Carvedilol Attenuates Inflammatory-Mediated Cardiotoxicity in Daunorubicin-Induced Rats

Abstract
: Cardiotoxicity, which results from intense cardiac oxidative stress and inflammation, is the main limiting factor of the anthracyclines. Carvedilol, a beta blocker that is used as a multifunctional neurohormonal antagonist, has been shown to act not only as an anti-oxidant, but also as an anti-inflammatory drug. This study was designed to evaluate whether carvedilol exerts a protective role against inflammation-mediated cardiotoxicity in the daunorubicin (DNR)-induced rats. Carvedilol was administered orally to the rats every day for 6 weeks at a cumulative dose of 9 mg/kg body weight DNR. DNR significantly induced cardiac damage and worsened cardiac function as well as increased cardiac mast cell density, elevating the myocardial protein and mRNA expression levels of tumor necrosis factor-α, vascular cell adhesion molecule-1, inter-cellular adhesion molecule-1, nuclear factor kappa-B, cyclooxygenase-2, monocyte chemotactic protein -1 and interleukin -6 compared to that in the control group. Cotreatment with carvedilol significantly attenuated the myocardial protein and mRNA expression levels of these inflammatory markers, decreased cardiac mast cell density, improved histological cardiac damage and cardiac functions. In conclusion, inflammation plays a significant role in DNR-induced cardiotoxicity, and carvedilol contributes to cardioprotection against inflammation-mediated cardiotoxicity in DNR-induced rats through its anti-inflammatory mechanism.1. Introduction
Anthracyclines are a group of antibiotics that are among some of the most active chemotherapeutic agents. They are highly effective against a spectrum of malignancies, including both hematological and solid tumors (lymphoma, gastric cancer, small cell lung cancer, sarcoma, and breast cancer). Some of the commonly used anthracycline antibiotics include doxorubicin, daunorubicin (DNR), and epirubicin [1]. However, the clinical use of anthracyclines is largely limited by the adverse reactions accompanying their use. Besides their reversible and manageable adverse effects (e.g., nausea, myelosuppression, etc.), severe complications can also occur, such as carditoxicity, including congestive heart failure [2], increased interstitial myocardial fibrosis, and myocarditis/pericarditis [3]. These cumulative myocardial toxicities prevent the use of anthracyclines at the maximum myelotoxic doses for the optimal number of the courses [4].
The mechanism of anthracycline-induced cardiomyopathy is largely unknown, but a solid body of evidence indicates that oxidative stress and cardiac inflammation are involved. In patients that have undergone epirubicin treatment, early contractility impairment was significantly associated with high levels of reactive oxygen species (ROS) and markers of inflammation including interleukin (IL)-6 and its soluble receptor (sIL-6R) [5]. Earlier studies have illustrated that doxorubicin causes inflammatory reactions in the vicinity of heart tissues where it was found to increase the incidence of thrombus formation in the atria of mice [6]. In vitro data showed that doxorubicin affected both the viability and neutrophil adhesion of endothelial cells with clinically achievable concentrations [7]. It also induces inflammatory effects in the vasculature and the myocardium and elevates the levels of proinflammatory cytokines [7]. These inflammatory reactions may be mediated through the elevation of the expression levels of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [8], vascular cell adhesion molecule (VCAM)-1 [6], and tumor necrosis factor (TNF)-α [9]. We have previously reported that the expression of Fas ligand (Fas-L) was significantly increased in DNR-induced rats, which also displayed significant increases in apoptosis and cardiotoxicity [10]. Fas-L itself has been well-characterized as a death factor and plays an important role as an upstream molecule of many inflammatory factors that induce the expression of a variety of inflammatory cytokines in vivo [11]. Thus, previous studies have indicated that inflammatory reactions are involved in DNR-induced cardiotoxicity. However, further experimental studies are needed to verify and to elucidate these possibilities.
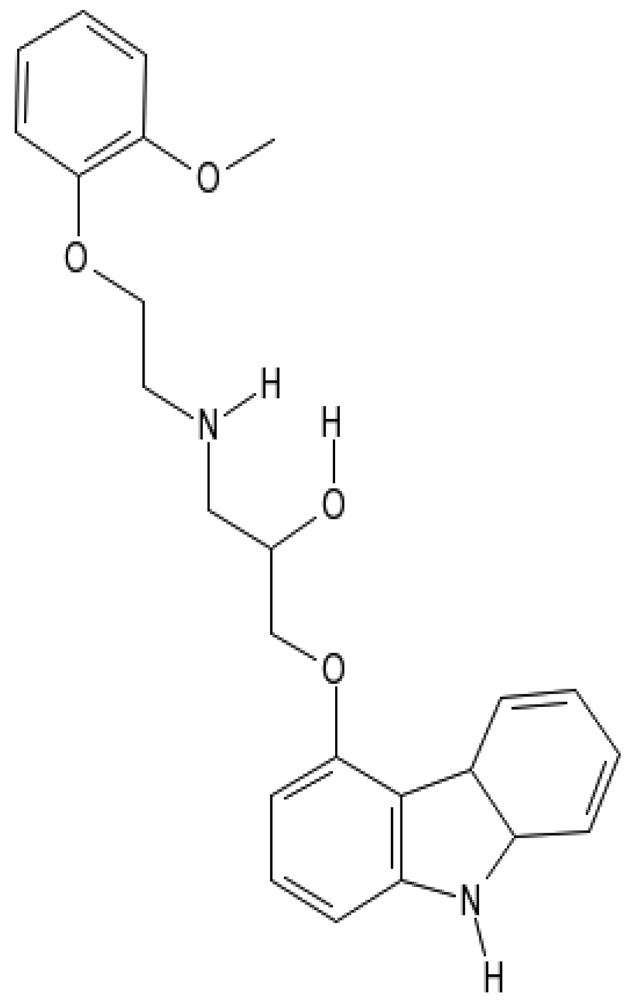
Carvedilol (Coreg™, Dilatrend™, Eucardic™, Carloc™, (±)-[3-(9H-carbazol-4-yloxy)-2-hydroxypropyl][2-(2-methoxyphenoxy)ethyl]amine, Figure 1), a multifunctional neurohormonal antagonist, has been shown to provide greater benefit than traditional β-blockers because of its antioxidant actions, which act in synergy with its nonspecific β- and α1- blocking effects [12]. Furthermore, carvedilol has been reported to have an anti-inflammatory effect by inhibiting IL-10 and IL-18 in patients with heart failure [13] and by inhibiting the expressions of VCAM-1, inter-cellular adhesion molecule (ICAM)-1, and IL-8 via NF-κB in the human endothelial cells [14]. Accumulating evidences have shown that carvedilol protects against anthracycline-induced cardiotoxicity through its anti-oxidant properties [15-17]. However, whether carvedilol exerts a protective role against inflammation-mediated cardiotoxicity in DNR-induced rats should be further studied. Thus, the present study was conducted to assess the possible role of inflammation-mediated cardiotoxicity in DNR-induced rats and to verify the anti-inflammatory effect of carvedilol against DNR-induced cardiotoxicity. In this study, we investigated the changes in left ventricular (LV) function, histopathology, and the levels of inflammatory markers in rats given DNR and DNR in combination with carvedilol.
2. Experimental
2.1. Drugs and Chemicals
Unless otherwise stated all reagents were of analytical grade and purchased from Sigma (Tokyo, Japan). DNR was kindly donated by Meiji Seika Kaisha Ltd, Tokyo, Japan. Carvedilol was donated by Daichi-Sankyo Pharmaceutical (Tokyo, Japan).
