Monoaminergic Antidepressants in the Relief of Pain: Potential Therapeutic Utility of Triple Reuptake Inhibitors (TRIs)

Abstract
: Over 75% of depressed patients suffer from painful symptoms predicting a greater severity and a less favorable outcome of depression. Imaging, anatomical and functional studies have demonstrated the existence of common brain structures, neuronal pathways and neurotransmitters in depression and pain. In particular, the ascending serotonergic and noradrenergic pathways originating from the raphe nuclei and the locus coeruleus; respectively, send projections to the limbic system. Such pathways control many of the psychological functions that are disturbed in depression and in the perception of pain. On the other hand, the descending pathways, from monoaminergic nuclei to the spinal cord, are specifically implicated in the inhibition of nociception providing rationale for the use of serotonin (5-HT) and/or norepinephrine (NE) reuptake inhibitors (SSRIs, NRIs, SNRIs), in the relief of pain. Compelling evidence suggests that dopamine (DA) is also involved in the pathophysiology and treatment of depression. Indeed, recent insights have demonstrated a central role for DA in analgesia through an action at both the spinal and suprasinal levels including brain regions such as the periaqueductal grey (PAG), the thalamus, the basal ganglia and the limbic system. In this context, dopaminergic antidepressants (i.e., containing dopaminergic activity), such as bupropion, nomifensine and more recently triple reuptake inhibitors (TRIs), might represent new promising therapeutic tools in the treatment of painful symptoms with depression. Nevertheless, whether the addition of the dopaminergic component produces more robust effects than single- or dual-acting agents, has yet to be demonstrated. This article reviews the main pathways regulating pain transmission in relation with the monoaminergic systems. It then focuses on the current knowledge regarding the in vivo pharmacological properties and mechanism of action of monoaminergic antidepressants including SSRIs, NRIs, SNRIs and TRIs. Finally, a synthesis of the preclinical studies supporting the efficacy of these antidepressants in analgesia is also addressed in order to highlight the relative contribution of 5-HT, NE and DA to nociception.1. Neurobiology of Pain and Its Modulation by Monoamines
Pain is a subjective experience that results from transfer and brain analysis of various information such as the nature, location, intensity and duration of a stimulus. It also involves adaptation and modulation of the nociceptive messages by various neuromediators and related receptors. Since these neuromediators are present in the central network of brain structures that process or regulate nociceptive information, it is difficult to dissociate the affective dimension of pain from its sensory dimension [1]. Anatomically, several brain regions have been implicated in both depressive disorder and pain.
At the peripheral level, the pain comes from direct or indirect stimulation and sensitization of nociceptors by various endogenous signalling molecules, including ions, prostaglandins and/or leukotriens, histamine, bradykinines, but also monoamines [2]. The activation of nociceptors creates action potentials that are transmetted by Aδ fibers (fast-conducting, location) and/or C fibers (delayed transmission, a feeling duller and less localized) leading to a more diffuse pain. Both types of pain fibers terminate in the superficial layers of the dorsal horn of the spinal cord where several neuropeptides but also 5-HT and NE play a major role in antinociception [3,4]. Fibers from the transmission cells of substantia gelatinosa convey impulses to the thalamus, the main brain region responsible for the integration of pain input. From the thalamus, third-order neurons transmit pain impulses to the cerebral cortex where further processing occurs resulting in pain awareness [5]. In this review, particular attention will be focused on the central brain monoaminergic regions and their pathways that regulate the nociceptive information at the central level. Indeed, given the close anatomical relationship between areas involved in pain and emotion and the emotional nature of pain, it is possible that treatment of mood disorders with monoaminergic antidepressants display a powerful impact on pain by regulating the affective, emotional and sensory dimensions of pain.
1.1. Spinal Cord, Ascending and Descending Pathways
At the spinal level, the central terminal of the nociceptor forms synapses with neurons of the superficial dorsal horn of the spinal cord. Glutamate seems to be the main neurotransmitter released in response to a nociceptive stimulus. Glutamate then acts on post-synaptic receptors present in: (i) the projection of cells whose axons convey information to various parts of the brain; and (ii) interneurons (both excitatory and inhibitory) that all contribute to the local modulatory circuit in the spinal cord. Thus, the ascending pathways distribute spinal action potentials to brain areas related to the two dimensions of pain perception, sensory and affective: the somatosensory cortex, the periaqueductal grey (PAG), hypothalamus and basal ganglia. Spreading from central projections, corticolimbic pathways are also activated. These sites which process noxious activation include the thalamus, insular cortex, anterior and posterior cingulated cortex, prefrontal cortex [6-8] but also the amygdala [9] and the hippocampus [10]. All these brain regions are endowed with a rich serotonergic, noradrenergic and/or dopaminergic innervations suggesting the role of monoamines in the modulation of pain.
Descending inhibitory or facilitatory pathways from brain areas converge at the dorsal horn, controlling peripheral inputs from nociceptors. Monoaminergic fibers originating from various brainstem nuclei control pain perception through the release of 5-HT and NE in the superficial dorsal horn via the dorsolateral funiculus (DLF) [11]. DLF fibers from descending pathway are thus comprised of serotonergic projections from the raphe nuclei and noradrenergic projections from the locus coeruleus (LC) [12]. Both 5-HT and NE contribute to the modulation of pain, constituting a gating mechanism that control impulse transmission in the dorsal horn (Figure 1). In this figure the limbic system (blue structures) includes various cortical subregions such as the somatosensory (SI and SII), anterior cingulate (ACC), prefrontal and insular cortex but also the amygdala (AMY), hippocampus (HIPP) and basal ganglia. All these structures, involved in the initiation of the descending controls of nociceptive information, are innervated by serotonergic, noradrenergic and dopaminergic neurons originating in the dorsal raphe nucleus (DRN), locus coeruleus (LC) and Ventral Tegmental Area (VTA) (purple structures); respectively. Distinct populations of monoaminergic neurons, via the dorsolateral funiculus (DLF) borrow descending pathways (green circuits) to exert a strong inhibitory effect on pain transmission in the dorsal horn (mediated by 5-HT, NE and likely DA, which may produce a local release of opioids). In particular, serotonergic inputs to the dorsal horn originate in neurons of the rotstral ventromedial medulla (RVM), including the raphe magnus and the nucleus reticularis magnocellularis. The noradrenergic innervation of the dorsal horn originates from several cell groups in the pontine tegmentum, including the A5 group. The main source of descending dopaminergic innervation of the dorsal horn is the A11 neurons of the periventricular posterior hypothalamus [13]. On the contrary, ascending pathways (red circuit) via the spino-thalamic tract, excites neurones in the periaqueductal grey matter (PAG: yellow structure) and thalamus (grey structure), which have direct and/or indirect interactions with the limbic system and monoaminergic nuclei. Interestingly, all the three monoaminergic nuclei display anatomical and functional reciprocal interactions (black arrows) regulating the release of 5-HT, NE and DA in their projections areas and thereby the sensory and emotional dimensions of pain.
