The Role of Transport Mechanisms in Mycobacterium Tuberculosis Drug Resistance and Tolerance

Abstract
:Abbreviations
| ABC | ATP-Binding Cassette transporters |
| MFS | Major Facilitator Superfamily transporters |
| RND | Resistance-Nodulation-Cell Division transporters |
| ATP | Adenosine triphosphate |
| CCCP | Carbonyl cyanide m-chlorophenyl hydrazone |
| PMF | Proton Motive Force |
1. Introduction
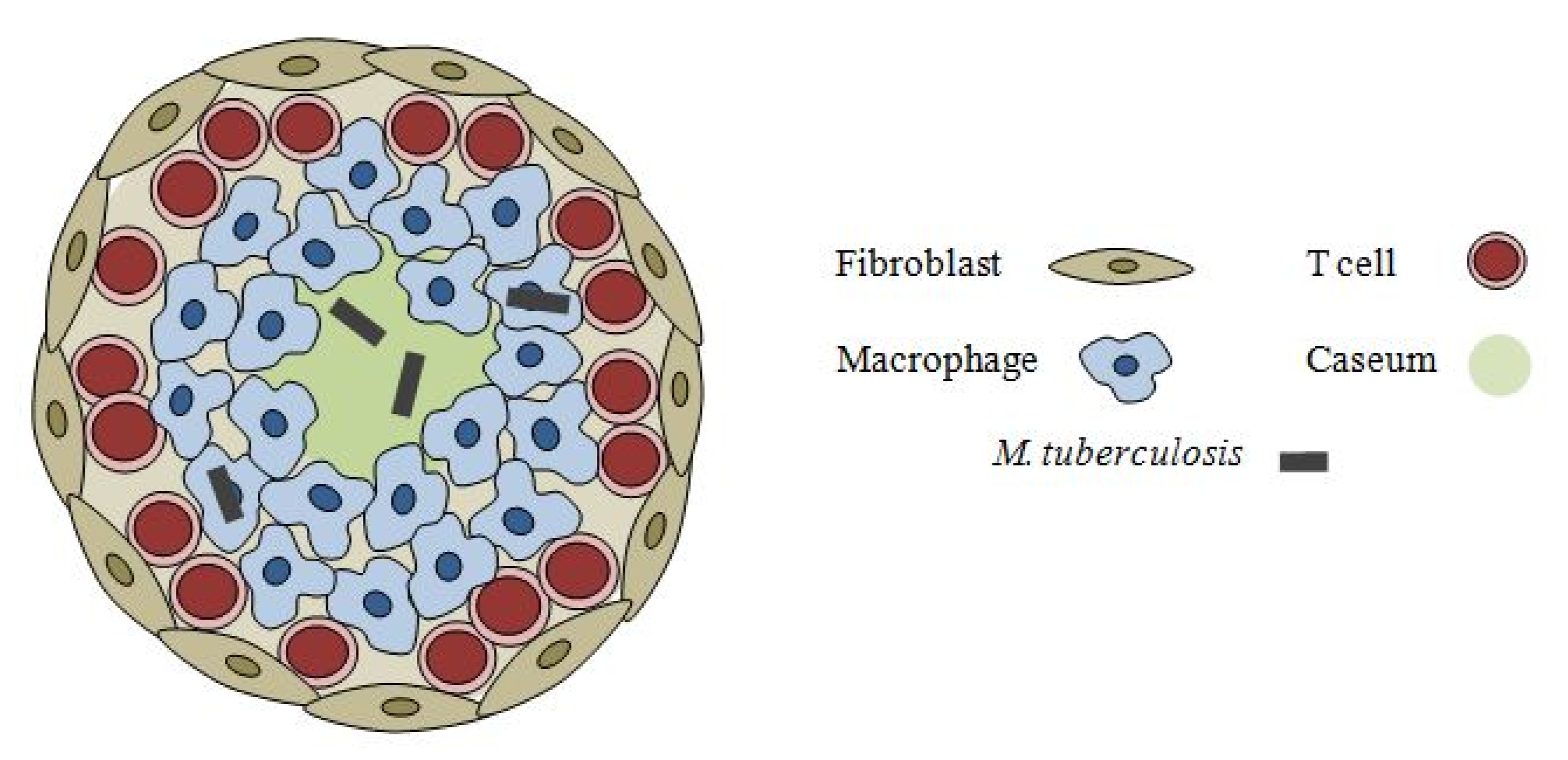
2. Mycobacterial Cell Wall: The Permeability Barrier
3. Passive Diffusion: The Hydrophobic Pathway
4. Facilitated Diffusion: The Porins

{kind=link}
| Species | Deleted Porin | Drug | Fold-reduction in Drug Uptake | Fold-increase in MIC | Reference |
|---|---|---|---|---|---|
| M. smegmatis | MspA & C double deletion | Ampicillin | - | 16 | [25] |
| Cephaloridine | - | 8 | |||
| Chloramphenicol | 1–2 | 4 | |||
| Norfloxacin | 4 | 2 | |||
| MspA | Ampicillin | - | 16 | [31] | |
| Cephaloridine | 9 | 8 | |||
| Vancomycin | - | 10 | |||
| MspA | Cephaloridine | 9 | - | [30] |
| Species | Porin | Channel Width (nm) | Single-Channel Conductance (nS) | Exclusion Limit (Da) | Reference |
|---|---|---|---|---|---|
| M. tuberculosis | OmpATb | 1.4–1.8 | 0.7 | Undetermined | [35] |
| M. smegmatis | MspA | 2.2–2.4 | 4.6 | Undetermined | [44,45] |
| E. coli | OmpA | 0.6–0.7 | 0.14 (at 37 °C) | 550* | [46,47,48] |
| OmpF | 1.2 | 0.82 | |||
| P. aeruginosa | OprF | 2.2 | 5 | 6000 | [49,50] |
| S. typhimurium | Not specified | 1.4 | 2.3 | 700 | [46,51] |
5. Active Transport Processes: Influx and Efflux
5.1. Influx Transporters
5.2. Efflux Pumps
5.2.1. Resistance Phenotype I—Natural Abundance
| Pump | Gene | Transporter Family | Known Substrates | Known Inhibitors | Energy Source | Mycobacteria | Reference | |
|---|---|---|---|---|---|---|---|---|
| - | rv2686c- | ABC | Fluoroquinolones | Verapamil | ATP | M. tuberculosis | [69] | |
| rv2687c- | Reserpine | |||||||
| rv2688c | CCCP | |||||||
| - | rv1218c | ABC | Novobiocins | Verapamil | ATP | M. tuberculosis | [70] | |
| Pyrazolones | Reserpine | |||||||
| Pyrroles | CCCP | |||||||
| - | rv0194 | ABC | Ampicillin | Reserpine | ATP | M. tuberculosis | [25] | |
| Chloramphenicol | ||||||||
| Streptomycin | ||||||||
| Novobiocin | ||||||||
| DrrAB | drrA-drrB | ABC | Doxorubicin | Verapamil | ATP | M. tuberculosis | [71] | |
| Reserpine | ||||||||
| MmpL7 | mmpL7 | RND | Isoniazid | ReserpineCCCP | PMF | M. tuberculosis | [72] | |
| Tap | rv1258c | MFS | Tetracycline | Piperine | PMF | M. tuberculosis | [73,74,75] | |
| Rifampicin | M. fortuitum | |||||||
| P55b | rv1410c | MFS | Rifampicin | CCCP | PMF | M. tuberculosis | [76,77] | |
| Clofazimine | ||||||||