2.2. Animals and Medication
Eight-week-old male Sprague-Dawley rats were obtained from Charles River Japan Inc. (Kanagawa, Japan). On day 0, each animal received a single intravenous injection of DNR at a dose of 3 mg/kg (i.v.). The drug was administered in three equal injections at 48 h intervals for a period of a week to achieve an accumulative dose of 9 mg/kg, which is well documented to achieve cardiotoxicity and nephrotoxicity [10,18,19]. Twenty DNR-treated rats were randomly divided into two groups and received oral administration of carvedilol (30 mg/kg/day; group Carv; n = 10) or vehicle (group DNR; n = 10). Age-matched rats were injected with corresponding volumes of 0.9% NaCl and used as a control (group Control; n = 4). The dose of carvedilol was chosen on the basis of our preliminary study (data not shown). Administration of carvedilol was started on the same day as DNR administration and continued for 5 additional weeks after cessation of DNR administration (6 weeks total period). On day 41, the body weight (BW) was measured. Throughout the study, all animals were cared for in accordance with the guidelines of our institute and the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health.
2.3. Hemodynamic and Echocardiographic Study
After the end of the study period (6 weeks), rats were anesthetized with 2% halothane in O2 and subjected to surgical procedures to measure hemodynamic parameters. After the instrumentation, the concentration of halothane was reduced to 0.5% to record steady-state hemodynamic data. Hemodynamic parameters such as mean blood pressure (MBP), peak left ventricular pressure (LVP), LV end-diastolic pressure (LVEDP), and the rate of intra-ventricular pressure rise and decline (± dP/dt) were recorded as previously described [20].
Two-dimensional echocardiographic studies were performed under 0.5% halothane anesthesia using an echocardiographic machine equipped with a 7.5-MHz transducer (SSD-5500; Aloka, Tokyo, Japan). M-mode tracings were recorded from the epicardial surface of the right ventricle; the short axis view of the LV was recorded to measure the LV dimension in diastole (LVDd) and LV dimension in systole (LVDs). LV fractional shortening (FS) and ejection fraction (EF) were calculated and expressed as percentages. The study was performed in a blinded manner.
2.4. Histopathological Analysis
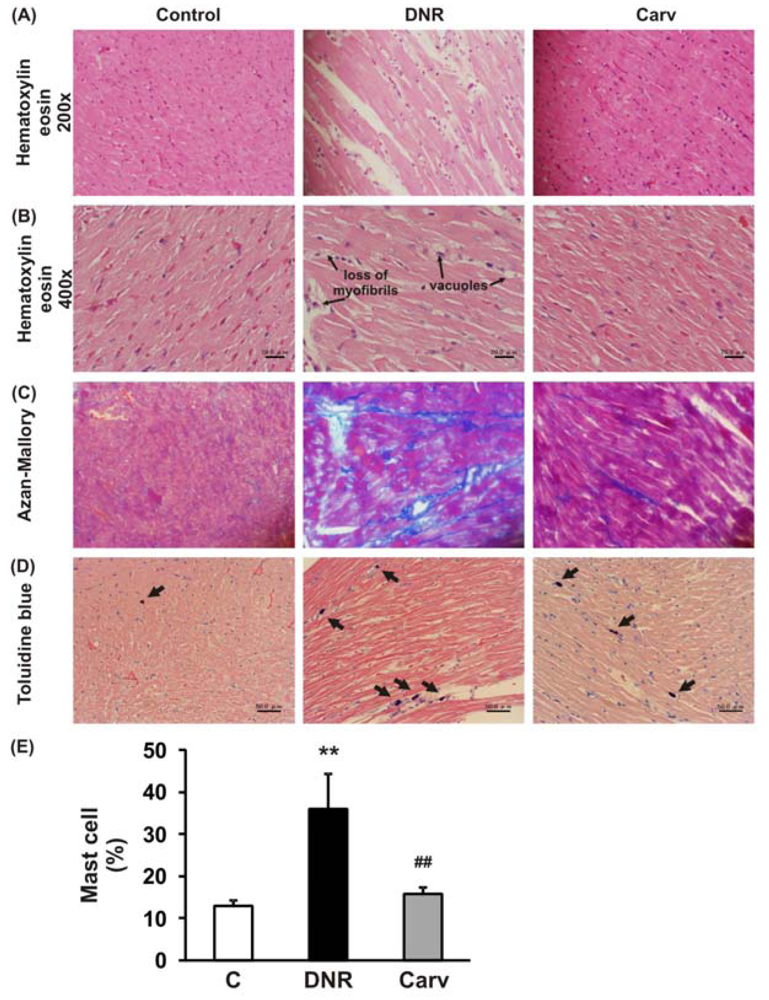
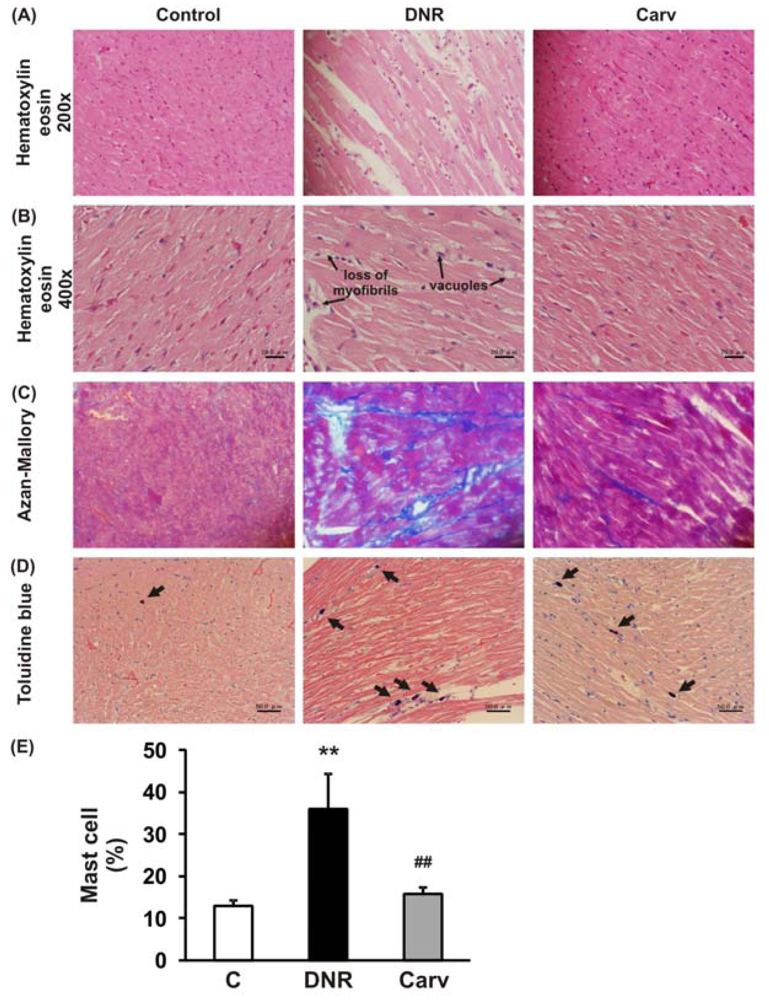
After the measurement of echocardiographic parameters, hearts were excised and weighed immediately (HW), and their ratios to BW (HW/BW) were calculated. Half of each heart was immediately snap-frozen in liquid nitrogen for subsequent protein extraction and enzymatic assays. The remaining excised hearts were cut into about 2-mm-thick transverse slices and fixed in 10% formalin. After being embedded in paraffin, several transverse sections were obtained from the ventricle and stained with hematoxylin-eosin (HE) for histological evaluation, and also stained with Azan-Mallory to demonstrate fibrosis in cardiac tissues. The frequency and the severity of lesions in the hearts were assessed semi-quantitatively as previously reported [21,22] by light microscopy using a scale where score 0, normal; 1, mild; 2, moderate; and 3, severe. The scoring criteria for myocardial lesions included the degree of myocyte vacuolization with respect to size and the number of vacuoles, degree of fibrosis, myocardial degeneration and interstitial edema and loss of myofibrils.