Compared with the enormous literature devoted to 5-HT and NE, the spinal action of DA has received less attention. It has been proposed that the dopaminergic innervation of the spinal cord may originate in the substantia nigra and hypothalamus [14]. Further, the purported existence of a small population of DA-synthesising cells in the dorsal root has been confirmed [14]. It is also possible that NE neurons themselves constitute an important source of DA in the dorsal horn to control pain. Although this property has yet to be determined in the spinal cord, several studies reported that the clearance of DA in various brain regions, may be mediated, at least in part, by the selective NE transporter NET [15-18].
1.2. The Brain
The periaqueductal grey (PAG) is an important nociception modulation site where the emotional and cognitive sensations from thalamic or anterior cortical areas meet the vegetative aspects from the hypothalamus [19,20]. Although, the PAG is indirectly connected to the dorsal horn of the spinal cord through adjacent regions of the pont and the medulla [21], it initiates descending and ascending inhibition resulting in the reduction of pain. Consistent with this observation, it has been demonstrated that stimulation of the PAG produces a profound antinociception [22,23] whereas its electrolytic lesion reduced the analgesic effect of morphine [24]. These data suggest that this brain region is a major site of action of opiates in producing analgesia. However, descending facilitatory projections from the PAG to the RVM may enhance spinal nociceptive transmission of peripheral inputs.
The dorsal raphe nucleus (DRN) is interconnected and functionally related to the PAG. Although both brain regions display strong anatomical interactions [25-28], the mechanism underlying their role in the modulation of pain is not fully elucidated. The PAG modulates incoming pain information by activating DRN in the rostral ventromedial medulla, which in turn causes the 5-HT release in the dorsal spinal cord to inhibit incoming sensory stimuli [29]. It was proposed that substance P (SP), which is increased in response to a nociceptive stimulus, regulates both enkephalin and 5-HT neurotransmission in the PAG and the DRN [30]. Interestingly, a subpopulation of non-serotonergic neurons arising from the DRN could release SP in the PAG to produce a robust antinociception [31-34], particularly by evoking a release of enkephalin [35]. In turn, the PAG might also send SPergic projections to the DRN. Multiple sources of evidence suggests that SP activates serotonergic neurons in the DRN [36,37] suggesting that reciprocal interactions between the serotonergic and tachykininergic systems might be an important substrate for reducing pain.
The LC and the A7 catecholamine cell groups, known to contain spinally noradrenergic neurons, are connected to the PAG through a monosynaptic pathway [38,39]. This provides direct anatomical evidence that this pathway may mediate at least some of the effects produced by activation of neurons in the PAG. For example, activation of projection neurons in the PAG has a predominantly inhibitory effect on LC neurons [40], this action contributing to the antinociception produced by PAG stimulation. Two neurochemicals have been suggested to be involved in the modulation of LC NE neuronal activity such as corticotropin-releasing factor (CRF) and the endogenous opioid enkephalin [41]. CRF has been shown to increase the spontaneous discharge rate of LC neurons [42] whereas enkephalins exert mostly inhibitory effects on LC neurons [43]. The balance between opioids and CRF influences in the LC regulate noradrenergic transmission and likely pain through the stimulation of descending pathways.
Dopaminergic neurons from the ventral tegmental area (VTA) have no clear anatomical and direct functional interactions with the PAG. Interestingly, several studies have localized the antinociceptive effects of morphine to the PAG [24,44-46]. Together with the observation that the lesion of DA neurons by the neurotoxin 6-OHDA (injected with a norepinephrine reuptake inhibitor to prevent depletion of NE neurons), caused a decrease in the effect of morphine [24], these results suggest that an intact DA system is necessary to the antinociceptive effect of morphine, particularly in the PAG [47]. In agreement with this hypothesis, a subpopulation of neurons within the PAG is dopaminergic. These neurons project to the central nucleus of the amygdala, ventral striatum, and locally within the PAG [48,49]. Since there is substantial overlap in the neural systems containing opioid and dopamine receptors [50-52], it has been proposed that the antinociceptive effect of morphine results from the release of DA in the PAG, which in turn would facilitate the local action of opioids [53].
The thalamus is the main relay site for nociceptive inputs to cortical and subcortical structures. It includes several nociceptive nuclei of the somatosensory and intralaminar thalamus. Thalamocortical networks that produce both sensory discriminative and affective components of the pain response generate conscious pain. Initial studies have indicated that the ablation of the parafascicular nucleus (PFN) selectively reduces the emotional suffering associated with acute and chronic pain in humans, and reduces responses to noxious stimulation in animals [54].
The DRN projects axons to the thalamus including the PFN, to suppress the pain sensations. Stimulation of the DRN has been found to effectively inhibit the responses induced by noxious stimulation of neurons in the PFN [55]. In contrast, the lesion of 5-HT neurons by the specific neurotoxin 5,7-DHT abolished the effect of DRN stimulation on pain-induced excitation of the PFN. These results indicate that 5-HT has a tonic inhibitory influence on responses to noxious stimulation. It was further found that, similar to DRN stimulation itself, iontophoretic application of 5-HT in the PFN inhibits changes caused by noxious stimulation [56] suggesting that the DRN is involved in pain modulation in this ascending pathway.
The LC, but also the VTA, send projections to the somatosensory thalamus [57-59]. The involvement of ascending noradrenergic innervation of the somatosensory thalamus in pain processing is supported by a recent study showing that nociceptive stimulation activates LC neurons projecting to the thalamus [60,61]. About the role of DA, recent study reported that the local application of DA in the PFN modulates the frequencies of pain-excited and pain-inhibited neurons [62], raising the possibility that DA in this brain region play an important role in the modulation of the nociceptive response. However, it seems that DA produces dual modulatory effects depending on the DA receptor subtypes [63].