| Aminoglycosides | Valinomycin | M. bovis | ||||||
| Tetracycline | ||||||||
| JefA | rv2459 | MFS | Isoniazid | Verapamil | Not speculated | M. tuberculosis | [78] | |
| Ethambutol | ||||||||
| CCCP | ||||||||
| Streptomycin | ||||||||
| EfpA | rv2846c | MFS | Not determined | - | PMF | M. tuberculosis | [67,79] | |
| M. smegmatis | ||||||||
| M. leprae | ||||||||
| M. avium | ||||||||
| IniAa | iniA | - | Isoniazid | Reserpine | Not speculated | M. tuberculosis | [80] | |
| Ethambutol | ||||||||
| Mmr | rv3065 | SMR | CCCP | PMF | M. tuberculosis | [81,82] | ||
| Erythromycin | ||||||||
| Thioridazine | ||||||||
| Tet(V) | tet(V) | MFS | Tetracycline | CCCP | PMF | M. smegmatis | [81] | |
| M. fortuitum | ||||||||
| LfrA | lfrA | MFS | Fluoroquinolones | CCCP | PMF | M. smegmatis | [83] | |
| Doxorubicin |
5.2.2. Resistance Phenotype II—Induction of Expression
5.2.3. Resistance Phenotype III—Efflux Pump Mutations
6. Phenotypic Drug Tolerance
6.1. The NRP State
6.2. Cell Wall Thickening
6.3. Intracellular M. tuberculosis
7. Accumulation of Selected Drugs in M. tuberculosis
| Antibiotic | Molecular Weight | CLogP * | PSA (Å2) * | Target | IC50 (mg/L) | MIC90 (mg/L) | Accumulation Factor a | Hypothesized Transport Mechanism | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Pyrazinamide | 123.12 | −0.676 | 68.87 | Fatty acid sysnthase Ib | N.A. | 16–50 (pH5.5) | 5.4–6.2 | ATP-dependent | [121,127] |
| Isoniazid | 137.14 | −0.668 | 68.01 | Enoyl-acyl carrier protein reductase | N.A. | 0.02–0.2 | 4–5 | Passive Diffusion | [121,127] |
| Ciprofloxacin | 331.35 | −0.725 | 77.04 | DNA Gyrase | 3.2(M. smeg) | 1.0 | 3.3–4.1 | Passive Diffusion | [120,128] |
| Levofloxacin | 361.38 | −0.508 | 77.48 | DNA Gyrase | 3.0(M. smeg) | 0.5 | 1.1–1.3 | Passive Diffusion | [120,128] |
| Ofloxacin | 361.38 | −0.508 | 77.48 | DNA Gyrase | 7.9(M. smeg) | 0.5 | 2.2–2.7 | Passive Diffusion | [120,128] |
| Norfloxacin | 319.34 | −0.780 | 77.04 | DNA Gyrase | Information unavailable | 2 | 1.8–2.2 | Passive Diffusion | [120] |
| Moxifloxacin | 401.44 | −0.082 | 86.27 | DNA Gyrase | Information unavailable | 0.5 | 1–1.3 | Passive Diffusion | [120] |
| Ethambutol | 204.32 | 0.119 | 64.52 | Arabinosyl-transferase | Information unavailable | 1– 5 | <1 | Passive Diffusion | [127,129] |
| Rifampicin | 822.96 | 3.710 | 220.15 | RNA polymerase | 0.07(M. avium) | 0.05–1 | 22.3–27.1 | Passive Diffusion | [120] |
8. Future Perspectives
References
- World Health Organization, Global Tuberculosis Control; WHO Press: Geneva, Switzerland, 2011.
- World Health Organization, Tuberculosis MDR-TB & XDR-TB Progress report; WHO Press: Geneva, Switzerland, 2011.
