The Biological Role of PI3K Pathway in Lung Cancer

Abstract
:1. Introduction
2. PI3K Signaling Pathway
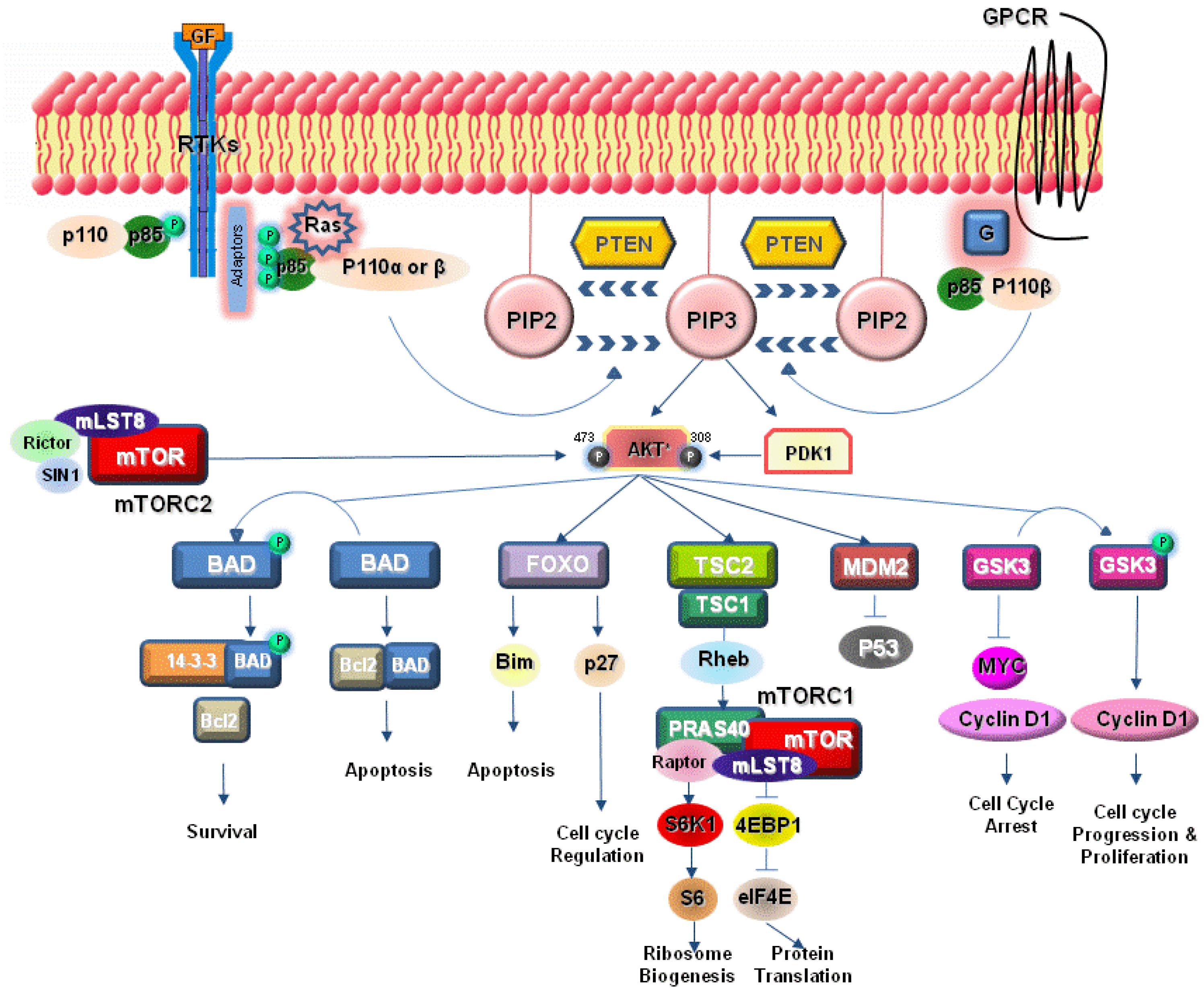
3. Activation of PI3K Pathway in Lung Cancer
4. PI3K Pathway Inhibitors in Lung Cancer
4.1. PI3K Inhibitors
4.2. Dual PI3K-mTOR Inhibitors
4.3. Akt Inhibitors
4.4. mTOR Inhibitors

{kind=link}
| Title | Phase | Protocol ID | Cancer type | Compounds | Mechanism |
|---|---|---|---|---|---|
| Study of PX-866 and Docetaxel in Solid Tumors [76] | Phase I + Phase II | NCT01204099 | Solid tumors(NSCLC) | PX-866 + Docetaxel | PI3K inhibitor |
| A Study of the Safety and Pharmacology Of PI3-Kinase Inhibitor GDC-0941 In Combination With Either Paclitaxel And Carboplatin (With or Without Bevacizumab) or Pemetrexed, Cisplatin, And Bevacizumab in Patients With Advanced Non Small Cell Lung Cancer [80] | Phase I | NCT00974584 | NSCLC | GDC-0941 + Paclitaxel + Carboplatin(with or without Bevacizumab) or Pemetrexed + Cisplatin + Bevacizumab | PI3K inhibitor |
| A Study of the Safety and Pharmacology of GDC-0941 in Combination With Erlotinib in Patients With Advanced Solid Tumors [81] | Phase I | NCT00975182 | Solid tumors | GDC-0941 + Erlotinib | PI3K inhibitor |
| Study Evaluating the Safety and Efficacy Of Carboplatin/Paclitaxel And Carboplatin/Paclitaxel/Bevacizumab With and Without GDC-0941 in Patients With Previously Untreated Advanced Or Recurrent Non-small Cell Lung [82] | Phase II | NCT01493843 | NSCLC | Carboplatin + Paclitaxel or Carboplatin + Paclitaxel + Bevacizumab with and without GDC-0941 | PI3K inhibitor |
| Safety Study of XL147 (SAR245408), in Combination With Paclitaxel and Carboplatin in Adults With Solid Tumors [84] | Phase I | NCT00756847 | Solid tumors | XL-147 + Paclitaxel + Carboplatin | PI3K inhibitor |
| A Trial of Gefitinib in Combination With BKM120 in Patients With Advanced Non-Small Cell Lung Cancer, With Enrichment for Patients Whose Tumors Harbour Molecular Alterations of PI3K Pathway and Known to Overexpress EGFR [88] | Phase I | NCT01570296 | NSCLC | BKM120 + Gefitinib | PI3K inhibitor |
| Trial of Erlotinib and BKM120 in Patients With Advanced Non Small Cell Lung Cancer Previously Sensitive to Erlotinib [89] | Phase I+Phase II | NCT01487265 | NSCLC | BKM120 + Erlotinib | PI3K inhibitor |
| Safety and Efficacy of BKM120 in Patients With Metastatic Non-small Cell Lung Cancer [90] | Phase II | NCT01297491 | NSCLC | BKM120 + Docetaxel orDocetaxel + Pemetrexed | PI3K inhibitor |
| A Phase I Study of BKM120 and Everolimus in Advanced Solid Malignancies [91] | Phase I | NCT01470209 | Solid tumors | BKM120 + Everolimus | PI3K inhibitor |
| BKM120 in Cancers With PIK3CA Activating Mutations [92] | Phase II | NCT01501604 | Solid tumors with PIK3CA mutations | BKM120 | PI3K inhibitor |
| Dose Defining Study For MK-2206 Combined With Gefitinib In Non Small Cell Lung Cancer (NSCLC) [113] | Phase I | NCT01147211 | NSCLC | MK-2206 + Gefitinib | Akt inhibitor |
| MK2206 and Erlotinib Hydrochloride in Treating Patients With Advanced Non-Small Cell Lung Cancer Who Have Progressed After Previous Response to Erlotinib Hydrochloride Therapy [114] | Phase II | NCT01294306 | NSCLC | MK-2206 + Erlotinib | Akt inhibitor |
| Temsirolimus and Pemetrexed for Recurrent or Refractory Non-Small Cell Lung Cancer [136] | Phase I + Phase II | NCT00921310 | NSCLC | Temsirolimus + Pemetrexed | mTOR inhibitor |
| Temsirolimus and Vinorelbine Ditartrate in Treating Patients With Unresectable or Metastatic Solid Tumors [137] | Phase I | NCT01155258 | Solid tumors | Temsirolimu + Vinorelbine | mTOR inhibitor |
| Phase I Study of Docetaxel and Temsirolimus in Resistant Solid Malignancies [138] | Phase I | NCT00703625 | Solid tumors | Temsirolimus + Docetaxel | mTOR inhibitor |
| Phase 1b Trial of RAD001 in Patients With Operable Non-Small Cell Lung Cancer (NSCLC) [154] | Phase I | NCT00401778 | NSCLC | Everolimus | mTOR inhibitor |
| Combination of RAD001 With Carboplatin, Paclitaxel and Bevacizumab in Non-small-cell Lung Cancer (NSCLC) Patients [155] | Phase I | NCT00457119 | NSCLC | Everolimus + Carboplatin + Paclitaxel + Bevacizumab | mTOR inhibitor |
| RAD001 With Paclitaxel and Carboplatin in First Line Treatment of Patients With Advanced Large Cell Lung Cancer With Neuroendocrine Differentiation [160] | Phase II | NCT01317615 | LCLC | Everolimus + Paclitaxel + Carboplatin | mTOR inhibitor |
| Combination Anticancer Therapy of Paclitaxel and Everolimus for Relapsed or Refractory Small Cell Lung Cancer [161] | Phase I | NCT01079481 | SCLC | Everolimus + Paclitaxel | mTOR inhibitor |
| Everolimus, Carboplatin, and Etoposide in Treating Patients With Small Cell Lung Cancer or Other Advanced Solid Tumors [162] | Phase I | NCT00807755 | SCLC(Solid tumors) | Everolimus + Carboplatin + Etoposide | mTOR inhibitor |
| Safety of RAD001 in Combination With Cisplatin and Etoposide in Lung Cancer Patients [163] | Phase I | NCT00466466 | SCLC | Everolimus + Cisplatin + Etoposide | mTOR inhibitor |
| A Study of Ridaforolimus in Non-Small Cell Lung Cancer (NSCLC) Patients With Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) Mutations (MK-8669-021 AM1) [170] | Phase II | NCT00818675 | NSCLC | Ridaforolimus | mTOR inhibitor |