Apart from the well-known involvement in motoric circuitry of the basal ganglia, these brain nuclei are involved in many neuronal pathways having emotional, motivational, associative and cognitive functions as well. This brain region contains several nuclei including the putamen, caudate nucleus, globus pallidus, subthalamic nucleus, and nucleus accumbens that receive multimodal input from all sensory systems and thereby serve as a gating station for continuous sensory information, including pain. Several studies have suggested that basal ganglia may be involved in the sensory-discriminative aspect of pain, the affective and cognitive aspect of pain but also the modulation of nociceptive information and sensory gating of nociceptive information to higher motor areas, because they are the main link between the thalamus and the cerebral cortex [64]. Data supporting a role for the basal ganglia in pain and analgesia processing have been derived from numerous clinical and preclinical studies [65]. For example, lesions of the basal ganlgia in patients suffering from Parkinson's disease have offered further insights into the potential role of this brain region in pain and analgesia. Indeed, infarction of the lenticular nucleus (composed of the putamen and globus pallidus) may result in sensory deficits including pain in some patients [66], whereas both unilateral and bilateral deep brain stimulation of the globus pallidus have been reported to relieve pain [67]. The basal ganglia receive inputs from all cortical areas (including medial and orbital, prefrontal, dorsolateral, premotor and motor cortex, sensorimotor and parietal cortex) and the thalamus, which are endowed with a rich innervation composed of serotonergic, noradrenergic and dopaminergic nerve terminals.
The DRN heavily innervates the nucleus accumbens [68], a brain region receiving β-endorphin containing nerve terminals originating from the arcuate nucleus [69]. In humans, rats, and many other species, injection of β-endorphin into the nucleus accumbens exerts an analgesic effect [70-73]. It was hypothesized that the effect of 5-HT on chronic pain might be due to an interaction with endogenous opioid systems [74,75]. In line with this hypothesis, it has been shown that local application of 5-HT can facilitate the release of β-endorphin in the arcuate nucleus and nucleus accumbens [76]. The involvement of 5-HT in the nucleus accumbens in mediating the antinociceptive effect was further suggested by the finding that the local application of cinanserin, a 5-HT2 receptor antagonist, attenuated the antinociceptive effect of morphine [77]. Several possibilities has been raised regarding the interaction between 5-HT and β-endorphin: (i) the existence of enkephalins in about one third of the neurons located in the DRN [78] suggests that 5-HT and enkephalins may act as co-transmitters in the synaptic events with one playing a regulatory function for the other; (ii) 5-HT released in the nucleus accumbens may activate the enkephalinergic interneurons within the nucleus, as demonstrated in the caudate putamen [79]; (iii) enkephalins may accelerate the release of 5-HT, although no evidence is yet available in favor of this possibility.
The LC send projection to the striatum [or caudate-putamen (CPu) in human] as suggested by immunohistochemical and fluorescence histochemical studies [80]. The CPu is one of the important components of the basal ganglia, and is recognized as one of the several sites involved in the modulation of nociceptive sensory input through descending controls to the level of the spinal cord. It is rich in monoamine neurotransmitters, including NE and DA [80,81]. Neurons in the CPu respond to noxious thermal, mechanical and electrical stimulation [64,82]. Interestingly, the stimulation of the CPu induces analgesia [83] while the CPu display two types of neurons named PEN and PIN (“on-cells” and “off-cells”), which are excited or inhibited respectively by nociceptive stimulation. NE potentiated the electric activities of the evoked discharges of PEN and simultaneously attenuated those of PIN, i.e. exhibiting the hyperalgesic effects of NE [84]. The latter study illustrate the fact that NE is involved in the modulation of nociceptive information transmission through an action in the CPu [85].
The VTA and substantia nigra (SN) send dense projection to the nucleus accumbens and basal ganglia [86]. Clinical and behavioral data indicate that dopaminergic pathways are involved in central pain processing. Data from electrical and chemical (i.e., DA receptor agonists and antagonists) stimulation or electrolytic and chemical lesions of the CPu, GP and substantia nigra (SN) provide evidence that the basal ganglia can modify behavioral responses to noxious stimulation. For example, it has been shown that a unilateral lesion of the nigrostriatal pathways causes hyperalgesic responses to painful stimuli at contralateral side [64,87-89]. As well, DA depletion by 6-OHDA injection into the medial forebrain bundle, CPu and SN results in hypersensitivity to mechanical [90,91], electrical [92] and thermal stimulation [91]. A recent study found hyperalgesic responses to painful chemical stimulation of the hind paw ipsilateral to intrastriatal 6-OHDA administration [88]. The role of DA in pain needs to be further investigated. Nevertheless, it seems that the activation of nigro-striatal dopaminergic transmission is associated with individual variations in subjective ratings of sensory and affective qualities of pain, whereas mesolimbic activation appears associated with variations in emotional responses during pain challenge [20].
The limbic system mainly consists of the anterior cingulated cortex (ACC), the insular cortex (IC), the prefrontal cortex (PFC), the amygdala and the hippocampus. Monoaminergic neurons project into these various brain regions and are involved in the regulation of pain, mood and in the affective dimension of pain [93]. Dysfunction of these ascending projections from the DRN, LC and VTA may contribute to the classical symptoms of depression. The activation of cortical structures has been shown in humans using imaging studies in response to pain [94-98]. Lesions of cortical region such as the ACC significantly reduced acute nociceptive responses [99]. The involvement of the ACC in pain modulation may be attributable to the activities of a variety of neurotransmitters as DA and glutamate [7]. Indeed, it has been reported that increased activity of glutamatergic projections intensifies nociception whereas dopaminergic projections into the ACC inhibits nociception [100]. DA also appears to be key neurotransmitters in nociception modulation in the IC that displays a high density of DA fibers arising principally from the VTA and SN [101].
The role of 5-HT and NE in these cortical regions is also well documented. For example, the increase in 5-HT extracellular levels induced by the inhibition of the 5-HT transporter (5-HTT) in the primary somatosensory cortex produces anti-hyperalgesic and anti-allodynic effects [102].
About the insular cortex, essentially considered as the anatomical substrate for integration and processing information from different functional systems, the mid-posterior part has been involved in somato and viscerosensory painful stimuli. Dense connections and interconnections between the different cortical areas allow multimodal integration of both informations [103].
The amygdala performs a primary role in the formation and storage of memories associated with emotional and affective events [104] and plays a key role in attaching emotional significance to pain [105]. Amygdala receives input from LC noradrenergic projections [19] and is involved in defense response, i.e. analgesia, associated with intense fear and dangerous situations [106]. Imaging studies showed an activation of the amygdala in response to different painful stimuli [107]. Changes in 5-HT receptor function in the amygdala were observed under a chronic pain-like state [102]. Apart interactions with hypothalamus and brainstem, it has been described that amygdala is involved in cognitive effects of pain through amygdala-cortical interactions. In addition, pain-related decision-making deficits involve increased GABAergic synaptic inhibition in prefrontal cortex [9].