Histochemical staining with toluidine blue was performed to identify mast cells. For toluidine blue staining, slides of paraffinized sections of the mid-ventricles were dewaxed, rehydrated and incubated with 0.05% w/v toluidine blue for 30 min followed by counterstaining with 0.01% w/v eosin for 15 min. Mast cell density was quantified by counting the number of toluidine-blue positive mast cells per field (200×) in the whole heart section and shown as a percentage.
2.5. Protein Analysis by Western Blotting
Protein lysate was prepared from heart tissues as described previously [23]. The total protein concentration in samples was measured by the bicinchoninic acid method. For the determination of protein levels of TNF-α, VCAM-1, ICAM-1, cyclooxygenase (COX)-2, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology Inc., CA, USA) and NF-κB (Cell Signaling Technology, Inc., Beverly, MA, USA) equal amounts of protein extracts (30 μg) were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (Bio-Rad, Richmond, CA, USA) and transferred electrophoretically to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk in Tris-buffered saline Tween (20 mM Tris, pH 7.6, 137 mM NaCl, and 0.1% Tween 20). All antibodies were used at a dilution of 1:1000. The membrane was incubated overnight at 4 °C with the primary antibody, and the bound antibody was visualized using the respective horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology Inc.) and chemiluminescence developing agents (Amersham Biosciences, Buckinghamshire, U.K.). The level of GAPDH was estimated in every sample to check for equal loading of samples. Films were scanned, and band densities were quantified with densitometric analysis using Scion Image program (Epson GT-X700, Tokyo, Japan). All values were normalized by setting the density of control samples as 1.0.
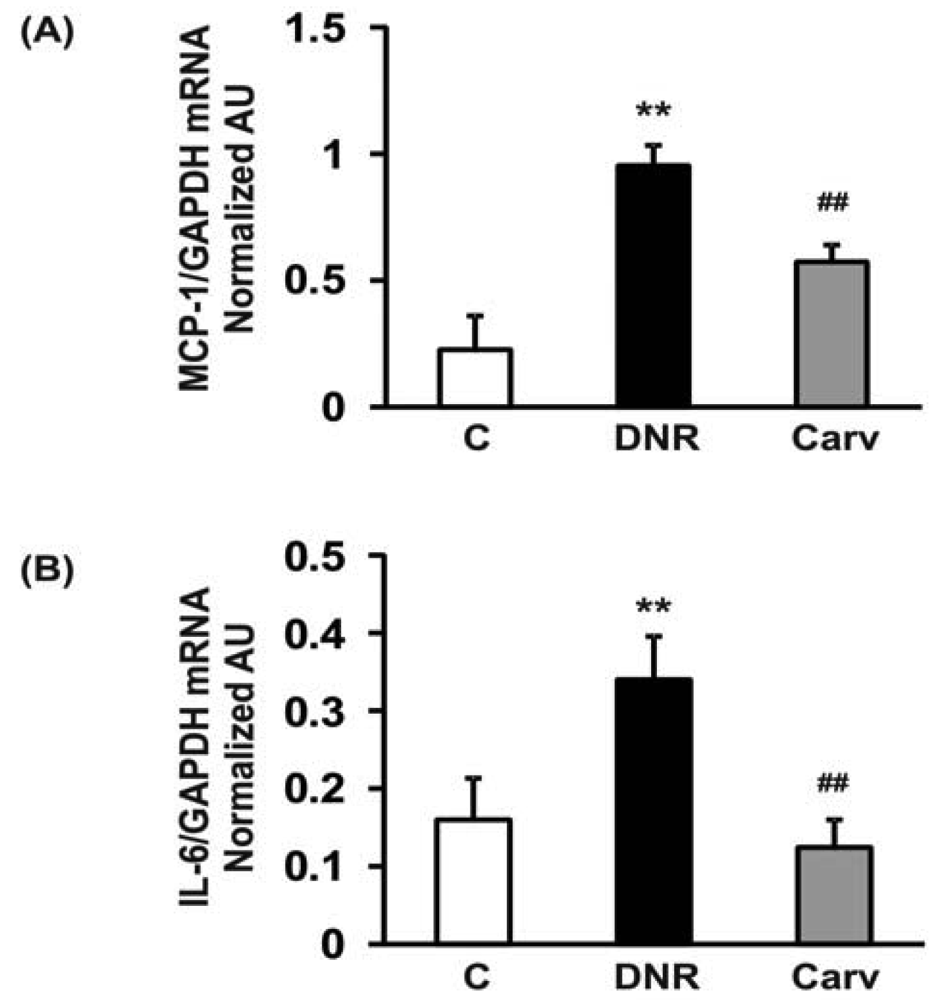
2.6. Quantitative Real-Time Polymerase Chain Reaction (RT-PCR) to Detect Monocyte Chemotactic Protein (MCP)-1 and IL-6
Total RNA was extracted from heart tissues with Trizol (Gibco BRL) in accordance with the standard protocol and reverse-transcribed. Thereafter, cDNA was amplified using the ABI 7700 sequence-detector system (Applied Biosystems, Foster City, CA, USA) with a set of primers and probes corresponding to MCP-1, IL-6 and GAPDH as previously described [24].
2.7. Statistical Analysis
Data are presented as mean ± SEM and were analyzed using one-way analysis of variance (ANOVA) followed by Tukey or Bonferroni methods for post hoc analysis and two-tailed t-test when appropriate. A value of p < 0.05 was considered statistically significant. For statistical analysis GraphPad Prism 5 software (GraphPad Software, San Diego, CA, USA) was used.
3. Results and Discussion
3.1. Effects of Carvedilol on BW and Survival Rate
As depicted in Table 1, BW was significantly decreased in the DNR and carvedilol groups of rats compared with the control group. Carvedilol treatment tended to increase the BW compared with that in the DNR group, although the effect did not attain statistical significance. The mortality rate was higher in the DNR group than that of the control group. Conversely, the mortality rate was lower in carvedilol group than that in the DNR group. In brief, five of 10 (50%) and two of 10 (20%) rats in the DNR and carvedilol groups, respectively, died between days 28 and 42 (Table 1). Furthermore, in prematurely dead animals in both DNR and carvedilol groups, a necropsy examination revealed marked sign of blood congestion such as hydrothorax and ascites. None of the rats in the control group died.
3.2. Effect of Carvedilol on Daunorubicin-Induced Cardiotoxicity
The definition of anthracycline-induced cardiotoxicity has expanded from the clinical events of cardiac failure to include a wide spectrum of predefined laboratory values, even in asymptomatic patients [1]. These include histological changes in the cardiomyocytes [25] and changes in LVEF [26]. As depicted in Table 2 and Figure 2A, 2B, HE staining showed that DNR induced significant cardiac damage as recognized by the presence of marked interstitial edema, focal subendocardial fibrosis, perinuclear vacuolation, and disorganization and degeneration of the myocardium compared to the control group. The morphological changes in the myocardia of the DNR-induced animals were also characterized by focal myocardial interstitial fibrosis of varying intensity [27]. The Azan-Mallory staining shown in Figure 2C demonstrated significant interstitial myocardial fibrosis in the DNR group compared with the control group. Carvedilol treatment significantly improved these lesions (Figure 2 and Table 2).