| Ridaforolimus With Cetuximab for Patients With Advanced Head and Neck Cancer, Non-Small Cell Lung Cancer and Colon Cancer [171] | Phase I | NCT01212627 | Solid tumors | Ridaforolim + Cetuximab | mTOR inhibitor |
5. Conclusions
Conflict of Interest
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Robinson, C.G.; Bradley, J.D. The treatment of early-stage disease. Semin. Radiat. Oncol. 2010, 20, 178–185. [Google Scholar] [CrossRef]
- Burdett, S.; Stephens, R.; Stewart, L.; Tierney, J.; Auperin, A.; Le Chevalier, T.; Le Pechoux, C.; Pignon, J.P.; Arriagada, R.; Higgins, J.; et al. Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: A systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J. Clin. Oncol. 2008, 26, 4617–4625. [Google Scholar]
- Govindan, R.; Page, N.; Morgensztern, D.; Read, W.; Tierney, R.; Vlahiotis, A.; Spitznagel, E.L.; Piccirillo, J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: Analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006, 24, 4539–44. [Google Scholar] [CrossRef]
- Jackman, D.M.; Johnson, B.E. Small-cell lung cancer. Lancet 2005, 366, 1385–1396. [Google Scholar] [CrossRef]
- Demedts, I.K.; Vermaelen, K.Y.; van Meerbeeck, J.P. Treatment of extensive-stage small cell lung carcinoma: current status and future prospects. Eur. Respir. J. 2010, 35, 202–215. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A.; Jett, J.R. Advances in chemotherapy of non-small cell lung cancer. Chest 2006, 130, 1211–1219. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. The role of the Akt/MTOR pathway in tobacco carcinogen-induced lung tumorigenesis. Clin. Cancer Res. 2010, 16, 4–10. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-Kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Kurosu, H.; Maehama, T.; Okada, T.; Yamamoto, T.; Hoshino, S.; Fukui, Y.; Ui, M.; Hazeki, O.; Katada, T. Heterodimeric phosphoinositide 3-kinase consisting of P85 and P110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J. Biol. Chem. 1997, 272, 24252–24256. [Google Scholar]
- Roche, S.; Downward, J.; Raynal, P.; Courtneidge, S.A. A function for phosphatidylinositol 3-Kinase Beta (P85alpha-P110beta) in fibroblasts during mitogenesis: Requirement for insulin and lysophosphatidic acid-mediated signal transduction. Mol. Cell Biol. 1998, 18, 7119–7129. [Google Scholar]
- Vanhaesebroeck, B.; Waterfield, M.D. Signaling by distinct classes of phosphoinositide 3-Kinases. Exp. Cell Res. 1999, 253, 239–254. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-Kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and MTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Grant, S.; Qiao, L.; Dent, P. Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front. Biosci. 2002, 7, d376–d389. [Google Scholar] [CrossRef]
- Krasilnikov, M.A. Phosphatidylinositol-3 kinase dependent pathways: The role in control of cell growth, survival, and malignant transformation. Biochemistry (Mosc.) 2000, 65, 59–67. [Google Scholar]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Currie, R.A.; Walker, K.S.; Gray, A.; Deak, M.; Casamayor, A.; Downes, C.P.; Cohen, P.; Alessi, D.R.; Lucocq, J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 1999, 337, 575–583. [Google Scholar] [CrossRef]
- Duronio, V. The life of a cell: Apoptosis regulation by the PI3K/PKB pathway. Biochem. J. 2008, 415, 333–344. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005, 65, 4562–4567. [Google Scholar] [CrossRef]
- Samuels, Y.; Velculescu, V.E. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004, 3, 1221–1224. [Google Scholar] [CrossRef]
- Shayesteh, L.; Lu, Y.; Kuo, W.L.; Baldocchi, R.; Godfrey, T.; Collins, C.; Pinkel, D.; Powell, B.; Mills, G.B.; Gray, J.W. PIK3CA Is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef]
- Yamamoto, H.; Shigematsu, H.; Nomura, M.; Lockwood, W.W.; Sato, M.; Okumura, N.; Soh, J.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008, 68, 6913–6921. [Google Scholar]
- Shibata, T.; Kokubu, A.; Tsuta, K.; Hirohashi, S. Oncogenic Mutation of PIK3CA in small cell lung carcinoma: A potential therapeutic target pathway for chemotherapy-resistant lung cancer. Cancer Lett. 2009, 283, 203–211. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Invest 2006, 116, 2695–2706. [Google Scholar] [CrossRef]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of Trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef]
- Massion, P.P.; Taflan, P.M.; Shyr, Y.; Rahman, S.M.; Yildiz, P.; Shakthour, B.; Edgerton, M.E.; Ninan, M.; Andersen, J.J.; Gonzalez, A.L. Early involvement of the phosphatidylinositol 3-kinase/akt pathway in lung cancer progression. Am. J. Respir. Crit Care Med. 2004, 170, 1088–1094. [Google Scholar] [CrossRef]
- Voortman, J.; Lee, J.H.; Killian, J.K.; Suuriniemi, M.; Wang, Y.; Lucchi, M.; Smith, W.I., Jr.; Meltzer, P.; Wang, Y.; Giaccone, G. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 13040–13045. [Google Scholar]
