12, 2822–2828. [Google Scholar] [CrossRef]
- Pandian, G.N.; Ohtsuki, A.; Bando, T.; Sato, S.; Hashiya, K.; Sugiyama, H. Development of programmable small DNA-binding molecules with epigenetic activity for induction of core pluripotency genes. Bioorg. Med. Chem. 2012, 20, 2656–2660. [Google Scholar] [CrossRef]
- Pandian, G.N.; Nakano, Y.; Sato, S.; Morinaga, H.; Bando, T.; Nagase, H.; Sugiyama, H. A synthetic small molecule for rapid induction of multiple pluripotency genes in mouse embryonic fibroblasts. Sci. Rep. 2012, 2, e544. [Google Scholar]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef]
- Matsui, T.; Takano, M.; Yoshida, K.; Ono, S.; Fujisaki, C.; Matsuzaki, Y.; Toyama, Y.; Nakamura, M.; Okano, H.; Akamatsu, W. Neural stem cells directly differentiated from partially reprogrammed fibroblasts rapidly acquire gliogenic competency. Stem Cells 2012. [Google Scholar] [CrossRef]
- Lister, R.; Gregory, B.D.; Ecker, J.R. Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond. Curr. Opin. Plant. Biol. 2009, 12, 107–118. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Gluckman, P.D.; Hanson, M.A. Developmental origins of metabolic disease: life course and intergenerational perspectives. Trends Endocrinol. Metabol. 2010, 21, 199–205. [Google Scholar] [CrossRef]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef]
- Franklin, T.B.; Mansuy, I.M. Epigenetic inheritance in mammals: evidence for the impact of adverse environmental effects. Neurobiol. Dis. 2010, 39, 61–65. [Google Scholar] [CrossRef]
- Meaney, M.J. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci. 2001, 24, 1161–1192. [Google Scholar] [CrossRef]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonte, B.; Szyf, M. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Hamm, C.A.; Costa, F.F. The impact of epigenomics on future drug design and new therapies. Drug Discov. Today 2011, 16, 626–635. [Google Scholar] [CrossRef]
- Covington, H.E., 3rd; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J., III; Wu, E.Y.; et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar]
- Harper, L.V. Epigenetic inheritance and the intergenerational transfer of experience. Psychol. Bull. 2005, 131, 340–360. [Google Scholar] [CrossRef]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar]
- Minoshima, M.; Bando, T.; Sasaki, S.; Fujimoto, J.; Sugiyama, H. Pyrrole-Imidazole hairpin polyamides with high affinity at 5'-CGCG-3' DNA sequence; Influence of cytosine methylation on binding. Nucleic Acids Res. 2008, 36, 2889–2894. [Google Scholar] [CrossRef]
- Vaijayanthi, T.; Bando, T.; Pandian, G. N.; Sugiyama, H. Progress and prospects of pyrrole-imidazole polyamide-fluorophore conjugates as sequence-selective DNA probes. ChemBioChem 2012, 13, 2170–2185. [Google Scholar] [CrossRef]
- Petronis, A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010, 465, 721–727. [Google Scholar]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar]
- Bohacek, J.; Mansuy, I.M. Epigenetic inheritance of disease and disease risk. Neuropsychopharmacol. Rev. 2012, 1–17. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pandian, G.N.; Sugiyama, H. Strategies To Modulate Heritable Epigenetic Defects in Cellular Machinery: Lessons from Nature. Pharmaceuticals 2013, 6, 1-24. https://doi.org/10.3390/ph6010001
Pandian GN, Sugiyama H. Strategies To Modulate Heritable Epigenetic Defects in Cellular Machinery: Lessons from Nature. Pharmaceuticals. 2013; 6(1):1-24. https://doi.org/10.3390/ph6010001
Chicago/Turabian StylePandian, Ganesh N., and Hiroshi Sugiyama. 2013. "Strategies To Modulate Heritable Epigenetic Defects in Cellular Machinery: Lessons from Nature" Pharmaceuticals 6, no. 1: 1-24. https://doi.org/10.3390/ph6010001
APA StylePandian, G. N., & Sugiyama, H. (2013). Strategies To Modulate Heritable Epigenetic Defects in Cellular Machinery: Lessons from Nature. Pharmaceuticals, 6(1), 1-24. https://doi.org/10.3390/ph6010001