- Mitchison, D.A. The action of antituberculosis drugs in short-course chemotherapy. Tubercle. 1985, 66, 219–225. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; Rubin, E.J.; Freundlich, J.S. Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2008, 6, 41–52. [Google Scholar] [CrossRef]
- Chao, M.C.; Rubin, E.J. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu. Rev. Microbiol. 2010, 64, 293–311. [Google Scholar] [CrossRef]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996, 64, 2062–2069. [Google Scholar]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar]
- Kjellsson, M.C.; Via, L.E.; Goh, A.; Weiner, D.; Low, K.M.; Kern, S.; Pillai, G.; Barry, C.E., III.; Dartois, V. Penetration of anti-tuberculosis agents in rabbit pulmonary lesions: a pharmacokinetic evaluation. Antimicrob. Agents Chemother. 2011, 56, 446–457. [Google Scholar]
- Coates, A.; Hu, Y.; Bax, R.; Page, C. The future challenges facing the development of new antimicrobial drugs. Nat. Rev. Drug Discov. 2002, 1, 895–910. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Bostian, K.A. Practical applications and feasibility of efflux pump inhibitors in the clinic--a vision for applied use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar]
- Jarlier, V.; Nikaido, H. Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett. 1994, 123, 11–18. [Google Scholar]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb). 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Daffe, M.; Draper, P. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 1998, 39, 131–203. [Google Scholar]
- Barry, C.E.; Crick, D.C.; McNeil, M.R. Targeting the formation of the cell wall core of M.tuberculosis. Infect. Disord. Drug Targets. 2007, 7, 182–202. [Google Scholar] [CrossRef]
- Barry, C.E., 3rd. Interpreting cell wall 'virulence factors' of Mycobacterium tuberculosis. Trends Microbiol. 2001, 9, 237–241. [Google Scholar] [CrossRef]
- Mishra, A.K.; Driessen, N.N.; Appelmelk, B.J.; Besra, G.S. Lipoarabinomannan and related glycoconjugates: structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol. Rev. 2011, 35, 1126–1157. [Google Scholar] [CrossRef]
- Jarlier, V.; Nikaido, H. Permeability barrier to hydrophilic solutes in Mycobacterium chelonei. J. Bacteriol. 1990, 172, 1418–1423. [Google Scholar]
- Connell, N.D.; Nikaido, H. In Tuberculosis: Pathogenesis, Protection, and Control; Bloom, B.R., Ed.; ASM Press: Washington, DC, USA, 1994; pp. 333–352, Chapter 22. [Google Scholar]
- Bhamidi, S.; Scherman, M.S.; Jones, V.; Crick, D.C.; Belisle, J.T.; Brennan, P.J.; McNeil, M.R. Detailed structural and quantitative analysis reveals the spatial organization of the cell walls of in vivo grown Mycobacterium leprae and in vitro grown Mycobacterium tuberculosis. J. Biol. Chem. 2011, 286, 23168–23177. [Google Scholar]
- Brennan, P.J.; Nikaido, H. The envelope of mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Daffe, M.; Etienne, G. The capsule of Mycobacterium tuberculosis and its implications for pathogenicity. Tuber. Lung Dis. 1999, 79, 153–169. [Google Scholar] [CrossRef]
- Mailaender, C.; Reiling, N.; Engelhardt, H.; Bossmann, S.; Ehlers, S.; Niederweis, M. The MspA porin promotes growth and increases antibiotic susceptibility of both Mycobacterium bovis BCG and Mycobacterium tuberculosis. Microbiology. 2004, 150, 853–864. [Google Scholar] [CrossRef]
- Liu, J.; Barry, C.E., 3rd; Besra, G.S.; Nikaido, H. Mycolic acid structure determines the fluidity of the mycobacterial cell wall. J. Biol. Chem. 1996, 271, 29545–29551. [Google Scholar]
- Danilchanka, O.; Pavlenok, M.; Niederweis, M. Role of porins for uptake of antibiotics by Mycobacterium smegmatis. Antimicrob. Agents Chemother. 2008, 52, 3127–3134. [Google Scholar]
- Trias, J.; Jarlier, V.; Benz, R. Porins in the cell wall of mycobacteria. Science. 1992, 258, 1479–1481. [Google Scholar]
- Nikaido, H.; Rosenberg, E.Y. Effect on solute size on diffusion rates through the transmembrane pores of the outer membrane of Escherichia coli. J. Gen. Physiol. 1981, 77, 121–135. [Google Scholar] [CrossRef]
- Nikaido, H.; Vaara, M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [Google Scholar]
- Niederweis, M.; Ehrt, S.; Heinz, C.; Klocker, U.; Karosi, S.; Swiderek, K.M.; Riley, L.W.; Benz, R. Cloning of the mspA gene encoding a porin from Mycobacterium smegmatis. Mol. Microbiol. 1999, 33, 933–945. [Google Scholar] [CrossRef]
- Stahl, C.; Kubetzko, S.; Kaps, I.; Seeber, S.; Engelhardt, H.; Niederweis, M. MspA provides the main hydrophilic pathway through the cell wall of Mycobacterium smegmatis. Mol. Microbiol. 2001, 40, 451–464. [Google Scholar] [CrossRef]