Finally, activation of the hippocampus has been demonstrated in healthy volunteers in response to a pain stimulus [108], and preclinical studies have reported changes in the hippocampal morphology, cell proliferation and gene expression in response to chronic pain [109,110]. Since the hippocampus receives a dense monoaminergic innervation, it is possible that the increase in extracellular levels of 5-HT, NE and DA, each monoamine known to stimulate neurogenesis and the expression of neurotrophic factors in the hippocampus [111-113], may produce antinociceptive effects. This is in agreement with a recent study showing that chronic pain suppresses the increase in the immunoreactivity of doublecortin-positive cells (a marker of neuron maturation) induced by an enriched environment [114].
2. Pharmacological Properties of Monoamines Reuptake Inhibitors
For many years, studies mainly focused on the serotonergic and the noradrenergic systems because of the efficacy of selective 5-HT or NE reuptake inhibitors (SSRIs/NRIs) in the treatment of major depressive disorder. SSRIs and NRIs block the 5-HT or NE transporter (5-HTT or NET), respectively; thereby increasing extracellular concentrations of these monoamines in the synapse and prolonging their duration of action at postsynaptic level. Despite the variety of SSRIs (citalopram, escitalopram, fluovoxamine, fluoxetine, paroxetine and sertraline) and NRIs (atomoxetine, desipramine, reboxetine), their binding property towards monoamine transporters may vary [115]. In addition, since close anatomical and functional interactions between monoaminergic systems exist, any action on one system may reverberate in the other system [116]. A corollary of this cross-modulation is that the in vivo net effect of SSRIs or NRIs on 5-HT or NE neurotransmission is difficult to anticipate. Functional in vitro and in vivo approaches have thus been applied to characterize the pharmacological properties of these antidepressants. Inhibition of [3H]-5-HT or [3H]-NE reuptake in synaptosomes, is one of the most widespread method to assess the in vitro potency of reuptake inhibitors [117] and to predict indirectly, their affinity and selectivity on biogenic amines transporters. Intracerebral electrophysiology and microdialysis have proven to be sensitive methods to assess the in vivo inhibitory potency of various drugs on reuptake. Indeed, at presynaptic level, when 5-HTT or NET are blocked on the serotonergic or noradrenergic cell bodies, respectively, there results an accumulation of 5-HT or NE in the vicinity of somatodendritic 5-HT1A or α2 autoreceptors in the dorsal raphe (DR) or locus coeruleus (LC). This lead to an attenuating firing DR 5-HT and LC NE neurons in a dose-dependent manner due to the activation of these neuronal elements exerting a negative feedback influence [118]. This parameter can be used to characterize the pharmacological profile of reuptake inhibitors. At nerve terminals, an accumulation of 5-HT or NE also occurs in response to the inactivation of the 5-HTT or the NET by SSRIs or NRIs, and the enhancement of extracellular levels of monoamines can be probed by microdialysis in various brain regions [119] and constitutes another parameter to study the functional activity of reuptake blockers. Nevertheless, since microdialysis methodology may vary between laboratories, the electrophysiological approach seems to be the most appropriate approach to establish relevant comparisons between compounds.
2.1. Single- and Dual-Acting Monoamine Reuptake Inhibitors
The six approved SSRIs in the treatment of depression are all potent 5-HT reuptake inhibitors in vitro as well as in vivo [117,120]. Paroxetine is the most potent inhibitor of 5-HT transporter, whereas citalopram and escitalopram are the most selective ones [115,117]. However, the 5-HT/NE ratios vary considerably between SSRIs (Table 1). Multiple sources of evidence from electrophysiological studies indicate that SSRIs reduce the firing activity of DR 5-HT neurons in rats with ED50 ranging between (60 and 600 μg/kg; i.v.). It is important to note that some of them also produce a weak, but significant decrease in LC NE and VTA DA neuronal activities after acute or sub-chronic administration (Table 2). As expected, microdialysis studies showed that all SSRIs enhance the extracellular levels of 5-HT. Interestingly, SSRIs may also enhance the extracellular levels of NE in the frontal cortex and hippocampus after acute or chronic administration in rodents [15,121-129]. Different hypotheses have been raised to explain such effects of 5-HT reuptake inhibitors on the noradrenergic system. It is possible that an increase in 5-HT is a prerequiste to stimulate the central NE transmission. Accordingly, the release of NE in serotonergic nerve terminals areas was observed in response to the activation of postsynaptic 5-HT1A [130] or 5-HT3 receptor types [131]. Together with the observation that SSRIs decreased the spontaneous neuronal activity of NE neurons in LC [132-134], this suggests that the enhancing property of SSRIs on the noradrenergic system may involve a local excitatory mechanism at nerve terminals, independent from the inhibition of the NET. Given the high degree of homology between monoamine transporters, another possibility would be that SSRIs exert non-selective effect through the blockade of the NET. In line with this assumption it was shown that paroxetine blocks the NET [135,136]. In addition, we have recently shown that the enhancement of cortical extracellular levels of NE induced by escitalopram remained intact in 5-HTT−/− mice, while the increase in cortical extracellular levels of 5-HT was suppressed (unsubmitted data), confirming the possibility that SSRIs may also inhibit, at least partially, the NET. Aberrant uptake of NE from serotonergic nerve terminals might also explain that SSRIs increase the extracellular levels of NE [137] but this latter point is still debate of matter. Finally, the pharmacological profile of SSRIs and more paticularly their monoaminergic receptor-profile may also play a significant role in the modulation of NE neurotransmission. As an example, the 5-HT2C blocking activity of fluoxetine [138] may prevent SSRI-induced decrease in LC NE neuronal activity [139] and consequently may participate in stimulating NE at nerve terminals.