- Soung, Y.H.; Lee, J.W.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutational analysis of AKT1, AKT2 and AKT3 genes in common human carcinomas. Oncology 2006, 70, 285–289. [Google Scholar] [CrossRef]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/Protein Kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar]
- Chun, K.H.; Kosmeder, J.W.; Sun, S.; Pezzuto, J.M.; Lotan, R.; Hong, W.K.; Lee, H.Y. Effects of deguelin on the phosphatidylinositol 3-Kinase/Akt pathway and apoptosis in premalignant human bronchial epithelial cells. J. Natl. Cancer Inst. 2003, 95, 291–302. [Google Scholar] [CrossRef]
- David, O.; Jett, J.; LeBeau, H.; Dy, G.; Hughes, J.; Friedman, M.; Brody, A.R. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin. Cancer Res. 2004, 10, 6865–6871. [Google Scholar] [CrossRef]
- Tsurutani, J.; Fukuoka, J.; Tsurutani, H.; Shih, J.H.; Hewitt, S.M.; Travis, W.D.; Jen, J.; Dennis, P.A. Evaluation of two phosphorylation sites improves the prognostic significance of akt activation in non-small-cell lung cancer tumors. J. Clin. Oncol. 2006, 24, 306–314. [Google Scholar]
- Blackhall, F.H.; Pintilie, M.; Michael, M.; Leighl, N.; Feld, R.; Tsao, M.S.; Shepherd, F.A. Expression and prognostic significance of kit, protein kinase b, and mitogen-activated protein kinase in patients with small cell lung cancer. Clin. Cancer Res. 2003, 9, 2241–2247. [Google Scholar]
- Kraus, A.C.; Ferber, I.; Bachmann, S.O.; Specht, H.; Wimmel, A.; Gross, M.W.; Schlegel, J.; Suske, G.; Schuermann, M. In vitro chemo- and radio-resistance in small cell lung cancer correlates with cell adhesion and constitutive activation of AKT and MAP kinase pathways. Oncogene 2002, 21, 8683–8695. [Google Scholar]
- Han, S.Y.; Kato, H.; Kato, S.; Suzuki, T.; Shibata, H.; Ishii, S.; Shiiba, K.; Matsuno, S.; Kanamaru, R.; Ishioka, C. Functional evaluation of pten missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 2000, 60, 3147–3151. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Sun, X.; Huang, J.; Homma, T.; Kita, D.; Klocker, H.; Schafer, G.; Boyle, P.; Ohgaki, H. Genetic alterations in the PI3K pathway in prostate cancer. Anticancer Res. 2009, 29, 1739–1743. [Google Scholar]
- Teng, D.H.; Hu, R.; Lin, H.; Davis, T.; Iliev, D.; Frye, C.; Swedlund, B.; Hansen, K.L.; Vinson, V.L.; Gumpper, K.L.; et al. MMAC1/PTEN mutations in primary tumor specimens and tumor cell lines. Cancer Res. 1997, 57, 5221–5225. [Google Scholar]
- Forgacs, E.; Biesterveld, E.J.; Sekido, Y.; Fong, K.; Muneer, S.; Wistuba, I.I.; Milchgrub, S.; Brezinschek, R.; Virmani, A.; Gazdar, A.F.; et al. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene 1998, 17, 1557–1565. [Google Scholar]
- Forgacs, E.; Zöchbauer-Müller, S.; Oláh, E.; Minna, J.D. Molecular genetic abnormalities in the pathogenesis of human lung cancer. Pathol Oncol Res 2001, 7, 6–13. [Google Scholar] [CrossRef]
- Yokomizo, A.; Tindall, D.J.; Drabkin, H.; Gemmill, R.; Franklin, W.; Yang, P.; Sugio, K.; Smith, D.I.; Liu, W. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene 1998, 17, 475–479. [Google Scholar]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN Expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar]
- García, J.M.; Silva, J.; Peña, C.; Garcia, V.; Rodríguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; España, P.; Bonilla, F. Promoter methylation of the pten gene is a common molecular change in breast cancer. Genes Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; Bertagnolli, M.M.; Boland, C.R. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004, 64, 3014–3021. [Google Scholar] [CrossRef]
- Balsara, B.R.; Pei, J.; Mitsuuchi, Y.; Page, R.; Klein-Szanto, A.; Wang, H.; Unger, M.; Testa, J.R. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis 2004, 25, 2053–2059. [Google Scholar] [CrossRef]
- Han, S.; Khuri, F.R.; Roman, J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of akt/mammalian target of rapamycin/s6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006, 66, 315–323. [Google Scholar] [CrossRef]
- Conde, E.; Angulo, B.; Tang, M.; Morente, M.; Torres-Lanzas, J.; Lopez-Encuentra, A.; Lopez-Rios, F.; Sanchez-Cespedes, M. Molecular context of the EGFR mutations: evidence for the activation of MTOR/S6K signaling. Clin. Cancer Res. 2006, 12, 710–717. [Google Scholar] [CrossRef]
- Wislez, M.; Spencer, M.L.; Izzo, J.G.; Juroske, D.M.; Balhara, K.; Cody, D.D.; Price, R.E.; Hittelman, W.N.; Wistuba, I.I.; Kurie, J.M. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-Ras. Cancer Res. 2005, 65, 3226–3235. [Google Scholar]
- Anagnostou, V.K.; Bepler, G.; Syrigos, K.N.; Tanoue, L.; Gettinger, S.; Homer, R.J.; Boffa, D.; Detterbeck, F.; Rimm, D.L. High expression of mammalian target of rapamycin is associated with better outcome for patients with early stage lung adenocarcinoma. Clin. Cancer Res. 2009, 15, 4157–4164. [Google Scholar]
- Kokubo, Y.; Gemma, A.; Noro, R.; Seike, M.; Kataoka, K.; Matsuda, K.; Okano, T.; Minegishi, Y.; Yoshimura, A.; Shibuya, M.; et al. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br. J. Cancer 2005, 92, 1711–1719. [Google Scholar] [CrossRef]