- Stephan, J.; Mailaender, C.; Etienne, G.; Daffe, M.; Niederweis, M. Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob. Agents Chemother. 2004, 48, 4163–4170. [Google Scholar] [CrossRef]
- Niederweis, M. Mycobacterial porins--new channel proteins in unique outer membranes. Mol. Microbiol. 2003, 49, 1167–1177. [Google Scholar] [CrossRef]
- Mahfoud, M.; Sukumaran, S.; Hulsmann, P.; Grieger, K.; Niederweis, M. Topology of the porin MspA in the outer membrane of Mycobacterium smegmatis. J. Biol. Chem. 2006, 281, 5908–5915. [Google Scholar]
- Niederweis, M. The Mycobacterial Cell Envelope; Daffe, M., Reyrat, J.M., Eds.; ASM Press: Washington, DC, U.S.A, 2008; pp. 153–166, Chapter 9. [Google Scholar]
- Senaratne, R.H.; Mobasheri, H.; Papavinasasundaram, K.G.; Jenner, P.; Lea, E.J.; Draper, P. Expression of a gene for a porin-like protein of the OmpA family from Mycobacterium tuberculosis H37Rv. J. Bacteriol. 1998, 180, 3541–3547. [Google Scholar]
- Teriete, P.; Yao, Y.; Kolodzik, A.; Yu, J.; Song, H.; Niederweis, M.; Marassi, F.M. Mycobacterium tuberculosis Rv0899 adopts a mixed alpha/beta-structure and does not form a transmembrane beta-barrel. Biochemistry. 2010, 49, 2768–2777. [Google Scholar]
- Yang, Y.; Auguin, D.; Delbecq, S.; Dumas, E.; Molle, G.; Molle, V.; Roumestand, C.; Saint, N. Structure of the Mycobacterium tuberculosis OmpATb protein: a model of an oligomeric channel in the mycobacterial cell wall. Proteins 2011, 79, 645–661. [Google Scholar] [CrossRef]
- Song, H.; Huff, J.; Janik, K.; Walter, K.; Keller, C.; Ehlers, S.; Bossmann, S.H.; Niederweis, M. Expression of the ompATb operon accelerates ammonia secretion and adaptation of Mycobacterium tuberculosis to acidic environments. Mol. Microbiol. 2011, 80, 900–918. [Google Scholar] [CrossRef]
- Rohde, K.; Yates, R.M.; Purdy, G.E.; Russell, D.G. Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 2007, 219, 37–54. [Google Scholar] [CrossRef]
- Marassi, F.M. Mycobacterium tuberculosis Rv0899 defines a family of membrane proteins widespread in nitrogen-fixing bacteria. Proteins 2011, 79, 2946–2955. [Google Scholar] [CrossRef]
- Song, H.; Sandie, R.; Wang, Y.; Andrade-Navarro, M.A.; Niederweis, M. Identification of outer membrane proteins of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2008, 88, 526–544. [Google Scholar] [CrossRef]
- Siroy, A.; Mailaender, C.; Harder, D.; Koerber, S.; Wolschendorf, F.; Danilchanka, O.; Wang, Y.; Heinz, C.; Niederweis, M. Rv1698 of Mycobacterium tuberculosis represents a new class of channel-forming outer membrane proteins. J. Biol. Chem. 2008, 283, 17827–17837. [Google Scholar]
- Sharbati, S.; Schramm, K.; Rempel, S.; Wang, H.; Andrich, R.; Tykiel, V.; Kunisch, R.; Lewin, A. Characterisation of porin genes from Mycobacterium fortuitum and their impact on growth. BMC Microbiol. 2009, 9, 31. [Google Scholar]
- Heinz, C.; Niederweis, M. Selective extraction and purification of a mycobacterial outer membrane protein. Anal. Biochem. 2000, 285, 113–120. [Google Scholar]
- Engelhardt, H.; Heinz, C.; Niederweis, M. A tetrameric porin limits the cell wall permeability of Mycobacterium smegmatis. J. Biol. Chem. 2002, 277, 37567–37572. [Google Scholar]
- Nakae, T. Identification of the outer membrane protein of E. coli that produces transmembrane channels in reconstituted vesicle membranes. Biochem. Biophys. Res. Commun. 1976, 71, 877–884. [Google Scholar] [CrossRef]
- Benz, R.; Orlik, F. Bacterial and Eukaryotic Porins: Structure, Function, Mechanism; Benz, R., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 183–212, Chapter 8. [Google Scholar]
- Saint, N.; Lou, K.L.; Widmer, C.; Luckey, M.; Schirmer, T.; Rosenbusch, J.P. Structural and functional characterization of OmpF porin mutants selected for larger pore size. II. Functional characterization. J. Biol. Chem. 1996, 271, 20676–20680. [Google Scholar]
- Hancock, R.E.; Decad, G.M.; Nikaido, H. Identification of the protein producing transmembrane diffusion pores in the outer membrane of Pseudomonas aeruginosa PA01. Biochim. Biophys. Acta. 1979, 554, 323–331. [Google Scholar] [CrossRef]
- Benz, R.; Hancock, R.E. Properties of the large ion-permeable pores formed from protein F of Pseudomonas aeruginosa in lipid bilayer membranes. Biochim. Biophys. Acta. 1981, 646, 298–308. [Google Scholar]
- Benz, R.; Ishii, J.; Nakae, T. Determination of ion permeability through the channels made of porins from the outer membrane of Salmonella typhimurium in lipid bilayer membranes. The J. Membr. Biol. 1980, 56, 19–29. [Google Scholar]
- Naenna, P.; Noisumdaeng, P.; Pongpech, P.; Tribuddharat, C. Detection of outer membrane porin protein, an imipenem influx channel, in Pseudomonas aeruginosa clinical isolates. Southeast Asian J. Trop. Med. Public Health. 2010, 41, 614–624. [Google Scholar]