Atomoxetine, desipramine and reboxetine are potent and selective NET inhibitors [150]. Their capacity to inhibit the NET in vitro is in the nanomolar range, whereas data from synaptosomes studies indicate that their potency to inhibit 5-HT reuptake is at least 50 times weaker (Table 1). In term of electrophysiological property, desipramine and reboxetine inhibit the firing activity of rats LC NE neurons with an ED50 of 110 and 480 μg/kg (i.v.), respectively. Accordingly, in vivo microdialysis experiments have revealed that this class of antidepressants is very potent at enhancing brain NE transmission in the rat frontal cortex, hippocampus and nucleus accumbens either after acute or chronic administration [15,151-156]. In contrast, these drugs had no effect on the DR 5-HT and VTA DA neuronal activities (Table 2), confirming their selectivity towards the NET. Nevertheless, the effects of reboxetine on the serotonergic system remains equivocal since an inhibition [157] or an increase [144,158] in 5-HT firing activity in rats was described after its systemic administration. Moreover, although additional works are required to elucidate the impact of NRIs on the serotonergic system, some studies failed to demonstrate that reboxetine increased 5-HT outflow [156,159]. Svensson and colleagues found that in rats it enhanced the extracellular levels of 5-HT despite their apparent low affinity for 5-HT reuptake sites [158]. Hence, NRIs may cause a secondary enhancement of central serotonergic activity by a mechanism separate from 5-HT reuptake inhibition. Accordingly, it has been proposed that the enhanced levels of synaptic NE in postynaptic regions likely enhanced serotonergic activity through the stimulation of postsynaptic α1-adrenoceptors located on 5-HT cell bodies in the DR [160,161]. Interestingly, since prolonged administration of desipramine or reboxetine desensitize α2-adrenergic receptors present on 5-HT terminals, it is possible that such a mechanism lead to an increase in synaptic availability of endogenous 5-HT as observed in the rat hippocampus [158].
A second generation of antidepressants targeting both 5-HT and NE, named serotonin and norepinephrine reuptake inhibitors (SNRIs), has then been developed with the hope to produce more robust therapeutic effects in depression. Among these antidepressants, duloxetine, milnacipran and venlafaxine have provided one of the first opportunities to propose that a specific dual-acting antidepressant would be significantly more effective than SSRIs in depressed patients [174,175]. Indeed, it is currently believed that the therapeutic efficacy of antidepressant drugs may depend on their capacity to enhance simultaneously brain 5-HT and NE neurotransmissions. Although SNRIs inhibit both 5-HTT and NET, there are considerable differences in their affinity, selectivity and potency [115] (Tables 1). In vitro, duloxetine and venlafaxine have a high affinity for the 5-HTT compared to the NET whereas milnacipran has a more balanced affinity for these transporters (Table 1). Electrophysiological experiments in rats have confirmed these pharmacological properties since duloxetine and venlafaxine reduce the firing activity of DR 5-HT neurons with an ED50 lower than that obtained for LC NE neurons (Table 2). Surprisingly, although high doses of milnacipran are required to inhibit the discharge of 5-HT neurons in rats, it does not modify the neuronal activity of NE neurons (Table 2). The effects of the three SNRIs on the extracellular levels of 5-HT and NE have been extensively studied. In freely moving rats duloxetine produced a dose-dependent increase in the extracellular levels of both 5-HT and NE in the frontal cortex or hypothalamus [176-178]. Venlafaxine enhanced the levels of NE but not 5-HT in the cortex [179,180]. Other studies, however, found a dose-dependent increased of both 5-HT and NE concentrations in the frontal cortex and hippocampus [124,144,181,182] with a greater increase of 5-HT output compared with that of NE [122]. About milnacipran, microdialysis studies revealed that it produced a dose-related similar increased in both extracellular 5-HT and NE in the guinea pig hypothalamus [141,183] in agreement with its in vitro pharmacological profile [184]. It is interesting to note that, in agreement with their pharmacological profile, neither venlafaxine nor milnacipran increased the extracellular level of DA in the rat hippocampus or frontal cortex [182,185]. However, a microdialysis study reported an enhancement of extracellular level of DA in the rat frontal cortex and nucleus accumbens in response to the administration of duloxetine [178]. These neurochemical data are surprising but agree with the observation that duloxetine displays a higher affinity for the DA transporter (DAT) [186,187], albeit weak, than the two other SNRIs.
The recent observation that pure dopaminergic drugs, such as the D2/D3 receptor agonist pramipexole, a drug without apparent affinity for either NE or 5-HT neuronal elements, are effective antidepressants [188], suggested that enhancing DA function may underlie, at least in part, a therapeutic response in major depressive disorder. So far, no selective DA reuptake inhibitors are available in clinic. Bupropion and nomifensine remain the sole drug used in the treatment of depression and sharing the property of inhibiting the DAT [118]. Nevertheless, both agents are not selective and display a noradrenergic component. Bupropion is an effective antidepressant when used alone or in combination with SSRIs that would exert its therapeutic effect through the selective blockade of the DA, but also NE transporter (Table 1). Despite this pharmacological profile, acute administration of bupropion had no effect on the firing activity of VTA DA and LC NE neuronal activities (Table 2). However, it has been shown to enhance extracellular levels of DA in the striatum and frontal cortex of mice and rats [189-192]. Together, these data indicate that bupropion display an original mechanism of action. In line with this hypothesis, positron emission tomography (PET) scan studies reported that clinically effective doses of bupropion produce very low occupancy of DA reuptake sites [193]. In preclinical studies, bupropion was hypothesized to be a NE releaser in LC and at the level of NE terminals in the DR [171]. Hence, administration of sub-acute bupropion dose increases firing of 5-HT neurons whereas NRIs do not. This effect was no longer present in NE-lesioned rats suggesting the involvement of the noradrenergic system in its mechanism of action. It is thus possible that the effect of bupropion on the serotonergic system would be mediated via α1-adrenoceptor, which exerts an excitatory action on 5-HT neurons activity [171,194-196].
Nomifensine is an antidepressant with potent NE and DA reuptake inhibiting properties (Table 1). Animal studies showed that an acute administration of nomifensine had no effect on the neuronal activity in the LC but it markedly decreased the firing rate of NE neurons after 2 days of treatment [172] as previously found with the NRI reboxetine. Similar to NE neurons, the firing rate of DA neurons in the VTA was significantly decreased with a short-term nomifensine regimen (Table 2) while increasing the firing rate of 5-HT neurons when the treatment was prolonged [172]. It is well documented that increases in synaptically available NE and DA, a property that was observed by using microdialysis in response to the administration of nomifensine [197]. Such neurochemical effects could also account for the activation of 5-HT neurons via excitatory α1-adrenoceptors [160] and D2 receptors [198-200] in the DR.