Our present study indicated that the cumulative administration of DNR resulted in cardiotoxicity as evidenced by worsening myocardial function. LVEDP was significantly higher (10.8 ± 0.2 vs. 7 ± 1 mmHg, p < 0.05) and LVP and ± dP/dt were significantly lower in the DNR group compared with the control group (105 ± 7 vs. 125.3 mmHg, p < 0.05; and 4635 ± 351 vs. 7123 ± 544 mmHg/s, p < 0.05; 3906 ± 329 vs. 7851 ± 656 mmHg/s, p < 0.05, respectively), indicating that systolic and diastolic dysfunction occurred in the DNR rats. Carvedilol treatment improved this myocardial dysfunction by significantly reducing LVEDP (8.2 ± 1.2 vs. 10.8 ± 0.2 mmHg, p < 0.05) and significantly elevating in the LVP and + dP/dt (120.5 ± 11 vs. 105 ± 7 mmHg, p < 0.05; and 6229 ± 581 vs. 4635 ± 351 mmHg/s, p < 0.05, respectively) compared with those in the DNR group (Table 3). Echocardiographic data showed that LV systolic function, as assessed by FS and EF was significantly reduced in the DNR group compared with that in the control group (28.4 ± 1.1 vs. 43.8 ± 1.5%, p < 0.05; and 59.4 ± 1.4 vs. 79.9 ± 1.8%, p < 0.05, respectively). Carvedilol significantly attenuated the reductions in both FS and EF compared with the DNR group (38.8 ± 3.5 vs. 28.4 ± 1.1%, p < 0.05; and 73.6 ± 4.4 vs. 59.4 ± 1.4%, p < 0.05, respectively) (Table 3).
3.3. Effect of Carvedilol on Inflammatory-Mediated Cardiotoxicity
The mechanism of anthracycline-induced cardiomyopathy have been the subject of considerable controversy, nevertheless the iron-mediated formation of reactive oxygen species and promotion of myocardial oxidative stress remains by the far the most frequently proposed mechanism. Similarly, there is a strong association between oxidative stress and the cardiac inflammatory response, including cytokine release after doxorubicin treatment [28]. In the clinical setting, it would be difficult to measure the inflammatory process in anthracycline-based therapy in the context where stimulation has activated the inflammatory process. In patients receiving chronic administration of anthracycline, endomyocardial biopsy data showed no inflammatory cells were found in the endocardial zone [29]. On the contrary, Mills et al. found that markers on inflammation (i.e VEGF, sICAM-1, sP-selectin, vWf) were significantly increased after four-3 weeks treatment of anthracycline compared with before treatment in the breast cancer patient [30]. Caddedu et al. found that IL-6 and TNF-α were significantly up-regulated in the patient received epirubicin treatment [31]. These results have shown the possibility that inflammation process appeared from the progression of the primary disease. This matter has become the limitation of our study since carvedilol was given in the same timing with the DNR. Therefore, we cannot evaluate further the inflammation process at the timing “after DNR” but “before carvedilol”. However, Zhu et al. demonstrated further that IL-1 family is closely connected with doxorubicin-induced cardiotoxicity. At day seven after doxorubicin treatment, IL-1β and IL-1Ra were both highly induced after different dosages of doxorubicin treatments. In the separated experiments, they found that administration of recombinant human interleukin-1 receptor antagonist (rhIL-1Ra) prevents heart damage, protects cardiac function loss and decreases cardiomyocyte apoptosis in doxorubicin-treated mice [32].
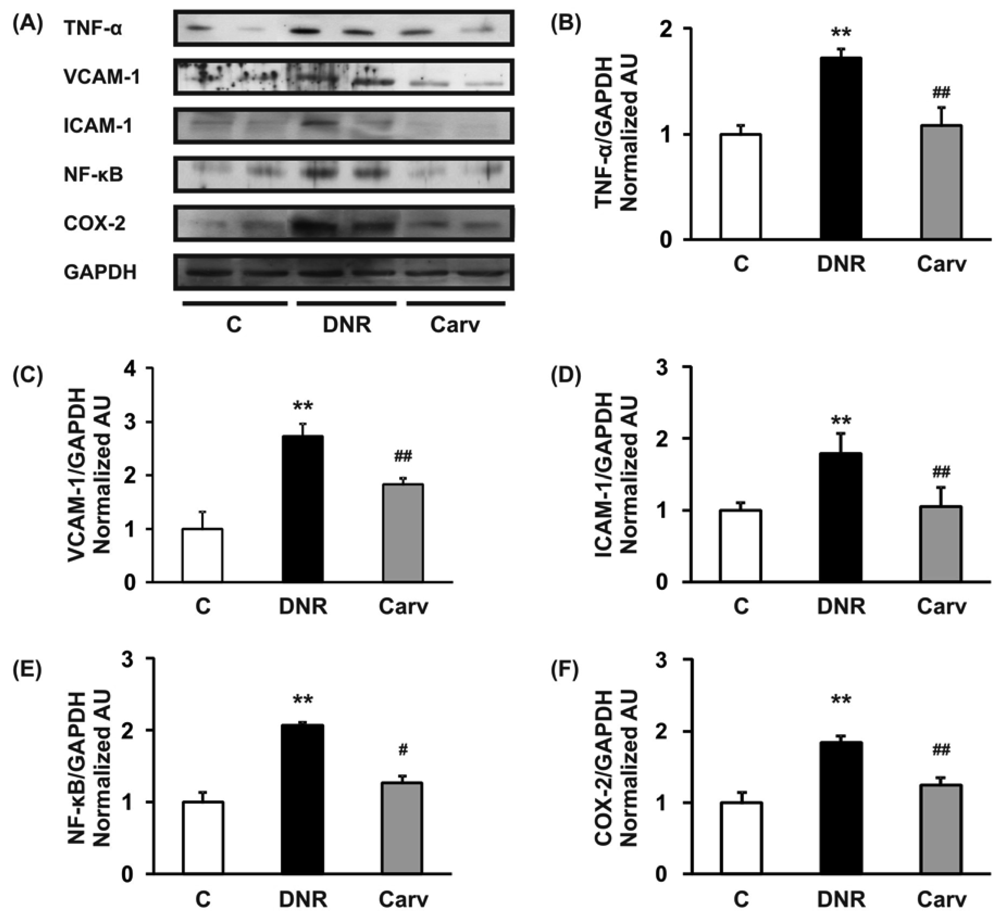
We have previously reported that the ROS level and Fas-L expression level were significantly up-regulated in the DNR-induced rats, which also displayed significant increases in apoptosis and cardiotoxicity [10,18]. Even though Fas-L has been well characterized as a death factor, it also plays a pivotal role as an upstream inflammatory factor that induces the expression of a variety of other inflammatory cytokines in vivo [11]. In mice received doxorubicin treatment, the activation of Fas signaling is also a key mediator of carditoxicity, including cardiomyocyte apoptosis and myocardial inflammation and inhibiting the Fas expression with sFas gene therapy prevents the progression of cardiotoxicity caused by Fas signaling [33]. Fas expression is strongly induced in mouse embryonic fibroblasts by the inflammatory cytokine TNF-α [34]. In this study, we analyzed the toluidine blue staining to identify mast cells and found that mast cells were significantly increased in the DNR rats compared with control rat and carvedilol treatment significantly reverses this condition (Figures 2D and E). This result suggests that inflammatory process may involve in the anthracycline-induced cardiomyopathy. As depicted in Figures 3A and B, we further found that DNR produced an increase in cardiac TNF-α expression compared with the control group. It is important to note that carvedilol treatment significantly decreased the cardiac expression of TNF-α protein compared with that in the DNR group. TNF-α has also been reported to mediate cardiac damage in the doxorubicin-induced mice [9]. In accordance with our result, in the clinical setting, it has been reported that carvedilol decreased the plasma levels of TNF-α in patients with ischemic dilated cardiomyopathy [35].