- Kishii, R.; Takei, M. Relationship between the expression of ompF and quinolone resistance in Escherichia coli. J. Infect. Chemother. 2009, 15, 361–366. [Google Scholar] [CrossRef]
- Tavio, M.M.; Vila, J.; Ruiz, J.; Martin-Sanchez, A.M.; Jimenez de Anta, M.T. Mechanisms involved in the development of resistance to fluoroquinolones in Escherichia coli isolates. J. Antimicrob. Chemother. 1999, 44, 735–742. [Google Scholar] [CrossRef]
- Davin-Regli, A.; Bolla, J.M.; James, C.E.; Lavigne, J.P.; Chevalier, J.; Garnotel, E.; Molitor, A.; Pages, J.M. Membrane permeability and regulation of drug "influx and efflux" in enterobacterial pathogens. Curr. Drug Targets. 2008, 9, 750–759. [Google Scholar] [CrossRef]
- Braibant, M.; Gilot, P.; Content, J. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 2000, 24, 449–467. [Google Scholar]
- Louw, G.E.; Warren, R.M.; Gey van Pittius, N.C.; McEvoy, C.R.; Van Helden, P.D.; Victor, T.C. A balancing act: efflux/influx in mycobacterial drug resistance. Antimicrobial. Agents Chemother. 2009, 53, 3181–3189. [Google Scholar]
- Davidson, A.L.; Chen, J. ATP-binding cassette transporters in bacteria. Annu. Rev. Biochem. 2004, 73, 241–268. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S. V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998, 393, 537–544. [Google Scholar]
- Linton, K.J.; Higgins, C.F. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 1998, 28, 5–13. [Google Scholar]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: an update. Drugs. 2009, 69, 1555–1623. [Google Scholar] [CrossRef]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria. Drugs. 2004, 64, 159–204. [Google Scholar]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007, 446, 749–757. [Google Scholar] [CrossRef]
- Hollenstein, K.; Dawson, R.J.; Locher, K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [Google Scholar]
- Mazurkiewicz, P.; Driessen, A.J.; Konings, W.N. What do proton motive force driven multidrug resistance transporters have in common? Curr. Issues Mol. Biol. 2005, 7, 7–21. [Google Scholar]
- Paulsen, I.T.; Nguyen, L.; Sliwinski, M.K.; Rabus, R.; Saier, M.H., Jr. Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes. J. Mol. Biol. 2000, 301, 75–100. [Google Scholar]
- De Rossi, E.; Ainsa, J.A.; Riccardi, G. Role of mycobacterial efflux transporters in drug resistance: an unresolved question. FEMS Microbiol. Rev. 2006, 30, 36–52. [Google Scholar] [CrossRef]
- Zimic, M.; Fuentes, P.; Gilman, R.H.; Gutierrez, A.H.; Kirwan, D.; Sheen, P. Pyrazinoic acid efflux rate in Mycobacterium tuberculosis is a better proxy of pyrazinamide resistance. Tuberculosis (Edinb). 2012, 92, 84–91. [Google Scholar] [CrossRef]
- Pasca, M.R.; Guglierame, P.; Arcesi, F.; Bellinzoni, M.; De Rossi, E.; Riccardi, G. Rv2686c-Rv2687c-Rv2688c, an ABC fluoroquinolone efflux pump in Mycobacterium tuberculosis. Antimicro. b Agents Chemother. 2004, 48, 3175–3178. [Google Scholar]
- Balganesh, M.; Kuruppath, S.; Marcel, N.; Sharma, S.; Nair, A.; Sharma, U. Rv1218c, an ABC transporter of Mycobacterium tuberculosis with implications in drug discovery. Antimicrob. Agents Chemother. 2010, 54, 5167–5172. [Google Scholar] [CrossRef]
- Choudhuri, B.S.; Bhakta, S.; Barik, R.; Basu, J.; Kundu, M.; Chakrabarti, P. Overexpression and functional characterization of an ABC (ATP-binding cassette) transporter encoded by the genes drrA and drrB of Mycobacterium tuberculosis. Biochem. J. 2002, 367, 279–285. [Google Scholar] [CrossRef]
- Pasca, M.R.; Guglierame, P.; De Rossi, E.; Zara, F.; Riccardi, G. mmpL7 gene of Mycobacterium tuberculosis is responsible for isoniazid efflux in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 2005, 49, 4775–4777. [Google Scholar]
- Ainsa, J.A.; Blokpoel, M.C.; Otal, I.; Young, D.B.; De Smet, K.A.; Martin, C. Molecular cloning and characterization of Tap, a putative multidrug efflux pump present in Mycobacterium fortuitum and Mycobacterium tuberculosis. J. Bacteriol. 1998, 180, 5836–5843. [Google Scholar]
- Ramon-Garcia, S.; Martin, C.; Ainsa, J.A.; De Rossi, E. Characterization of tetracycline resistance mediated by the efflux pump Tap from Mycobacterium fortuitum. J. Antimicrob. Chemother. 2006, 57, 252–259. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, M.; Nargotra, A.; Koul, S.; Khan, I.A. Piperine as an inhibitor of Rv1258c, a putative multidrug efflux pump of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2010, 65, 1694–1701. [Google Scholar]