In summary, a wide range of monoaminergic antidepressants is thus available for clinicians. These agents with either selective or dual actions have distinct mechanisms of action and it seems important to take into consideration the high degree of connectivity between monoaminergic systems [173] to predict their in vivo effects. Understanding the mode of action of drugs targeting these catecholaminergic neurotransmitters can improve their utilization in monotherapy and in combination with other compounds particularly the SSRIs. Due to the purported role of DA in the depression, triple reuptake inhibitors (TRIs) could be the next generation of antidepressants after SSRIs, NRIs, SNRIs, in the treatment of depressive disorders and other symptoms such as sexual dysfunction and/or chronic pain [115].
2.2. Pharmacological Properties of Triple Monoamines Reuptake Inhibitors
A number of compounds with the ability to bind and block all three monoamine transporters have been developed. To illustrate the growing interest for TRIs, it is interesting to note that just in 2010, the pharmacological profiles of seven compounds have been reported [201-207]. Experiments from rat cortical synaptosomal fractions indicate that the inhibition of monoaminergic transporters by TRIs is lower than that of single- or dual-acting agents (Table 1). This indicates that the novelty of this new class of antidepressant lies in their balanced profile at blocking 5-HTT, NET and DAT rather than in their in vitro potency. In an attempt to further characterize the functional activity of TRIs, recent electrophysiological and neurochemical studies have been conducted. In agreement with a low in vivo potency in comparison with single- or dual-reuptake inhibitors, relative high intraveinous doses of the TRIs SEP225289 and DOV216303 were required to inhibit the electrical activities of DR 5-HT, LC NE and VTA DA neurons. Although this may result from a lower affinity for the monoaminergic transporters than selective reupake inhibitors [115] or from a poor brain penetration, 5 mg/kg of DOV216303, produced an inhibition of 80% of LC NE neuronal activity but only of 30% and 40% of DR 5-HT and VTA DA neurons; respectively [173]. The observation that both TRIs exerted a predominant effect in the LC, while producing only a partial decrease in DR 5-HT firing activity was puzzling given the equal in vitro affinity and potency of the former drugs for all three transporters. The reciprocal interactions between monoaminergic neurons might have thus contributed to alter the functional in vivo activity of TRIs because the majority of SSRIs, NRIs and SNRIs produce a complete suppression of DR 5-HT neurons firing (Table 2). The possibility has been raised that the lesser than expected effect of SEP225289 or DOV216303 on the firing activity of 5-HT neurons resulted, at least in part, from the accumulation of DA and NE in the DR, which are supposed to be excitatory [160,198-200]. In line with this hypothesis, when SEP225289 was administered to rats following an acute intravenous administration of the 5-HT1A receptor antagonist WAY100635, the discharge of 5-HT neurons was blocked [173]. These results further demonstrate that the excitatory influence of catecholamines (i.e., NE and DA) specifically unveiled in response to 5-HT1A autoreceptors inactivation. The observation that bupropion or nomifensine reversed escitalopram-induced decrease in 5-HT neuronal activity [170,172] further supports the idea that the simultaneous increase in cathecholamines may counteract the electrophysiological effect of 5-HTT inhibition in the DR. Despite these results, in two recent microdialysis studies in rats DOV216303 (20–60 mg/kg) simultaneously increased all three monoamines but did not produce a greater increase in cortical 5-HT outflow in comparison to catecholamines [201,208]. It is noteworthy that JAZ-IV-22, a new TRI displaying a balanced profile close to that reported for DOV216303, produced similar neurochemical effects than the former compound [201]. PRC200-SS (5 and 10 mg/kg; i.p.) also stimulated the cortical extracellular levels of all three monoamines [149] with a marked effect on NE and 5-HT transmissions, in agreement with its in vitro pharmacological profile. Nevertheless, PRC200-SS failed to modify the extracellular levels of DA in the mPFC. This is somewhat surprising given the dense dopaminergic innervation and the high expression of DAT in this brain region in rats [209]. These neurochemical observations also contrast with the fact that catecholamine uptake blockers such as nomifensine, desipramine and reboxetine increased, although moderately, DA levels [210-214]. Interestingly, in the core of the nucleus accumbens (Nacc), PRC200-SS (10 mg/kg; i.p.) increased DA, and, to a lower extent, 5-HT outflow without affecting NE, probably due to the highest density of dopaminergic nerve terminals in this brain region. As a last example to emphasize the contrast between in vitro and in vivo functional activity of TRIs, JNJ7925476 produced a robust and dose-dependent increase in all three monoamines, with a maximal effect for DA (15-fold above basal level) compared to 5-HT and NE (5–7 fold above basal level) at the highest dose tested (10 mg/kg; s.c.) in the cortex of freely moving rats [215]. However, data indicate that JNJ7925476 displayed a better in vitro binding affinity and blocking activity for 5-HTT and NET than for DAT in rats [215]. Differences in transporter occupancy cannot explain these findings since this parameter followed the same trend observed with cortical extracellular monoamines levels. It was therefore proposed that the high cortical levels of DA might have resulted from the blockade of the NET by this drug, which displays a high affinity for the DAT [16,216]. Finally, it is interesting to note that among the recently synthesized TRIs, despite their overall balanced profile, their systemic administration in rodents resulted in considerable variations between 5-HT, NE and DA extracellular levels [205,206].
All these findings illustrate the fact that the in vivo activity of reuptake inhibitors will not necessarily reflect their in vitro functional activity, probably due in part, to the functional interactions between monoaminergic neurons. Despite such differences, all TRIs, depending on the brain region, are able to enhance monoaminergic neurotransmission to produce antidepressant-like effect in naïve or depressive-like animals. Thus, in generalized pain states in which mood changes and diffuse pain occur in relation with an attenuation of brain monoaminergic transmission, TRIs could modulate nociceptive systems and display a major role in its relief.
3. Monoamines Reuptake Inhibitors and Pain Preclinical Outcomes
Acute and chronic pains may in part, result from reduced levels of endogenous 5-HT, NE and DA activity, at both the spinal and supraspinal levels [217]. As described in the first part of this review and depicted in Figure 1, nociception is a bi-directional process of ascending and descending neuronal pathways involving monoaminergic systems whose activation may have an inhibitory influence on pain. Consequentially, it is presumed that 5-HT, NE and/or DA reuptake inhibitors may attenuate pain by preventing their presynaptic reuptake, leading to increased postsynaptic monoamines' levels and sustained activation of the descending pain inhibitory pathways [218]. In this part, we will describe the preclinical data supporting the role of currently available antidepressant drugs in the control of pain. Despite the complexity of pharmacological interactions between monoaminergic neurons that sometimes may attenuate monoaminergic neurotransmission (Figure 1), one would expect a better efficacy of dual-or triple-acting agents over selective 5-HT or NE reuptake inhibitors. Indeed, since all three monoamines are involved in antinociception, the recruitment of more than one system may produce beneficial effects. Although findings indicate that the antinociceptive potency of reuptake inhibitors varies according to their monoamine specificity, the nature of stimuli and the animal models of pain [219], this section may help determine the optimal choice of monoaminergic activity in the management of pain. In particular, it is important to consider the potency of monoaminergic reuptake inhibitors in animal models of pain in regard to their in vivo pharmacological properties towards SERT, NET and DAT and the reciprocal interactions between monoaminergic systems.