Our result further showed that the myocardial protein expression levels of adhesion molecules (VCAM-1 and ICAM-1) were significantly up-regulated in the DNR group compared with the control group (Figure 3A, C and D). These increases in the expression levels of adhesion molecules may be mediated through the regulation of TNF-α and the toxicity of anthracyclines. A previous study reported that TNF-α enhanced the expression of adhesion molecules on the membranes of endothelial cells [36]. Furthermore, doxorubicin has been reported to induce the expression of VCAM-1 [6]. Our result confirmed that carvedilol significantly reversed the expression of VCAM-1 and ICAM-1 compared with the DNR group (Figures 3A, C and D). These results support those of a previously published study that demonstrated that carvedilol inhibits TNF-α-induced adhesion molecule expression in human endothelial cells [14].
During the progression of cardiovascular diseases such as congestive heart failure and atherosclerosis, TNF-α may activate membrane-bound NADPH oxidase and increase intracellular ROS production, which could in turn activate redox-sensitive transcriptional pathways such as NF-κB and regulate the expression of downstream inflammatory molecules [37]. Doxorubicin has also been reported also to induce NF-κB-associated apoptosis and NF-κB-associated cytotoxicity in endothelial cells and human colon cancer HT29 cells, respectively [38,39].
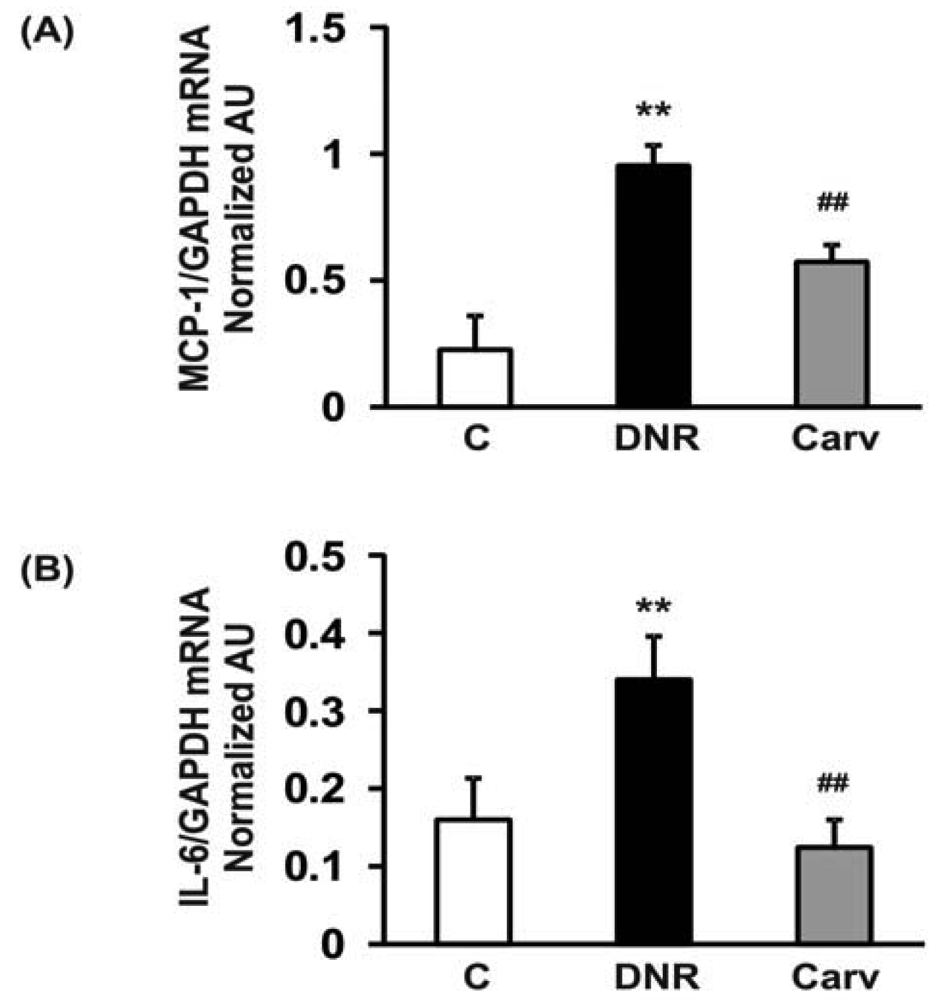
Therefore, NF-κB may mediate anthracyclines-induced cardiotoxicity through its role in the regulation of apoptosis and the expression of downstream inflammatory molecules. In accordance with these findings, we found that DNR significantly induced the expression of transcription factors related to inflammation, e.g., NF-κB, compared to the control group. Additionally, the myocardial protein expression levels of COX-2, which is regulated by NF-κB [40], were also significantly increased in the DNR group compared with the control group. Carvedilol significantly attenuated the myocardial expression of NF-κB and COX-2 compared to that in the DNR group (Figures 3A and F). These results are reasonable since carvedilol has been shown to inhibit the expression of TNF-α-induced NF-κB in human endothelial cells [14,37]. Accumulating evidence has shown that doxorubicin plays a pivotal role in the induction of MCP-1 expression in human lung carcinoma cells [41]. Furthermore, IL-6 has been shown to be involved in doxorubicin-induced cardiotoxicity [42]. To assess whether the expression of these cytokines was induced after DNR treatment, we performed RT-PCR analysis to evaluate MCP-1 and IL-6 mRNA expression in cardiac tissues. Our results showed that the expression levels of MCP-1 and IL-6 were significantly up-regulated in the DNR group compared to those in the control group (Figures 4A and B), and carvedilol significantly down-regulated the mRNA expression of these cytokines compared to their levels in the DNR group. These results support the findings of previous studies in which carvedilol was found to exert its anti-inflammatory effects by attenuating TNF-α induced MCP-1 expression [14].
4. Conclusions
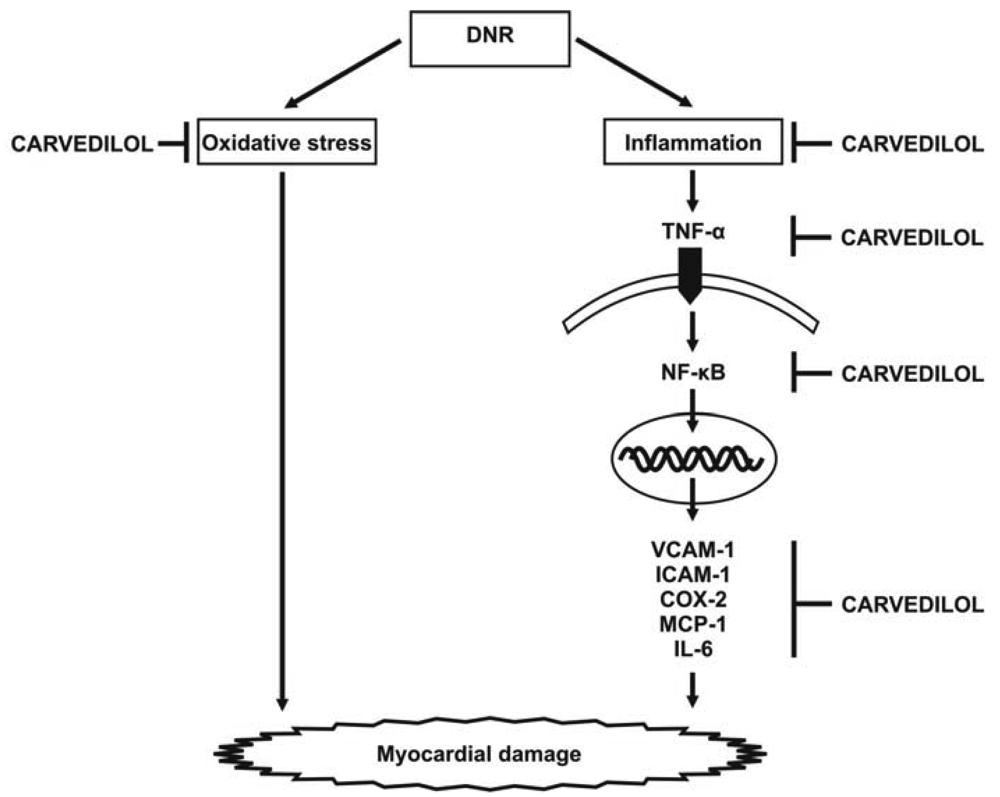
Cardiotoxicity, which may result from intense cardiac oxidative stress and inflammation, is the main limiting factor of anti-cancer therapy using anthracyclines. However, the mechanism by which inflammatory reactions elicit cardiotoxicity is largely unknown. Our recent report shows that DNR induced significant cardiac damage, as recognized by histological changes and worsening cardiac function, and significantly increased the expression levels of inflammatory cytokines and proinflammatory chemokines including TNF-α, VCAM-1, ICAM-1, NF-κB, COX-2, MCP-1 and IL-6 (Figure 5). Thus, the simultaneous induction of these inflammatory markers may augment carditoxicity in DNR-induced rats. These cytokines may produce deleterious effects on LV structure (remodeling) and endothelial function. In fact, in addition to the effects of inflammatory