- Ramon-Garcia, S.; Martin, C.; Thompson, C.J.; Ainsa, J.A. Role of the Mycobacterium tuberculosis P55 efflux pump in intrinsic drug resistance, oxidative stress responses, and growth. Antimicrob. Agents Chemother. 2009, 53, 3675–3682. [Google Scholar] [CrossRef]
- Silva, P.E.; Bigi, F.; Santangelo, M.P.; Romano, M.I.; Martin, C.; Cataldi, A.; Ainsa, J.A. Characterization of P55, a multidrug efflux pump in Mycobacterium bovis and Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2001, 45, 800–804. [Google Scholar]
- Gupta, A.K.; Katoch, V.M.; Chauhan, D.S.; Sharma, R.; Singh, M.; Venkatesan, K.; Sharma, V.D. Microarray analysis of efflux pump genes in multidrug-resistant Mycobacterium tuberculosis during stress induced by common anti-tuberculous drugs. Microb. Drug Resist. 2010, 16, 21–28. [Google Scholar] [CrossRef]
- Doran, J.L.; Pang, Y.; Mdluli, K.E.; Moran, A.J.; Victor, T.C.; Stokes, R.W.; Mahenthiralingam, E.; Kreiswirth, B.N.; Butt, J.L.; Baron, G.S.; Treit, J.D.; Kerr, V.J.; Van Helden, P.D.; Roberts, M.C.; Nano, F.E. Mycobacterium tuberculosis efpA encodes an efflux protein of the QacA transporter family. Clin. Diagn. Lab. Immunol. 1997, 4, 23–32. [Google Scholar]
- Colangeli, R.; Helb, D.; Sridharan, S.; Sun, J.; Varma-Basil, M.; Hazbon, M.H.; Harbacheuski, R.; Megjugorac, N.J.; Jacobs, W.R. , Jr.; Holzenburg, A.; Sacchettini, J.C.; Alland, D. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol. Microbiol. 2005, 55, 1829–1840. [Google Scholar] [CrossRef]
- De Rossi, E.; Branzoni, M.; Cantoni, R.; Milano, A.; Riccardi, G.; Ciferri, O. mmr, a Mycobacterium tuberculosis gene conferring resistance to small cationic dyes and inhibitors. J. Bacteriol. 1998, 180, 6068–6071. [Google Scholar]
- Dutta, N.K.; Mehra, S. ; Kaushal, D.A. Mycobacterium tuberculosis sigma factor network responds to cell-envelope damage by the promising anti-mycobacterial thioridazine. PLoS ONE. 2010, 5, e10069. [Google Scholar]
- Liu, J.; Takiff, H.E.; Nikaido, H. Active efflux of fluoroquinolones in Mycobacterium smegmatis mediated by LfrA, a multidrug efflux pump. J. Bbacteriol. 1996, 178, 3791–3795. [Google Scholar]
- Drage, M.G.; Tsai, H.C.; Pecora, N.D.; Cheng, T.Y.; Arida, A.R.; Shukla, S.; Rojas, R.E.; Seshadri, C.; Moody, D.B.; Boom, W. H.; Sacchettini, J.C.; Harding, C.V. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat. Struct. Mol. Biol. 2010, 17, 1088–1095. [Google Scholar]
- Bianco, M.V.; Blanco, F.C.; Imperiale, B.; Forrellad, M.A.; Rocha, R.V.; Klepp, L.I.; Cataldi, A.A.; Morcillo, N.; Bigi, F. Role of P27 -P55 operon from Mycobacterium tuberculosis in the resistance to toxic compounds. BMC Infect. Dis. 2011, 11, 195. [Google Scholar] [CrossRef]
- Nguyen, L.; Thompson, C.J. Foundations of antibiotic resistance in bacterial physiology: the mycobacterial paradigm. Trends Microbiol. 2006, 14, 304–312. [Google Scholar]
- de Steenwinkel, J.E.; de Knegt, G.J.; ten Kate, M.T.; van Belkum, A.; Verbrugh, H.A.; Kremer, K.; van Soolingen, D.; Bakker-Woudenberg, I.A. Time-kill kinetics of anti-tuberculosis drugs, and emergence of resistance, in relation to metabolic activity of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2010, 65, 2582–2589. [Google Scholar] [CrossRef]
- Machado, D.; Couto, I.; Perdigao, J.; Rodrigues, L.; Portugal, I.; Baptista, P.; Veigas, B.; Amaral, L.; Viveiros, M. Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis. PLoS ONE. 2012, 7, e34538. [Google Scholar]
- Rodrigues, L.; Machado, D.; Couto, I.; Amaral, L.; Viveiros, M. Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infect. Genet. Evol. 2012, 12, 695–700. [Google Scholar]
- Wilson, M.; DeRisi, J.; Kristensen, H.H.; Imboden, P.; Rane, S.; Brown, P.O.; Schoolnik, G.K. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 12833–12838. [Google Scholar]
- Jiang, X.; Zhang, W.; Zhang, Y.; Gao, F.; Lu, C.; Zhang, X.; Wang, H. Assessment of efflux pump gene expression in a clinical isolate Mycobacterium tuberculosis by real-time reverse transcription PCR. Microb. Drug Resist. 2008, 14, 7–11. [Google Scholar] [CrossRef]
- Adams, K.N.; Takaki, K.; Connolly, L.E.; Wiedenhoft, H.; Winglee, K.; Humbert, O.; Edelstein, P.H.; Cosma, C.L.; Ramakrishnan, L. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011, 145, 39–53. [Google Scholar]
- Louw, G.E.; Warren, R.M.; Gey van Pittius, N.C.; Leon, R.; Jimenez, A.; Hernandez-Pando, R.; McEvoy, C. R.; Grobbelaar, M.; Murray, M.; van Helden, P.D.; Victor, T.C. Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am. J. Respir. Crit. Care Med. 2011, 184, 269–276. [Google Scholar] [CrossRef]
- Nikaido, E.; Giraud, E.; Baucheron, S.; Yamasaki, S.; Wiedemann, A.; Okamoto, K.; Takagi, T.; Yamaguchi, A.; Cloeckaert, A.; Nishino, K. Effects of indole on drug resistance and virulence of Salmonella enterica serovar Typhimurium revealed by genome-wide analyses. Gut. pathogens. 2012, 4, 5. [Google Scholar]