3.1. Serotonin Selective Reuptake Inhibitors (SSRIs)
The idea that the inactivation of SERT and the related increase in extracellular levels of 5-HT [220] could be a relevant strategy in the relief of pain, is supported by the observation that morphine-induced analgesia is potentiated in 5-HTT deficient mice [221]. Although the spontaneous pain sensitivity is unaltered in these mutant mice compared to their wild-type littermates [221], multiple sources of evidence suggest that the pharmacological blockade of 5-HTT induced by SSRIs reduces acute pain in the hotplate and tail flick tests (Table 3). For example, citalopram produces antinociceptive effects in both rats [222-224] and mice [219,225]. Such antinociceptive behaviors were reported with other SSRIs such as fluoxetine [226,227], fluvoxamine [219,222,228], paroxetine and sertraline [229-232] (Table 3). Interestingly, the effects of fluoxetine are completely blunted in 5-HT depleted animals [227,233] suggesting that SSRIs-induced antinociception involves serotonergic pathways. In addition, fluoxetine but also fluvoxamine, paroxetine and sertraline significantly potentiated the analgesic effect of morphine [234-239] (Table 3). Although these effects were blocked by naloxone, fluoxetine did not alter the binding of [3H]-naloxone demonstrating the lack of affinity of this SSRI for opioid receptors [239]. The site of action of SSRIs remains poorly studied and somewhat equivocal. It was reported that the effects of SSRIs may involve supraspinal structures since the intracerebroventrical, but not intrathecal, injection of citalopram mimics the effect of its systemic administration [223]. Nevertheless, this contrasts with studies demonstrating that citalopram attenuated evoked glutamate release in the dorsal horn of the anesthetized rat [240] or studies reporting that the antinociceptive effects of SSRIs resulted from an increase in opioid transmission in the spinal cord [241,242].
It is thus possible that the antinociceptive effects of SSRIs in acute pain involves both spinal and supraspinal mechanisms. This hypothesis is supported by the fact that in “positive” studies comparing the effects of SSRIs in the tail flick and hot plate tests, these pharmacological agents produced analgesic responses in both tests (Table 3). Indeed, tail flick is known to result from a spinal reflex [243] whereas in the hot plate test, licking or jumping responses, are known to be the result of supraspinal sensory integration [244,245]. In a recent study comparing the effect of various SSRIs in the hotplate test, differences between citalopram, escitalopram, fluvoxamine and fluoxetine (all SSRIs administered intraperitoneally) were yielded [225]. There are several hypotheses that may explain such differences in the antinociceptive action of SSRIs. Since all three monoamines are involved in antinociception, it is possible that the differential interaction of SSRIs with the noradrenergic and/or the dopaminergic systems may mediate an additional effect. Another possibility may arise from the differential interaction of each SSRI with monoaminergic or non-monoaminergic receptors (e.g. 5-HT2C receptor for fluoxetine, sigma opioid receptor for fluvoxamine).
In chronic neuropathic and inflammatory models of pain, citalopram reduced hyperalgesia [247]. The observation that escitalopram produced more robust antinociceptive effects than citalopram [225], is in agreement with previous study showing that the R-enantiomer may antagonize and thus attenuate the effects of the active enantiomer [116,321,322]. All six SSRIs share this analgesic property in chronic models of pain in rodents since fluoxetine, paroxetine and sertraline also produced anti-hyperalgesic and/or anti-allodynic effects in streptozotocin (STZ)-induced diabetic neuropathy, sciatic nerve ligation (SNL) [241,251,254,255,257,266,268,323] or in inflammatory conditions such as formalin or acetic-acid injection [222,233,260,261,265,324] (Table 3). Surprisingly, the antinociceptive effects in chronic models of pain have been revealed after single administration of SSRIs. Despite all these positive studies, some studies failed to unveil antinociceptive effects of SSRIs in chronic pain [241,258,262,264,271]. This inconsistency of effects needs to be validated following chronic SSRIs treatments.
3.2. Norepinephrine Reuptake Inhibitors (NRIs)
Genetically modified mice lacking the NE transporter exhibit normal nociceptive threshold [325]. This is consistent with findings showing that two NRIs, desipramine and reboxetine, failed to modify the response to a thermal nociceptive stimulation in the hotplate test [273,278,326] (Table 3). Nevertheless, some studies have reported a weak analgesic effect of NRIs in the tail flick and hotplate tests [219,280,281,283]. This could be attributable to the systemic administration of the drugs. Indeed, it was reported that intrathecal injection of desipramine produced antinociceptive effect in the tail-flick [277], but also in other analgesic tests [284,327]. It is also possible that a prolonged administration of NRIs is required to produce antinociception. In line with this hypothesis, repeated administration of desipramine produced analgesia in the tail flick and hotplate tests [282] and potentiated the analgesic effect of morphine [277,328,329]. Another hypothesis would be that NRIs acted at the spinal level since the majority of studies on acute pain suggests that NRIs produce analgesic responses specifically in the tail flick test (Table 3).
In various models of neuropathic and inflammatory pains, both desipramine and reboxetine displayed robust antinociceptive effects by reversing allodynia and/or hyperalgesia either after acute or chronic administration in mice [256,272] and rats [262,274-276,279]. The observation that these antinociceptive behaviors were reversed by naloxone [279] strongly suggests the involvement of the opioid system in the analgesic effect of NRIs. Two others compounds, displaying a high selectivity for the NET were tested in chronic neuropathic pain in rat and appear to be promising candidate for development as novel analgesic drugs. The intrathecal administration of the conopeptide Xen2174 resulted in a long duration anti-allodynic responses in rats with chronic constriction injury (CCI) of the sciatic nerve or an L5/L6 spinal nerve injury [330] whereas WAY-318068 was shown to be efficacious in models of acute, visceral, inflammatory, neuropathic, diabetic and bone cancer pain [331].