mediators on cardiac structure and function, there is growing evidence that the concentration of inflammatory mediators that exist in heart failure are sufficient to contribute to endothelial dysfunction [43,44]. Therefore, the lack of evaluation the serum circulating levels of cytokines and endothelium dysfunction marker such as asymmetric dimethylarginine can be considered as limitations of present study. As carvedilol is known to have anti-inflammatory effects, we used this drug to improve DNR-induced cardiotoxicity in our model. We have shown that carvedilol significantly attenuated the protein and mRNA expression levels of these markers and improved cardiac function. Based on these results, we speculate that carvedilol contributes to cardioprotection against DNR-induced cardiotoxicity through its anti-oxidant and anti-inflammation properties. Since dexrazoxane (ICRF-187) is the only clinically approved cardioprotectant against anthracycline cardiotoxicity [45], it would be of interest to combine carvedilol with dexrazoxane to evaluate the possible potential synergistic or additive effect in preventing anthracycline-induced cardiotoxicity. Further investigation is warranted to address these issues.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control n = 4 | DNR n = 10 | Carv n = 10 | |
|---|---|---|---|
| No. of rats died | 0 | 5 | 2 |
| Survival rate (%) | 100 | 50 | 80 |
| BW (g) | 540 ± 11.5 | 373 ± 7.4 * | 405 ± 21 |
| HW (g) | 1.3 ± 0.02 | 1.1 ± 0.01 * | 0.9 ± 0.05 * |
| HW/BW (g/kg) | 2.2 ± 0.02 | 2.9 ± 0.05 | 2.4 ± 0.02 |
Results are presented as the mean ± SEM. BW, body weight; HW, heart weight; HW/BW, ratio of heart weight to body weight. Group Control, aged matched normal rats; group DNR, DNR rats treated with vehicle; group Carv, DNR rats treated with carvedilol (30 mg/kg/day).*p < 0.05 vs. group Control.
| Histopathological Finding | Control n = 4 | DNR n = 5 | Carv n = 8 |
|---|---|---|---|
| Cardiac tissue | |||
| Myocardial fibrosis | 0.0 ± 0.0 | 1.5 ± 0.21* | 0.4 ± 0.18# |
| Perinuclear vacuolization | 0.0 ± 0.0 | 0.5 ± 0.25* | 0.2 ± 0.2# |
| Myocardial degeneration | 0.0 ± 0.0 | 2.75 ± 0.21* | 1.4 ± 0.21# |
| Interstitial edema | 0.0 ± 0.0 | 2.75 ± 0.19* | 1.6 ± 0.22# |
Results are presented as the mean ± SEM. Group Control, aged matched normal rats; group DNR, DNR rats treated with vehicle; group Carv, DNR rats treated with carvedilol (30 mg/kg/day).*p < 0.05 vs. group Control and#p < 0.05 vs. group DNR.
| Parameter | Control n = 4 | DNR n = 10 | Carv n = 10 |
|---|---|---|---|
| CVP (mmHg) | -0.5 ± 0.04 | 0.44 ± 0.05 | 0.3 ± 0.05 |
| MBP (mmHg) | 96 ± 7.4 | 87 ± 6.4 | 82 ± 9.7 |
| LVP (mmHg) | 125.3 ± 5 | 105 ± 7* | 120.5 ± 11# |
| LVEDP (mmHg) | 7 ± 1 | 10.8 ± 0.2* | 8.2 ± 1.2# |
| +dP/dt (mmHg/s) | 7123 ± 544 | 4635 ± 351* | 6229 ± 581# |
| -dP/dt (mmHg/s) | 7851 ± 656 | 3906 ± 329* | 5034 ± 516* |
| HR (beats/min) | 362 ± 34 | 329 ± 11 | 317 ± 31 |
| LVDd (mm) | 6.5 ± 0.5 | 7.8 ± 0.1 | 7.05 ± 0.4 |
| LVDs (mm) | 4.2 ± 0.3 | 5.5 ± 0.2 | 4.9 ± 0.5 |
| FS (%) | 43.8 ± 1.5 | 28.4 ± 1.1* | 38.8 ± 3.5# |
| EF (%) | 79.9 ± 1.8 | 59.4 ± 1.4* | 73.6 ± 4.4# |
Results are presented as the mean ± SEM. CVP, central venous pressure; MBP, mean blood pressure; LVP, left ventricular pressure; LVEDP, left ventricular end-diastolic pressure; ±dP/dt, rate of intra-ventricular pressure rise and decline; HR, heart rate; LVDd, left ventricular dimension in diastole; LVDs, left ventricular dimension in systole; FS, fractional shortening; EF, ejection fraction; group Control, aged matched normal rats; group DNR, DNR rats treated with vehicle; group Carv, DNR rats treated with carvedilol (30 mg/kg/day).*p < 0.05 vs. group Control and#p < 0.05 vs. group DNR.
Acknowledgements
This research was supported by a Yujin Memorial Grant, Ministry of Education, Science, Sports and Culture, Japan and by a grant from the Promotion and Mutual Aid Corporation for Private Schools, Japan. We thank Sayaka Egawa, Vijayakumar Sukumaran, Yoshiyashu Kobayashi, and Arun Prasath Lakshmanan for their assistance in this research work.