- She, Q.B.; Solit, D.; Basso, A.; Moasser, M.M. Resistance to gefitinib in PTEN-Null HER-Overexpressing Tumor Cells Can Be Overcome Through Restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3'-Kinase/Akt pathway signaling. Clin. Cancer Res. 2003, 9, 4340–4346. [Google Scholar]
- La Monica, S.; Galetti, M.; Alfieri, R.R.; Cavazzoni, A.; Ardizzoni, A.; Tiseo, M.; Capelletti, M.; Goldoni, M.; Tagliaferri, S.; Mutti, A.; et al. Everolimus restores gefitinib sensitivity in resistant non-small cell lung cancer cell lines. Biochem. Pharmacol. 2009, 78, 460–468. [Google Scholar] [CrossRef]
- Donev, I.S.; Wang, W.; Yamada, T.; Li, Q.; Takeuchi, S.; Matsumoto, K.; Yamori, T.; Nishioka, Y.; Sone, S.; Yano, S. Transient PI3K inhibition induces apoptosis and overcomes hgf-mediated resistance to egfr-tkis in egfr mutant lung cancer. Clin. Cancer Res. 2011, 17, 2260–2269. [Google Scholar] [CrossRef]
- Land, H.; Parada, L.F.; Weinberg, R.A. Cellular oncogenes and multistep carcinogenesis. Science 1983, 222, 771–778. [Google Scholar]
- Bos, J.L. Ras oncogenes in human cancer: a review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal structure and functional analysis of ras binding to its effector phosphoinositide 3-kinase gamma. Cell 2000, 103, 931–943. [Google Scholar] [CrossRef]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of Ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef]
- Ihle, N.T.; Lemos, R., Jr.; Wipf, P.; Yacoub, A.; Mitchell, C.; Siwak, D.; Mills, G.B.; Dent, P.; Kirkpatrick, D.L.; Powis, G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic ras is a dominant predictor for resistance. Cancer Res. 2009, 69, 143–150. [Google Scholar] [CrossRef]
- Powis, G.; Bonjouklian, R.; Berggren, M.M.; Gallegos, A.; Abraham, R.; Ashendel, C.; Zalkow, L.; Matter, W.F.; Dodge, J.; Grindey, G. Wortmannin, A potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994, 54, 2419–2423. [Google Scholar]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-Morpholinyl)-8-Phenyl-4H-1-Benzopyran-4-One (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Gupta, A.K.; Soto, D.E.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Hahn, S.M.; Machtay, M.; Muschel, R.J.; McKenna, W.G. Signaling pathways in NSCLC as a predictor of outcome and response to therapy. Lung 2004, 182, 151–162. [Google Scholar]
- Ihle, N.T.; Williams, R.; Chow, S.; Chew, W.; Berggren, M.I.; Paine-Murrieta, G.; Minion, D.J.; Halter, R.J.; Wipf, P.; Abraham, R.; et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol. Cancer Ther. 2004, 3, 763–772. [Google Scholar]
- Ihle, N.T.; Paine-Murrieta, G.; Berggren, M.I.; Baker, A.; Tate, W.R.; Wipf, P.; Abraham, R.T.; Kirkpatrick, D.L.; Powis, G. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in a-549 human non-small cell lung cancer xenografts. Mol. Cancer Ther. 2005, 4, 1349–1357. [Google Scholar] [CrossRef]
- Ihle, N.T.; Lemos, R.; Schwartz, D.; Oh, J.; Halter, R.J.; Wipf, P.; Kirkpatrick, L.; Powis, G. Peroxisome proliferator-activated receptor gamma agonist pioglitazone prevents the hyperglycemia caused by phosphatidylinositol 3-kinase pathway inhibition by PX-866 without affecting antitumor activity. Mol. Cancer Ther. 2009, 8, 94–100. [Google Scholar]
- Yang, Y.; Iwanaga, K.; Raso, M.G.; Wislez, M.; Hanna, A.E.; Wieder, E.D.; Molldrem, J.J.; Wistuba, I.I.; Powis, G.; Demayo, F.J.; et al. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-Ras-induced lung cancer. PLoS. One. 2008, 3, e2220. [Google Scholar]
- Oncothyreon Inc. Study of PX-866 and Docetaxel in Solid Tumors. Available online: http://clinicaltrials.gov/show/NCT01204099 (accessed on 14 Novermber 2012).
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The identification of 2-(1H-Indazol-4-Yl)-6-(4-Methanesulfonyl-Piperazin-1-Ylmethyl)-4-Morpholin-4-Yl-t Hieno[3,2-d]Pyrimidine (GDC-0941) As a potent, selective, orally bioavailable inhibitor of class I PI3 Kinase for the treatment of cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar]
- Sarker, D.; Kristeleit, R.; Mazina, K.E.; Ware, J.A.; Yan, Y.; Dresser, M.; Derynck, M.K.; De-Bono, J. A phase I study evaluating the pharmacokinetics (PK) and pharmacodynamics (PD) of the oral Pan-Phosphoinositide-3 kinase (PI3K) inhibitor GDC-0941. J. Clin. Oncol. 2009, 27. abstr 3538. [Google Scholar]
- Wagner, A.J.; Von Hoff, D.H.; LoRusso, P.M.; Tibes, R.; Mazina, K.E.; Ware, J.A.; Yan, Y.; Derynck, M.K.; Demetri, G.D. A first-in-human phase I study to evaluate the Pan-PI3K inhibitor GDC-0941 administered QD or BID in patients with advanced solid tumors. J. Clin. Oncol. 2009, 27. abstr 3501. [Google Scholar]
- Genentech. A study of the safety and pharmacology of pi3-kinase inhibitor gdc-0941 in combination with either paclitaxel and carboplatin (with or without bevacizumab) or pemetrexed, cisplatin, and bevacizumab in patients with advanced non small cell lung cancer. Available online: http://clinicaltrials.gov/show/NCT00974584 (accessed on 14 Novermber 2012).
- Genentech. A study of the safety and pharmacology of GDC-0941 in combination with erlotinib in patients with advanced solid tumors. Available online: http://clinicaltrials.gov/show/NCT00975182 (accessed on 14 Novermber 2012).