- McPhee, J.B.; Tamber, S.; Brazas, M.D.; Lewenza, S.; Hancock, R.E.W. In Antimicrobial Drug Resistance; Mayers , D.L., Ed.; Humana Press: New York, NY, U.S.A, 2009; pp. 97–110, Chapter 9. [Google Scholar]
- Warner, D.M.; Shafer, W.M.; Jerse, A.E. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol. Microbiol. 2008, 70, 462–478. [Google Scholar] [CrossRef]
- Maseda, H.; Hashida, Y.; Shirai, A.; Omasa, T.; Nakae, T. Mutation in the sdeS gene promotes expression of the sdeAB efflux pump genes and multidrug resistance in Serratia marcescens. Antimicrob. Agents Chemother. 2011, 55, 2922–2926. [Google Scholar] [CrossRef]
- Kehrenberg, C.; Cloeckaert, A.; Klein, G.; Schwarz, S. Decreased fluoroquinolone susceptibility in mutants of Salmonella serovars other than Typhimurium: detection of novel mutations involved in modulated expression of ramA and soxS. J. Antimicrob. Chemother. 2009, 64, 1175–1180. [Google Scholar]
- Ramaswamy, S.V.; Reich, R.; Dou, S.J.; Jasperse, L.; Pan, X.; Wanger, A.; Quitugua, T.; Graviss, E.A. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 1241–1250. [Google Scholar] [CrossRef]
- Telenti, A.; Imboden, P.; Marchesi, F.; Lowrie, D.; Cole, S.; Colston, M.J.; Matter, L.; Schopfer, K.; Bodmer, T. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet. 1993, 341, 647–650. [Google Scholar]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Rao, S.P.; Pethe, K.; Dick, T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology. 2010, 156, 81–87. [Google Scholar] [CrossRef]
- Zhang, Y. Persistent and dormant tubercle bacilli and latent tuberculosis. Front. Biosci. 2004, 9, 1136–1156. [Google Scholar]
- Herbert, D.; Paramasivan, C.N.; Venkatesan, P.; Kubendiran, G.; Prabhakar, R.; Mitchison, D.A. Bactericidal action of ofloxacin, sulbactam-ampicillin, rifampin, and isoniazid on logarithmic- and stationary-phase cultures of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1996, 40, 2296–2299. [Google Scholar]
- Xie, Z.; Siddiqi, N.; Rubin, E.J. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 2005, 49, 4778–4780. [Google Scholar]
- Seiler, P.; Ulrichs, T.; Bandermann, S.; Pradl, L.; Jorg, S.; Krenn, V.; Morawietz, L.; Kaufmann, S.H.; Aichele, P. Cell-wall alterations as an attribute of Mycobacterium tuberculosis in latent infection. J. Infect. Dis. 2003, 188, 1326–1331. [Google Scholar] [CrossRef]
- Boon, C.; Dick, T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J. Bacteriol. 2002, 184, 6760–6767. [Google Scholar]
- Park, H.D.; Guinn, K.M.; Harrell, M.I.; Liao, R.; Voskuil, M.I.; Tompa, M.; Schoolnik, G.K.; Sherman, D.R. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 2003, 48, 833–843. [Google Scholar] [CrossRef]
- Boon, C.; Dick, T. How Mycobacterium tuberculosis goes to sleep: the dormancy survival regulator DosR a decade later. Future Microbiol. 2012, 7, 513–518. [Google Scholar]
- Muttucumaru, D.G.; Roberts, G.; Hinds, J.; Stabler, R.A.; Parish, T. Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberculosis (Edinb). 2004, 84, 239–246. [Google Scholar] [CrossRef]
- Ramon-Garcia, S.; Mick, V.; Dainese, E.; Martin, C.; Thompson, C.J.; De Rossi, E.; Manganelli, R.; Ainsa, J.A. Functional and genetic characterization of the tap efflux pump in Mycobacterium bovis BCG. Antimicrob. Agents Chemother. 2012, 56, 2074–2083. [Google Scholar]
- De la Cruz, M.A.; Calva, E. The complexities of porin genetic regulation. J. Mol. Microbiol. Biotechnol. 2010, 18, 24–36. [Google Scholar] [CrossRef]
- Hoffmann, C.; Leis, A.; Niederweis, M.; Plitzko, J.M.; Engelhardt, H. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3963–3967. [Google Scholar]
- Zuber, B.; Chami, M.; Houssin, C.; Dubochet, J.; Griffiths, G.; Daffe, M. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 2008, 190, 5672–5680. [Google Scholar]
- Cunningham, A.F.; Spreadbury, C.L. Mycobacterial stationary phase induced by low oxygen tension: cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J. Bacteriol. 1998, 180, 801–808. [Google Scholar]
- Velayati, A.A.; Farnia, P.; Masjedi, M.R.; Zhavnerko, G.K.; Merza, M.A.; Ghanavi, J.; Tabarsi, P.; Poleschuyk, N.N.; Ignatyev, G. Sequential adaptation in latent tuberculosis bacilli: observation by atomic force microscopy (AFM). Int. J. Clin. Exp. Med. 2011, 4, 193–199. [Google Scholar]
- Walburger, A.; Koul, A.; Ferrari, G.; Nguyen, L.; Prescianotto-Baschong, C.; Huygen, K.; Klebl, B.; Thompson, C.; Bacher, G.; Pieters, J. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science. 2004, 304, 1800–1804. [Google Scholar] [CrossRef]