References and Notes
- Rahman, A.M.; Yusuf, S.W.; Ewer, M.S. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomed. 2007, 2, 567–583. [Google Scholar]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar]
- Gaudin, P.B.; Hruban, R.H.; Beschorner, W.E.; Kasper, E.K.; Olson, J.L.; Baughman, K.L.; Hutchins, G.M. Myocarditis associated with doxorubicin cardiotoxicity. Am. J. Clin. Pathol. 1993, 100, 158–163. [Google Scholar]
- Danesi, R.; Foqli, S.; Gennari, A.; Conte, P.; Del Tacca, M. Pharmacokinetic-pharmacodynamic relationships of the anthracyline anticancer drugs. Clin. Pharmacokinet. 2002, 41, 431–444. [Google Scholar]
- Mercuro, G.; Cadeddu, C.; Piras, A.; Dessì, M.; Madeddu, C.; Deidda, M.; Serpe, R.; Massa, E.; Mantovani, G. Early epirubicin-induced myocardial dysfunction revealed by serial tissue Doppler echocardiography: correlation with inflammatory and oxidative stress markers. Oncologist 2007, 12, 1124–1133. [Google Scholar]
- Fujihira, S.; Yamamoto, T.; Matsumoto, M.; Yoshizawa, K.; Oishi, Y.; Fujii, T.; Noguchi, H.; Mori, H. The high incidence of atrial thrombosis in mice given doxorubicin. Toxicol. Pathol. 1993, 21, 362–368. [Google Scholar]
- Abou El Hassan, M.A.; Verheul, H.M.; Jorna, A.S.; Schalkwijk, C.; van Bezu, J.; van der Vijgh, W.J.; Bast, A. The new cardioprotector Monohydroxyethylrutoside protects against doxorubicin-induced inflammatory effects in vitro. Br. J. Cancer. 2003, 89, 357–362. [Google Scholar]
- Hou, G.; Dick, R.; Abrams, G.D.; Brewer, G.J. Tetrathiomolybdate protects against cardiac damage by doxorubicin in mice. J. Lab. Clin. Med. 2005, 146, 299–303. [Google Scholar]
- Riad, A.; Bien, S.; Westermann, D.; Becher, P.M.; Loya, K.; Landmesser, U.; Kroemer, H.K.; Schultheiss, H.P.; Tschöpe, C. Pretreatment with statin attenuates the cardiotoxicity of Doxorubicin in mice. Cancer. Res. 2009, 69, 695–699. [Google Scholar]
- Soga, M.; Kamal, F.A.; Watanabe, K.; Ma, M.; Palaniyandi, S.; Prakash, P.; Veeraveedu, P.; Mito, S.; Kunisaki, M.; Tachikawa, H.; Kodama, M.; Aizawa, Y. Effects of angiotensin II receptor blocker (candesartan) in daunorubicin-induced cardiomyopathic rats. Int. J. Cardiol. 2006, 110, 378–385. [Google Scholar]
- Umemura, M.; Kawabe, T.; Shudo, K.; Kidoya, H.; Fukui, M.; Asano, M.; Iwakura, Y.; Matsuzaki, G.; Imamura, R.; Suda, T. Involvement of IL-17 in Fas ligand-induced inflammation. Int. Immunol. 2004, 8, 1099–1108. [Google Scholar]
- Feuerstein, G.Z.; Ruffolo, R.R. Carvedilol, a novel vasodilating beta-blocker with the potential for cardiovascular organ protection. Eur. Heart. J. 1996, 17 Suppl B, 24–29. [Google Scholar]
- Alfieri, A.B.; Briceno, L.; Fragasso, G.; Spoladore, R.; Palloshi, A.; Bassanelli, G.; Montano, C.; Arioli, F.; Cuko, A.; Ruotolo, G.; Margonato, A. Differential long-term effects of carvedilol on proinflammatory and antiinflammatory cytokines, asymmetric dimethylarginine, and left ventricular function in patients with heart failure. J. Cardiovasc. Pharmacol. 2008, 52, 49–54. [Google Scholar]
- Kim, Y.S.; Ahn, Y.; Hong, M.H.; Joo, S.Y.; Jeong, M.H.; Kim, K.H.; Sohn, I.S.; Park, H.W.; Hong, Y.J.; Kim, J.H.; Kim, W.; Cho, J.G.; Park, J.C.; Kang, J.C. Carvedilol Inhibits Expressions of Vascular Cell Adhesion Molecule-1, Intercellular Adhesion Molecule-1, Monocyte Chemoattractant-1, and Interleukin-8 via NF-κB Inhibition in Human Endothelial Cells. Korean Circulation J. 2005, 35, 576–582. [Google Scholar]
- Matsui, H.; Morishima, I.; Numaguchi, Y.; Toki, Y.; Okumura, K.; Hayakawa, T. Protective effects of carvedilol against doxorubicin-induced cardiomyopathy in rats. Life Sci. 1999, 65, 1265–1274. [Google Scholar]
- Kalay, N.; Basar, E.; Ozdogru, I.; Er, O.; Cetinkaya, Y.; Dogan, A.; Inanc, T.; Oguzhan, A.; Eryol, N.K.; Topsakal, R.; Ergin, A. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 2258–2262. [Google Scholar]
- Oliveira, P.J.; Bjork, J.A.; Santos, M.S.; Leino, R.L.; Froberg, M.K.; Moreno, A.J.; Wallace, K.B. Carvedilol-mediated antioxidant protection against doxorubicin-induced cardiac mitochondrial toxicity. Toxicol. Appl. Pharmacol. 2004, 200, 159–168. [Google Scholar]
- Arozal, W.; Watanabe, K.; Veeraveedu, P.T.; Ma, M.; Thandavarayan, R.A.; Sukumaran, V.; Suzuki, K.; Kodama, M.; Aizawa, Y. Protective effect of carvedilol on daunorubicin-induced cardiotoxicity and nephrotoxicity in rats. Toxicology 2010, 274, 18–26. [Google Scholar]
- Dziegel, P; Jethon, Z.; Suder, E.; Sopel, M.; Rabczynski, L.; Surowiak, P.; Zabel, M. Role of exogenous melatonin in reducing the cardiotoxic effect of daunorubicin and doxorubicin in the rat. Exp. Toxicol. Pathol. 2002, 53, 433–439. [Google Scholar]
- Watanabe, K.; Ohta, Y.; Nakazawa, M.; Higuchi, H.; Hasegawa, G.; Naito, M.; Fuse, K.; Ito, M.; Hirono, S.; Tanabe, N.; Hanawa, H.; Kato, K.; Kodama, M.; Aizawa, Y. Low dose carvedilol inhibits progression of heart failure in rats with dilated cardiomyopathy. Br. J. Pharmacol. 2000, 130, 1489–1495. [Google Scholar]
- Yilmaz, S.; Atessahin, A.; Sahna, E.; Karaha, I.; Ozer, S. Protective effect of lycopene on adriamycin-induced cardiotoxicity and nephrotoxicity. Toxicology 2006, 218, 164–171. [Google Scholar]
- Ibrahim, M.A.; Ashour, O.M.; Ibrahim, Y.F.; El-Bitar, H.I.; Gomaa, W.; Abdel-Rahim, S.R. Angiotensin-converting enzyme inhibition and angiotensin AT(1)-receptor antagonism equally improve doxorubicin-induced cardiotoxicity and nephrotoxicity. Pharmacol. Res. 2009, 60, 378–381. [Google Scholar]
- Gurusamy, N.; Watanabe, K.; Ma, M.; Zhang, S.; Muslin, A.J.; Kodama, M.; Aizawa, Y. Dominant negative 14-3-3 promotes cardiomyocyte apoptosis in early stage of type I diabetes mellitus through activation of JNK. Biochem. Biophys. Res. Commun. 2004, 320, 773–780. [Google Scholar]
- Yndestad, A.; Ueland, T.; Øie, E.; Florholmen, G.; Halvorsen, B.; Attramadal, H.; Simonsen, S.; Frøland, S.S.; Gullestad, L.; Christensen, G.; Damås, J.K.; Aukrust, P. Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation 2004, 109, 1379–1385. [Google Scholar]
- Billingham, M.E.; Mason, J.W.; Bristow, M.R.; Daniels, J.R. Anthracycline cardiomyopathy monitored by morphologic changes. Cancer. Treat. Rep. 1978, 62, 865–872. [Google Scholar]
- Schwartz, R.G.; McKenzie, W.B.; Alexander, J.; Sager, P.; D'Souza, A.; Manatunga, A.; Schwartz, P.E.; Berger, H.J.; Setaro, J.; Surkin, L.; et al. Congestive heart failure and left ventricular dysfunction complicating doxorubicin therapy. Seven-year experience using serial radionuclide angiocardiography. Am. J. Med. 1987, 82, 1109–1118. [Google Scholar]