- Genentech. Study evaluating the safety and efficacy of carboplatin/paclitaxel and carboplatin/paclitaxel/bevacizumab with and without GDC-0941 in patients with previously untreated advanced or recurrent non-small cell lung cancer. Available online: http://clinicaltrials.gov/show/NCT01493843, (accessed on 14 Novermber 2012).
- Edelman, G.; Bedell, C.; Shapiro, G.; Pandya, S.S.; Kwak, E.L.; Scheffold, C.; Nguyen, L.T.; Laird, A.; Baselga, J.; Rodon, J. A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J. Clin. Oncol. (Meeting Abstracts) 2010, 28. abstr 3004. [Google Scholar]
- Sanofi-Aventis. Safety study of XL147 (SAR245408), in combination with paclitaxel and carboplatin in adults with solid tumors. Available online: http://clinicaltrials.gov/show/NCT00756847 (accessed on 14 Novermber 2012).
- Moldovan, C.; Soria, J.; LoRusso, P.; Guthrie, T.; Song, C.; Nguyen, L.T.; Martini, J.; Infante, J.R.; Burris, H.A. A phase I safety and pharmacokinetic (PK) study of the PI3K inhibitor XL147 (SAR245408) in combination with erlotinib in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2010, 28. abstr 3070. [Google Scholar]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef]
- Ren, H.; Chen, M.; Yue, P.; Tao, H.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R.; Sun, S.Y. The combination of RAD001 and NVP-BKM120 synergistically inhibits the growth of lung cancer in vitro and in vivo. Cancer Lett. 2012, 325, 139–146. [Google Scholar] [CrossRef]
- A Trial of gefitinib in combination with BKM120 in patients with advanced non-small cell lung cancer, with enrichment for patients whose tumours harbour molecular alterations of PI3K pathway and known to overexpress EGFR. Available online: http://clinicaltrials.gov/show/NCT01570296 (accessed on 14 Novermber 2012).
- Trial of erlotinib and BKM120 in patients with advanced non small cell lung cancer previously sensitive to erlotinib. Available online: http://clinicaltrials.gov/show/NCT01487265 (accessed on 14 Novermber 2012).
- Novartis Pharmaceuticals. Safety and Efficacy of BKM120 in Patients With Metastatic Non-small Cell Lung Cancer. Available online: http://clinicaltrials.gov/show/NCT01297491 (accessed on 14 Novermber 2012).
- A Phase I Study of BKM120 and Everolimus in Advanced Solid Malignancies. Available online: http://clinicaltrials.gov/show/NCT01470209 (accessed on 14 Novermber 2012).
- BKM120 in Cancers With PIK3CA Activating Mutations. Available online: http://clinicaltrials.gov/show/NCT01501604 (accessed on 14 Novermber 2012).
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De, P.A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef]
- Stauffer, F.; Maira, S.M.; Furet, P.; Garcia-Echeverria, C. Imidazo[4,5-c]Quinolines as inhibitors of the PI3K/PKB-pathway. Bioorg. Med. Chem. Lett. 2008, 18, 1027–1030. [Google Scholar] [CrossRef]
- Burris, H.; Rodon, J.; Sharma, S.; Herbst, R.S.; Tabernero, J.; Infante, J.R.; Silva, A.; Demanse, D.; Hackl, W.; Baselga, J. First-in-human phase I study of the oral PI3K inhibitor BEZ235 in patients (pts) with advanced solid tumors. J. Clin. Oncol. (Meeting Abstracts) 2010, 28. abstr 3005. [Google Scholar]
- Konstantinidou, G.; Bey, E.A.; Rabellino, A.; Schuster, K.; Maira, M.S.; Gazdar, A.F.; Amici, A.; Boothman, D.A.; Scaglioni, P.P. Dual phosphoinositide 3-Kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009, 69, 7644–7652. [Google Scholar] [CrossRef]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a dual PI3K/MTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar]
- Cho, D.C.; Cohen, M.B.; Panka, D.J.; Collins, M.; Ghebremichael, M.; Atkins, M.B.; Signoretti, S.; Mier, J.W. The efficacy of the novel dual PI3-kinase/MTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clin. Cancer Res. 2010, 16, 3628–3638. [Google Scholar] [CrossRef]
- Sos, M.L.; Fischer, S.; Ullrich, R.; Peifer, M.; Heuckmann, J.M.; Koker, M.; Heynck, S.; Stuckrath, I.; Weiss, J.; Fischer, F.; et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 18351–18356. [Google Scholar]
- Faber, A.C.; Li, D.; Song, Y.; Liang, M.C.; Yeap, B.Y.; Bronson, R.T.; Lifshits, E.; Chen, Z.; Maira, S.M.; Garcia-Echeverria, C.; et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 19503–19508. [Google Scholar]
- LoRusso, P.; Markman, B.; Tabernero, J.; Shazer, R.; Nguyen, L.; Heath, E.; Patnaik, A.; Papadopoulos, K. A Phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765, a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced solid tumors. J. Clin. Oncol. (Meeting Abstracts) 2009, 27. abstr 3502. [Google Scholar]
- Cohen, R.B.; Janne, P.A.; Engelman, J.A.; Martínez, P.; Nishida, Y.; Gendreau, S.; Wu, B.; Felip, E. A phase I safety and pharmacokinetic (PK) study of PI3K/TORC1/TORC2 inhibitor XL765 (SAR245409) in combination with erlotinib (E) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2010, 28, 3015. [Google Scholar]
- Zou, Z.Q.; Zhang, X.H.; Wang, F.; Shen, Q.J.; Xu, J.; Zhang, L.N.; Xing, W.H.; Zhuo, R.J.; Li, D. A novel dual PI3Kalpha/MTOR inhibitor PI-103 with high antitumor activity in non-small cell lung cancer cells. Int. J. Mol. Med. 2009, 24, 97–101. [Google Scholar]
- Prevo, R.; Deutsch, E.; Sampson, O.; Diplexcito, J.; Cengel, K.; Harper, J.; O'Neill, P.; McKenna, W.G.; Patel, S.; Bernhard, E.J. Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor pi-103 enhances tumor radiosensitivity. Cancer Res. 2008, 68, 5915–5923. [Google Scholar]
- Qayum, N.; Muschel, R.J.; Im, J.H.; Balathasan, L.; Koch, C.J.; Patel, S.; McKenna, W.G.; Bernhard, E.J. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res. 2009, 69, 6347–6354. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, G.D. Targeting serine/threonine protein kinase B/Akt and Cell-Cycle checkpoint kinases for treating cancer. Curr. Top. Med. Chem. 2002, 2, 939–971. [Google Scholar] [CrossRef]
- Kumar, C.C.; Madison, V. Drugs Targeted Against Protein Kinases. Expert. Opin. Emerg. Drugs 2001, 6, 303–315. [Google Scholar] [CrossRef]
- Cherrin, C.; Haskell, K.; Howell, B.; Jones, R.; Leander, K.; Robinson, R.; Watkins, A.; Bilodeau, M.; Hoffman, J.; Sanderson, P.; et al. An Allosteric Akt inhibitor effectively blocks akt signaling and tumor growth with only transient effects on glucose and insulin levels in vivo. Cancer Biol. Ther. 2010, 9, 493–503. [Google Scholar] [CrossRef]
- Luo, Y.; Shoemaker, A.R.; Liu, X.; Woods, K.W.; Thomas, S.A.; de Jong, R.; Han, E.K.; Li, T.; Stoll, V.S.; Powlas, J.A.; et al. Potent and selective inhibitors of akt kinases slow the progress of tumors in vivo. Mol. Cancer Ther. 2005, 4, 977–986. [Google Scholar] [CrossRef]
- Crouthamel, M.C.; Kahana, J.A.; Korenchuk, S.; Zhang, S.Y.; Sundaresan, G.; Eberwein, D.J.; Brown, K.K.; Kumar, R. Mechanism and management of AKT inhibitor-induced hyperglycemia. Clin. Cancer Res. 2009, 15, 217–225. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral Pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar]
- National Taiwan University Hospital. Dose defining study for MK-2206 combined with gefitinib in non small cell lung cancer (NSCLC). Available online: http://clinicaltrials.gov/show/NCT01147211 (acesseed on 14 November 2012).