- Meena, L.S.; Rajni. Survival mechanisms of pathogenic Mycobacterium tuberculosis H37Rv. FEBS J. 2010, 277, 2416–2427. [Google Scholar] [CrossRef]
- Piddock, L.J.; Ricci, V. Accumulation of five fluoroquinolones by Mycobacterium tuberculosis H37Rv. J. Antimicrob. Chemother. 2001, 48, 787–791. [Google Scholar]
- Raynaud, C.; Laneelle, M.A.; Senaratne, R.H.; Draper, P.; Laneelle, G.; Daffe, M. Mechanisms of pyrazinamide resistance in mycobacteria: importance of lack of uptake in addition to lack of pyrazinamidase activity. Microbiology. 1999, 145 ( Pt 6), 1359–1367. [Google Scholar]
- Bardou, F.; Raynaud, C.; Ramos, C.; Laneelle, M.A.; Laneelle, G. Mechanism of isoniazid uptake in Mycobacterium tuberculosis. Microbiology. 1998, 144, 2539–2544. [Google Scholar]
- Piddock, L.J.; Williams, K. J.; Ricci, V. Accumulation of rifampicin by Mycobacterium aurum, Mycobacterium smegmatis and Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2000, 45, 159–165. [Google Scholar]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000, 44, 235–249. [Google Scholar]
- Franklin, T.J.; Snow, G.A. In Biochemistry and Molecular Biology of Antimicrobial Drug Action; Franklin, T.J., Snow, G.A., Eds.; Springer: New York, NY, USA, 2005; pp. 121–134, Chapter 7. [Google Scholar]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature. 2011, 469, 483–490. [Google Scholar]
- Zhang, Y.; Yew, W.W. Mechanisms of drug resistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2009, 13, 1320–1330. [Google Scholar]
- Guillemin, I.; Sougakoff, W.; Cambau, E.; Revel-Viravau, V.; Moreau, N.; Jarlier, V. Purification and inhibition by quinolones of DNA gyrases from Mycobacterium avium, Mycobacterium smegmatis and Mycobacterium fortuitum bv. peregrinum. Microbiology. 1999, 145, 2527–2532. [Google Scholar]
- Beggs, W.H.; Auran, N.E. Uptake and binding of 14C-ethambutol by tubercle bacilli and the relation of binding to growth inhibition. Antimicrob. Agents Chemother. 1972, 2, 390–394. [Google Scholar]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef]
- Muehlbacher, J. InSilicoProfile, Version 3.2; Novartis: Basel, Switzerland, 2005. [Google Scholar]
- Vega, A.L.D.; Delcour, A.H. Polyamines decrease Escherichia coli outer membrane permeability. J. Bacteriol. 1996, 178, 3715–3721. [Google Scholar]
- Iyer, R.; Delcour, A.H. Complex inhibition of OmpF and OmpC bacterial porins by polyamines. J. Biol. Chem. 1997, 272, 18595–18601. [Google Scholar]
- Dela Vega, A.L.; Delcour, A.H. Cadaverine induces closing of E. coli porins. EMBO J. 1995, 14, 6058–6065. [Google Scholar]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011, 473, 216–220. [Google Scholar]
- Bryan, L.E.; Van den Elzen, H.M. Streptomycin accumulation in susceptible and resistant strains of Escherichia coli and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1976, 9, 928–938. [Google Scholar] [CrossRef]
- Bryan, L.E.; Van Den Elzen, H.M. Gentamicin accumulation by sensitive strains of Escherichia coli and Pseudomonas aeruginosa. J. Antibiot. 1975, 28, 696–703. [Google Scholar]
- Amaral, L.; Boeree, M.J.; Gillespie, S.H.; Udwadia, Z.F.; van Soolingen, D. Thioridazine cures extensively drug-resistant tuberculosis (XDR-TB) and the need for global trials is now! Int. J. Antimicrob. Agents. 2010, 35, 524–526. [Google Scholar] [CrossRef]
- Amaral, L.; Martins, M.; Viveiros, M.; Molnar, J.; Kristiansen, J.E. Promising therapy of XDR-TB/MDR-TB with thioridazine an inhibitor of bacterial efflux pumps. Curr. Drug Targets. 2008, 9, 816–819. [Google Scholar]
- Amaral, L.; Martins, M.; Viveiros, M. Enhanced killing of intracellular multidrug-resistant Mycobacterium tuberculosis by compounds that affect the activity of efflux pumps. J. Antimicrob. Chemother. 2007, 59, 1237–1246. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sarathy, J.P.; Dartois, V.; Lee, E.J.D. The Role of Transport Mechanisms in Mycobacterium Tuberculosis Drug Resistance and Tolerance. Pharmaceuticals 2012, 5, 1210-1235. https://doi.org/10.3390/ph5111210
Sarathy JP, Dartois V, Lee EJD. The Role of Transport Mechanisms in Mycobacterium Tuberculosis Drug Resistance and Tolerance. Pharmaceuticals. 2012; 5(11):1210-1235. https://doi.org/10.3390/ph5111210
Chicago/Turabian StyleSarathy, Jansy Passiflora, Véronique Dartois, and Edmund Jon Deoon Lee. 2012. "The Role of Transport Mechanisms in Mycobacterium Tuberculosis Drug Resistance and Tolerance" Pharmaceuticals 5, no. 11: 1210-1235. https://doi.org/10.3390/ph5111210
APA StyleSarathy, J. P., Dartois, V., & Lee, E. J. D. (2012). The Role of Transport Mechanisms in Mycobacterium Tuberculosis Drug Resistance and Tolerance. Pharmaceuticals, 5(11), 1210-1235. https://doi.org/10.3390/ph5111210