- Adamcová, M.; Potáčová, A.; Popelová, O.; Stěrba, M.; Mazurová, Y.; Aupperle, H.; Geršl, V. Cardiac remodeling and MMPs on the model of chronic daunorubicin-induced cardiomyopathy in rabbits. Physiol. Res. 2010, 59, 831–836. [Google Scholar]
- Bien, S.; Riad, A.; Ritter, C.A.; Gratz, M.; Olshausen, F.; Westermann, D.; Grube, M.; Krieg, T.; Ciecholewski, S.; Felix, S.B.; Staudt, A.; Schultheiss, H.P.; Ewert, R.; Völker, U.; Tschöpe, C.; Kroemer, H.K. The endothelin receptor blocker bosentan inhibits doxorubicin-induced cardiomyopathy. Cancer Res. 2007, 67, 10428–10435. [Google Scholar]
- Mortensen, S.A.; Olsen, H.S.; Baandrup, U. Chronic anthracycline cardiotoxicity: haemodynamic and histopathological manifestations suggesting a restrictive endomyocardial disease. Br. Heart. J. 1986, 55, 274–282. [Google Scholar]
- Mills, P.J.; Ancoli-Israel, S.; Parker, B.; Natarajan, L.; Hong, S.; Jain, S.; Sadler, G.R.; von Känel, R. Predictors of inflammation in response to anthracycline-based chemotherapy for breast cancer. Brain. Behav. Immun. 2008, 22, 98–104. [Google Scholar]
- Cadeddu, C.; Piras, A.; Mantovani, G.; Deidda, M.; Dessì, M.; Madeddu, C.; Massa, E.; Mercuro, G. Protective effects of the angiotensin II receptor blocker telmisartan on epirubicin-induced inflammation, oxidative stress, and early ventricular impairment. Am. Heart. J. 2010, 160, 487.e1–487.e7. [Google Scholar]
- Zhu, J.; Zhang, J.; Xiang, D.; Zhang, Z.; Zhang, L.; Wu, M.; Zhu, S.; Zhang, R.; Han, W. Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. Eur. J. Pharmacol. 2010, 643, 247–253. [Google Scholar]
- Miyata, S.; Takemura, G.; Kosai, K.; Takahashi, T.; Esaki, M.; Li, L.; Kanamori, H.; Maruyama, R.; Goto, K.; Tsujimoto, A.; Takeyama, T.; Kawaguchi, T.; Ohno, T.; Nishigaki, K.; Fujiwara, T.; Fujiwara, H.; Minatoguchi, S. Anti-Fas gene therapy prevents doxorubicin-induced acute cardiotoxicity through mechanisms independent of apoptosis. Am. J. Pathol. 2010, 176, 687–698. [Google Scholar]
- Zheng, Y.; Ouaaz, F.; Bruzzo, P.; Singh, V.; Gerondakis, S.; Beg, A.A. NF-kappa B RelA (p65) is essential for TNF-alpha-induced fas expression but dispensable for both TCR-induced expression and activation-induced cell death. J. Immunol. 2001, 166, 4949–4957. [Google Scholar]
- Cinquegrana, G.; D'Aniello, L.; Landi, M.; Spinelli, L.; Grande, G.; De Prisco, F.; Petretta, M. Effects of different degrees of sympathetic antagonism on cytokine network in patients with ischemic dilated cardiomyopathy. J. Card. Fail. 2005, 11, 213–219. [Google Scholar]
- Molet, S.; Gosset, P.; Vanhee, D.; Tillie-Leblond, I.; Wallaert, B.; Capron, M.; Tonnel, A.B. Modulation of cell adhesion molecules on human endothelial cells by eosinophil-derived mediators. J. Leukoc. Biol. 1998, 63, 351–358. [Google Scholar]
- Lin, P.Y.; Shen, H.C.; Chen, C.J.; Wu, S.E.; Kao, H.L.; Huang, J.H.; Wang, D.L.; Chen, S.C. The inhibition in tumor necrosis factor-alpha-induced attenuation in endothelial thrombomodulin expression by carvedilol is mediated by nuclear factor-kappaB and reactive oxygen species. J. Thromb. Thrombolysis 2010, 29, 52–59. [Google Scholar]
- Wang, S.; Kotamraju, S.; Konorev, E.; Kalivendi, S.; Joseph, J.; Kalyanaraman, B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem. J. 2002, 367, 729–740. [Google Scholar]
- Riganti, C.; Doublier, S.; Costamagna, C.; Aldieri, E.; Pescarmona, G.; Ghigo, D.; Bosia, A. Activation of nuclear factor-kappa B pathway by simvastatin and RhoA silencing increases doxorubicin cytotoxicity in human colon cancer HT29 cells. Mol. Pharmacol. 2008, 74, 476–484. [Google Scholar]
- Lee, K.M.; Kang, B.S.; Lee, H.L.; Son, S.J.; Hwang, S.H.; Kim, D.S.; Park, J.S.; Cho, H.J. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur. J. Neurosci. 2004, 19, 3375–3381. [Google Scholar]
- Niiya, M.; Niiya, K.; Kiguchi, T.; Shibakura, M.; Asaumi, N.; Shinagawa, K.; Ishimaru, F.; Kiura, K.; Ikeda, K.; Ueoka, H.; Tanimoto, M. Induction of TNF-alpha, uPA, IL-8 and MCP-1 by doxorubicin in human lung carcinoma cells. Cancer. Chemother. Pharmacol. 2003, 52, 391–398. [Google Scholar]
- Niu, J.; Azfer, A.; Wang, K.; Wang, X.; Kolattukudy, PE. Cardiac-targeted expression of soluble fas attenuates doxorubicin-induced cardiotoxicity in mice. J. Pharmacol. Exp. Ther. 2009, 328, 740–748. [Google Scholar]
- Mann, D.L. Inflammatory mediators and the failing heart: past, present, and in the foreseeable future. Circ. Res. 2002, 91, 988–998. [Google Scholar]
- Chong, E.Y.; Freestone, B.; Patel, J.; Lim, H.S.; Hughes, E.; Blann, A.D.; Lip, G.Y. Endothelial activation, dysfunction, and damage in congestive heart failure and the relation to brain natriuretic peptide and outcomes. Am. J. Cardiol. 2006, 97, 671–675. [Google Scholar]
- Wouters, K.A.; Kremer, L.C.; Miller, T.L.; Herman, E.H.; Lipshultz, S.E. Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. Br J Haematol. 2005, 131, 561–578. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sari, F.R.; Arozal, W.; Watanabe, K.; Harima, M.; Veeravedu, P.T.; Thandavarayan, R.A.; Suzuki, K.; Arumugam, S.; Soetikno, V.; Kodama, M. Carvedilol Attenuates Inflammatory-Mediated Cardiotoxicity in Daunorubicin-Induced Rats. Pharmaceuticals 2011, 4, 551-566. https://doi.org/10.3390/ph4030551
Sari FR, Arozal W, Watanabe K, Harima M, Veeravedu PT, Thandavarayan RA, Suzuki K, Arumugam S, Soetikno V, Kodama M. Carvedilol Attenuates Inflammatory-Mediated Cardiotoxicity in Daunorubicin-Induced Rats. Pharmaceuticals. 2011; 4(3):551-566. https://doi.org/10.3390/ph4030551
Chicago/Turabian StyleSari, Flori R., Wawaimuli Arozal, Kenichi Watanabe, Meilei Harima, Punniyakoti T. Veeravedu, Rajarajan A. Thandavarayan, Kenji Suzuki, Somasundaram Arumugam, Vivian Soetikno, and Makoto Kodama. 2011. "Carvedilol Attenuates Inflammatory-Mediated Cardiotoxicity in Daunorubicin-Induced Rats" Pharmaceuticals 4, no. 3: 551-566. https://doi.org/10.3390/ph4030551
APA StyleSari, F. R., Arozal, W., Watanabe, K., Harima, M., Veeravedu, P. T., Thandavarayan, R. A., Suzuki, K., Arumugam, S., Soetikno, V., & Kodama, M. (2011). Carvedilol Attenuates Inflammatory-Mediated Cardiotoxicity in Daunorubicin-Induced Rats. Pharmaceuticals, 4(3), 551-566. https://doi.org/10.3390/ph4030551