- MK2206 and erlotinib hydrochloride in treating patients with advanced non-small cell lung cancer who have progressed after previous response to erlotinib hydrochloride therapy. Available online: http://clinicaltrials.gov/show/NCT01294306 (acesseed on 14 November 2012).
- Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an Akt Kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar]
- Douros, J.; Suffness, M. New Antitumor Substances of Natural Origin. Cancer Treat. Rev. 1981, 8, 63–87. [Google Scholar] [CrossRef]
- Decker, T.; Hipp, S.; Ringshausen, I.; Bogner, C.; Oelsner, M.; Schneller, F.; Peschel, C. Rapamycin-induced G1 arrest in cycling B-CLL cells is associated with reduced expression of Cyclin D3, Cyclin E, Cyclin A, and Survivin. Blood 2003, 101, 278–285. [Google Scholar] [CrossRef]
- Albers, M.W.; Williams, R.T.; Brown, E.J.; Tanaka, A.; Hall, F.L.; Schreiber, S.L. FKBP-Rapamycin inhibits a Cyclin-dependent kinase activity and a Cyclin D1-Cdk association in early G1 of an osteosarcoma cell line. J. Biol. Chem. 1993, 268, 22825–22829. [Google Scholar]
- Luo, Y.; Marx, S.O.; Kiyokawa, H.; Koff, A.; Massague, J.; Marks, A.R. Rapamycin resistance tied to defective regulation of P27Kip1. Mol. Cell Biol. 1996, 16, 6744–6751. [Google Scholar]
- Zezula, J.; Sexl, V.; Hutter, C.; Karel, A.; Schutz, W.; Freissmuth, M. The cyclin-dependent Kinase inhibitor P21cip1 mediates the growth inhibitory effect of phorbol esters in human venous endothelial cells. J. Biol. Chem. 1997, 272, 29967–29974. [Google Scholar]
- Humar, R.; Kiefer, F.N.; Berns, H.; Resink, T.J.; Battegay, E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in Vitro via rapamycin (MTOR)-dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and EIF4E survival pathways by Rapamycin-mediated mammalian target of tapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar]
- Boffa, D.J.; Luan, F.; Thomas, D.; Yang, H.; Sharma, V.K.; Lagman, M.; Suthanthiran, M. Rapamycin inhibits the growth and metastatic progression of non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 293–300. [Google Scholar] [CrossRef]
- Wislez, M.; Spencer, M.L.; Izzo, J.G.; Juroske, D.M.; Balhara, K.; Cody, D.D.; Price, R.E.; Hittelman, W.N.; Wistuba, I.I.; Kurie, J.M. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K-Ras. Cancer Res. 2005, 65, 3226–3235. [Google Scholar]
- Sun, S.Y.; Fu, H.; Khuri, F.R. Targeting MTOR signaling for lung cancer therapy. J. Thorac. Oncol. 2006, 1, 109–111. [Google Scholar] [CrossRef]
- Zito, C.R.; Jilaveanu, L.B.; Anagnostou, V.; Rimm, D.; Bepler, G.; Maira, S.M.; Hackl, W.; Camp, R.; Kluger, H.M.; Chao, H.H. Multi-level targeting of the phosphatidylinositol-3-kinase pathway in non-small cell lung cancer cells. PLoS. One. 2012, 7, e31331. [Google Scholar]
- Dudkin, L.; Dilling, M.B.; Cheshire, P.J.; Harwood, F.C.; Hollingshead, M.; Arbuck, S.G.; Travis, R.; Sausville, E.A.; Houghton, P.J. Biochemical correlates of MTOR inhibition by the rapamycin Ester CCI-779 and tumor growth inhibition. Clin. Cancer Res. 2001, 7, 1758–1764. [Google Scholar]
- Ohara, T.; Takaoka, M.; Toyooka, S.; Tomono, Y.; Nishikawa, T.; Shirakawa, Y.; Yamatsuji, T.; Tanaka, N.; Fujiwara, T.; Naomoto, Y. Inhibition of MTOR by temsirolimus contributes to prolonged survival of mice with pleural dissemination of non-small-cell lung cancer cells. Cancer Sci. 2011, 102, 1344–1349. [Google Scholar] [CrossRef]
- Hidalgo, M.; Buckner, J.C.; Erlichman, C.; Pollack, M.S.; Boni, J.P.; Dukart, G.; Marshall, B.; Speicher, L.; Moore, L.; Rowinsky, E.K. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 5755–5763. [Google Scholar] [CrossRef]
- Buckner, J.C.; Forouzesh, B.; Erlichman, C.; Hidalgo, M.; Boni, J.P.; Dukart, G.; Berkenblit, A.; Rowinsky, E.K. Phase I, Pharmacokinetic study of temsirolimus administered orally to patients with advanced cancer. Invest. New Drugs 2010, 28, 334–342. [Google Scholar] [CrossRef]
- Pandya, K.J.; Dahlberg, S.; Hidalgo, M.; Cohen, R.B.; Lee, M.W.; Schiller, J.H.; Johnson, D.H. A randomized, phase II trial of two dose levels of temsirolimus (CCI-779) in patients with extensive-stage small-cell lung cancer who have responding or stable disease after induction chemotherapy: a trial of the eastern cooperative oncology group (E1500). J. Thorac. Oncol. 2007, 2, 1036–1041. [Google Scholar]
- Reungwetwattana, T.; Molina, J.R.; Mandrekar, S.J.; Allen-Ziegler, K.; Rowland, K.M.; Reuter, N.F.; Luyun, R.F.; Dy, G.K.; Marks, R.S.; Schild, S.E.; et al. Brief report: a phase II "window-of-opportunity" frontline study of the MTOR inhibitor, temsirolimus given as a single agent in patients with advanced NSCLC, an NCCTG study. J. Thorac. Oncol. 2012, 7, 919–922. [Google Scholar] [CrossRef]
- Bryce, A.H.; Rao, R.; Sarkaria, J.; Reid, J.M.; Qi, Y.; Qin, R.; James, C.D.; Jenkins, R.B.; Boni, J.; Erlichman, C.; et al. Phase I study of temsirolimus in combination With EKB-569 in patients with advanced solid tumors. Invest. New Drugs 2012, 30, 1934–1941. [Google Scholar] [CrossRef]
- Washington University School of Medicine. Temsirolimus and pemetrexed for recurrent or refractory non-small cell lung cancer. Available online: http://clinicaltrials.gov/show/NCT00921310 (accessed on 14 November 2012).
- Temsirolimus and vinorelbine ditartrate in treating patients with unresectable or metastatic solid tumors. Available online: http://clinicaltrials.gov/show/NCT01155258 (accessed on 14 November 2012).
- Washington University School of Medicine. Phase I study of docetaxel and temsirolimus in resistant solid malignancies. Available online: http://clinicaltrials.gov/show/NCT00703625 (accessed on 14 November 2012).
- Eisen, H.J.; Tuzcu, E.M.; Dorent, R.; Kobashigawa, J.; Mancini, D.; Valantine-von Kaeppler, H.A.; Starling, R.C.; Sorensen, K.; Hummel, M.; Lind, J.M.; et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. N. Engl. J. Med. 2003, 349, 847–858. [Google Scholar] [CrossRef]
- Lorber, M.I.; Mulgaonkar, S.; Butt, K.M.; Elkhammas, E.; Mendez, R.; Rajagopalan, P.R.; Kahan, B.; Sollinger, H.; Li, Y.; Cretin, N.; et al. Everolimus versus mycophenolate mofetil in the prevention of rejection in de novo renal transplant recipients: a 3-year randomized, multicenter, phase III study. Transplantation 2005, 80, 244–252. [Google Scholar] [CrossRef]
- Beuvink, I.; Boulay, A.; Fumagalli, S.; Zilbermann, F.; Ruetz, S.; O'Reilly, T.; Natt, F.; Hall, J.; Lane, H.A.; Thomas, G. The MTOR Inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of P21 translation. Cell 2005, 120, 747–759. [Google Scholar] [CrossRef]
- Mabuchi, S.; Altomare, D.A.; Cheung, M.; Zhang, L.; Poulikakos, P.I.; Hensley, H.H.; Schilder, R.J.; Ozols, R.F.; Testa, J.R. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin. Cancer Res. 2007, 13, 4261–4270. [Google Scholar] [CrossRef]
- Boulay, A.; Zumstein-Mecker, S.; Stephan, C.; Beuvink, I.; Zilbermann, F.; Haller, R.; Tobler, S.; Heusser, C.; O'Reilly, T.; Stolz, B.; et al. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 2004, 64, 252–261. [Google Scholar]
- Buck, E.; Eyzaguirre, A.; Brown, E.; Petti, F.; McCormack, S.; Haley, J.D.; Iwata, K.K.; Gibson, N.W.; Griffin, G. Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol. Cancer Ther. 2006, 5, 2676–2684. [Google Scholar] [CrossRef]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. MTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef]
- Soria, J.C.; Shepherd, F.A.; Douillard, J.Y.; Wolf, J.; Giaccone, G.; Crino, L.; Cappuzzo, F.; Sharma, S.; Gross, S.H.; Dimitrijevic, S.; et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann. Oncol. 2009, 20, 1674–1681. [Google Scholar] [CrossRef]
- Milton, D.T.; Riely, G.J.; Azzoli, C.G.; Gomez, J.E.; Heelan, R.T.; Kris, M.G.; Krug, L.M.; Pao, W.; Pizzo, B.; Rizvi, N.A.; et al. Phase 1 trial of everolimus and gefitinib in patients with advanced nonsmall-cell lung cancer. Cancer 2007, 110, 599–605. [Google Scholar] [CrossRef]
- Price, K.A.; Azzoli, C.G.; Krug, L.M.; Pietanza, M.C.; Rizvi, N.A.; Pao, W.; Kris, M.G.; Riely, G.J.; Heelan, R.T.; Arcila, M.E.; et al. Phase II trial of gefitinib and everolimus in advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1623–1629. [Google Scholar] [CrossRef]
- Campone, M.; Levy, V.; Bourbouloux, E.; Berton, R.D.; Bootle, D.; Dutreix, C.; Zoellner, U.; Shand, N.; Calvo, F.; Raymond, E. Safety and pharmacokinetics of paclitaxel and the oral MTOR inhibitor everolimus in advanced solid tumours. Br. J. Cancer 2009, 100, 315–321. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Harvey, R.D.; Saba, N.; Owonikoko, T.K.; Kauh, J.; Shin, D.M.; Sun, S.Y.; Strychor, S.; Tighiouart, M.; Egorin, M.J.; et al. Phase 1 and pharmacokinetic study of everolimus, a mammalian target of rapamycin inhibitor, in combination with docetaxel for recurrent/refractory nonsmall cell lung cancer. Cancer 2010, 116, 3903–3909. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Solomon, B.; Boyer, M.; Wolf, J.; Miller, N.; Di Scala, L.; Pylvaenaeinen, I.; Petrovic, K.; Dimitrijevic, S.; Anrys, B.; et al. Everolimus in combination with pemetrexed in patients with advanced non-small cell lung cancer previously treated with chemotherapy: a phase i study using a novel, adaptive bayesian dose-escalation model. J. Thorac. Oncol. 2011, 6, 2120–2129. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.; Blumenschein, G.R.; Leighl, N.B.; Bennouna, J.; Soria, J.C.; Burris, H.A.; Dimitrijevic, S.; Kunz, T.; Di Scala, L.; Johnson, B.E. a phase 1/2 study investigating the combination of RAD001 (R) (Everolimus) and Erlotinib (E) as 2nd and 3rd line therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC) Previously treated with chemotherapy (C): Phase 1 Results. J. Clin. Oncol. 2008, 26. abstr 8051. [Google Scholar]
- Leighl, N.B.; Soria, J.; Bennouna, J.; Blais, N.; Traynor, A.M.; Papadimitrakopoulou, V.; Klimovsky, J.; Jappe, A.; Jehl, V.; Johnson, B.E. Phase II study of everolimus plus erlotinib in previously treated patients with advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2010, 28. abstr 7524. [Google Scholar]
- Emory University. Phase 1b trial of RAD001 in patients with operable non-small cell lung cancer (NSCLC). Available online: http://clinicaltrials.gov/show/NCT00401778 (accessed on 14 November 2012).
- Novartis Pharmaceuticals. combination of RAD001 with carboplatin, paclitaxel and bevacizumab in non-small-cell lung cancer (NSCLC) patients. Available online: http://clinicaltrials.gov/show/NCT00457119 (accessed on 14 November 2012).
- Marinov, M.; Ziogas, A.; Pardo, O.E.; Tan, L.T.; Dhillon, T.; Mauri, F.A.; Lane, H.A.; Lemoine, N.R.; Zangemeister-Wittke, U.; Seckl, M.J.; et al. AKT/MTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin. Cancer Res. 2009, 15, 1277–1287. [Google Scholar] [CrossRef]
- Stracke, S.; Ramudo, L.; Keller, F.; Henne-Bruns, D.; Mayer, J.M. Antiproliferative and overadditive effects of everolimus and mycophenolate mofetil in pancreas and lung cancer cells in vitro. Transplant. Proc. 2006, 38, 766–770. [Google Scholar] [CrossRef]
- Tarhini, A.; Kotsakis, A.; Gooding, W.; Shuai, Y.; Petro, D.; Friedland, D.; Belani, C.P.; Dacic, S.; Argiris, A. Phase II Study of everolimus (RAD001) in previously treated small cell lung cancer. Clin. Cancer Res. 2010, 16, 5900–5907. [Google Scholar] [CrossRef]
- Bago-Horvath, Z.; Sieghart, W.; Grusch, M.; Lackner, A.; Hayden, H.; Pirker, C.; Komina, O.; Wesierska-Gadek, J.; Haitel, A.; Filipits, M.; et al. Synergistic effects of erlotinib and everolimus on bronchial carcinoids and large-cell neuroendocrine carcinomas with activated EGFR/AKT/MTOR pathway. Neuroendocrinology 2012, 96, 228–237. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. RAD001 with paclitaxel and carboplatin in first line treatment of patients with advanced large cell lung cancer with neuroendocrine differentiation. Available online: http://clinicaltrials.gov/show/NCT01317615 (accessed on 14 November 2012).
- Samsung Medical Center. Combination anticancer therapy of paclitaxel and everolimus for relapsed or refractory small cell lung cancer. Available online: http://clinicaltrials.gov/show/NCT01079481 (accessed on 14 November 2012).
- Everolimus, carboplatin, and etoposide in treating patients with small cell lung cancer or other advanced solid tumors. Available online: http://clinicaltrials.gov/show/NCT00807755 (accessed on 14 November 2012).
- Novartis Pharmaceuticals. Safety of RAD001 in combination with cisplatin and etoposide in lung cancer patients. Available online: http://clinicaltrials.gov/show/NCT00466466 (accessed on 14 November 2012).
- White, D.A.; Schwartz, L.H.; Dimitrijevic, S.; Scala, L.D.; Hayes, W.; Gross, S.H. Characterization of pneumonitis in patients with advanced non-small cell lung cancer treated with everolimus (RAD001). J. Thorac. Oncol. 2009, 4, 1357–1363. [Google Scholar] [CrossRef]
- Duran, I.; Siu, L.L.; Oza, A.M.; Chung, T.B.; Sturgeon, J.; Townsley, C.A.; Pond, G.R.; Seymour, L.; Niroumand, M. Characterisation of the lung toxicity of the cell cycle inhibitor temsirolimus. Eur. J. Cancer 2006, 42, 1875–1880. [Google Scholar] [CrossRef]
- Rivera, M.V.; Tang, H.; Metcalf, A.C.; Keenan, P.T.; Sundaramoorthi, R.; Liu, S.; Clackson, T. Anti-proliferative activity of the mTOR inhibitor AP23573 in combination with cytotoxic and targeted agents. Proc. Am. Assoc. Cancer. Res. 2004, 45. abstr 3887. [Google Scholar]
- Clackson, T.; Metcalf, A.C.; Rozamus, W.L.; Knowles, L.H.; Wardwell, D.S.; Roses, B.J.; Burns, D.K.; Graytock, C.; Pradeepan, S.; Notari, D.S.; et al. Regression of tumor xenografts in mice after oral administration of AP23573, a novel mTOR inhibitor that induces tumor starvation. Proc. Am. Assoc. Cancer. Res. 2002, 43. abstr LB95. [Google Scholar]
- Seki, Y.; Yamamoto, N.; Tamura, Y.; Goto, Y.; Shibata, T.; Tanioka, M.; Asahina, H.; Nokihara, H.; Yamada, Y.; Shimamoto, T.; et al. Phase I Study for ridaforolimus, an oral MTOR inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 69, 1099–1105. [Google Scholar] [CrossRef]
- Mita, M.M.; Mita, A.C.; Chu, Q.S.; Rowinsky, E.K.; Fetterly, G.J.; Goldston, M.; Patnaik, A.; Mathews, L.; Ricart, A.D.; Mays, T.; et al. Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J. Clin. Oncol. 2008, 26, 361–367. [Google Scholar]
- Merck. A study of ridaforolimus in non-small cell lung cancer (NSCLC) Patients with kirsten rat sarcoma viral oncogene homolog (KRAS) mutations (MK-8669–021 AM1). Available online: http://clinicaltrials.gov/show/NCT00818675/ (accessed on 14 November 2012).
- Isdale, D.G. Ridaforolimus with cetuximab for patients with advanced head and neck cancer, non-small cell lung cancer and colon cancer. Available online: http://clinicaltrials.gov/show/NCT01212627/ (accessed on 14 November 2012).
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of MTOR target rapamycin-resistant outputs of MTORC1 and MTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.; Taylor, R.; Kaye, S.; et al. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of AZD8055 in Advanced Solid Tumours and Lymphoma. Br. J. Cancer 2012, 107, 1093–1099. [Google Scholar] [CrossRef]
- Holt, S.V.; Logie, A.; Davies, B.R.; Alferez, D.; Runswick, S.; Fenton, S.; Chresta, C.M.; Gu, Y.; Zhang, J.; Wu, Y.L.; et al. Enhanced apoptosis and tumor growth suppression elicited by combination of mek (Selumetinib) and MTOR kinase inhibitors (AZD8055). Cancer Res. 2012, 72, 1804–1813. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sarris, E.G.; Saif, M.W.; Syrigos, K.N. The Biological Role of PI3K Pathway in Lung Cancer. Pharmaceuticals 2012, 5, 1236-1264. https://doi.org/10.3390/ph5111236
Sarris EG, Saif MW, Syrigos KN. The Biological Role of PI3K Pathway in Lung Cancer. Pharmaceuticals. 2012; 5(11):1236-1264. https://doi.org/10.3390/ph5111236
Chicago/Turabian StyleSarris, Evangelos G., Muhammad W. Saif, and Kostas N. Syrigos. 2012. "The Biological Role of PI3K Pathway in Lung Cancer" Pharmaceuticals 5, no. 11: 1236-1264. https://doi.org/10.3390/ph5111236
APA StyleSarris, E. G., Saif, M. W., & Syrigos, K. N. (2012). The Biological Role of PI3K Pathway in Lung Cancer. Pharmaceuticals, 5(11), 1236-1264. https://doi.org/10.3390/ph5111236