Although the increase in extracellular levels of 5-HT or NE induced by SSRIs or NRIs seems to participate, at least in part, in attenuating pain, the impact of both monoamines to nociception remains difficult to evaluate. In a recent study, the relative contribution of 5-HT and NE in the spinal nerve ligation (SNL) model of neuropathic pain has been examined. It was demonstrated that NRIs, desipramine and reboxetine, reversed allodynia, while the SSRI fluoxetine displayed a minimal activity suggesting that compounds with greater affinity for the NET are more effective in attenuating pain than compounds with a greater affinity for the SERT [256]. Such results could be explained by the serotonin's propensity to both facilitate and inhibit pain in contrast to NE, which is purely antinociceptive. Interestingly, in the latter study, paroxetine produced similar anti-allodynic effect to that observed with NRIs. This unexpected property of paroxetine, which is reported to be a selective SRI in vitro, may involve the noradrenergic property of paroxetine compared to fluoxetine (Table 1). This is in agreement with previous studies showing the capacity of paroxetine to block NET [135,136] and increase the extracellular levels of NE in the frontal cortex and hippocampus of rodents [118-126]. Another study comparing the antinociceptive effects of citalopram and duloxetine showed that in the hot plate test, duloxetine significantly increased the nociceptive response latency, whereas citalopram was ineffective [247], further supporting the fact that the simultaneous enhancement of 5-HT and NE neurotransmissions may induce synergistic effects [218]. SNRIs may thus produce more robust effect than SSRIs or NRIs in animal models of pain. Nevertheless, the efficacy of the highly selective NET inhibitors Xen2174 and WAY-318068 suggests that NRI activity alone was sufficient to produce analgesia.
3.3. Serotonin Norepinephrine Reuptake Inhibitors (SNRIs)
As observed with NRIs, duloxetine has no or modest effect in acute models of pain in mice [289] or rats [247,293]. Interestingly, when both tail flick and hot plate were tested in the same study, antinociceptive responses were only observed in the hot plate paradigm, suggesting an involvement of supraspinal mechanism. However, it clearly appears that in both species, duloxetine produced antinociceptive effects by reducing hyperalgesia and/or allodynia in chronic neuropathic [285,293] or inflammatory models of pain [247,289,293] (Table 3). Interestingly, duloxetine has a more pronounced antinociceptive potency than milnacipran or venlafaxine [290,293]. This may result, at least in part, from the fact that duloxetine, but not milnacipran and venlafaxine, increased DA transmission [178,182,185] in addition to their neurochemical effects on the serotonergic and noradrenergic systems. This is in agreement with the pharmacological profile of duloxetine, which is more potent at blocking SERT and/or NET than milnacipran or venlafaxine (Table 1 and Table 2). Milnacipran also displays robust anti-hyperalgesic and anti-allodynic effects in various models of chronic pain such as the ligature of the sciatic nerve or the fifth lumbar nerve [102,241,271,296,299,301], or inflammatory pain [228,290,300]. The demonstration of the contribution of both serotonergic and noradrenergic systems in the antinociceptive property of milnacipran or venlafaxine comes from a recent study showing that the effects of these SNRIs can be prevented by the inhibitor of 5-HT or NE synthesis, parachlorophenylalanine or alpha-methyl-para-tyrosine; respectively [301,332]. The observation that naloxone also attenuated the antinociceptive effect of milnacipran in these models [301], emphasizes the importance of the opioid system in its behavioral effects. Although the site of action of milnacipran has yet to be determined, evidence suggests that it produces antinociceptive effect not only at the spinal but also at the supraspinal level. Indeed, the effects of the systemic administration of milnacipran were reproduced after its intrathecal administration [241,271,298]. At the supraspinal level, milnacipran attenuates the increase in c-fos expression in the ACC in response to noxious stimulation [299]. Surprisingly, in contrast to duloxetine and milnacipran, venlafaxine produced antinociceptive effects in mice and rats in acute pain assessed in the hotplate test [303,304,311]. Although venlafaxine has a weak affinity for 5-HT and NE transporters in comparison with duloxetine and milnacipran, the reasons of such difference remain obscure. It is possible that the metabolism of these drugs play an important role in their pharmacological activity. Indeed, the fact that venlafaxine is metabolized into desmethylvenlafaxine may potentiate its antinociceptive property. In rats, it has been shown that venlafaxine may act as an SSRI at low doses (<10 mg/kg; i.p.) and begin to exhibit dual reuptake activity at doses >30 mg/kg; i.p. Consequently, it was proposed that the maximal antinociceptive effect of venlafaxine was observed when it was acting as a dual re-uptake inhibitor. Finally, as observed with duloxetine and milnacipran, venlafaxine attenuated hyperalgesia and/or allodynia in various models of neuropathic [265,272,309,310,315,332,333] or inflammatory pain [307,313] (Table 3). In extension of these findings, venlafaxine dose-dependently attenuated formalin-induced nociceptive behaviors [265], an effect antagonized by naloxone [306].
Thus, although limited, studies comparing the analgesic effect of monoamines reuptake inhibitors in rodents suggest that dual acting antidepressants are more active in alleviating acute or chronic pain than selective NE reuptake inhibitors, which themselves appear more potent than SSRIs [334]. The rank of order of analgesic effects of these antidepressants is thus: SNRIs > NRIs > SSRIs.
3.4. Norepinephrine and Dopamine Reuptake Inhibitors
The role of DA reuptake inhibitor has been poorly studied in animal models of pain. This is likely due to the lack of selective DA reuptake inhibitors currently available. However, it was reported that the DA reuptake inhibitor, GBR12909 did not affect nociception in thermal or mechanical tests [335,336] but decreased buspirone-induced analgesia in the thermal test [336]. It was also shown that microinjection of the DA reuptake inhibitor GBR12935 into the rostral agranular insular cortex increased pain thresholds to radiant heat at baseline and produced analgesia to sustained pain from subsequent intraplanar formalin [337].
The contribution of DA and the putative interest of DA reuptake inhibitors in relieving pain must be estimated also from the behavioral effects of agents displaying a blocking DAT activity such as bupropion or nomifensine. In rats, bupropion decreased allodynia induced by CCI and spinal nerve ligation models of neuropathic pain [265]. The inability of reboxetine to reverse mechanical allodynia, in combination with in vitro and in vivo pharmacological property demonstrates that bupropion is a much weaker inhibitor of NE reuptake than reboxetine [338-340] (Table 1 and Table 2), might suggest that inhibition of DA reuptake accounted for the anti-allodynic action of bupropion. Correspondingly, the inability of bupropion, in contrast to reboxetine, to attenuate formalin-induced nociceptive behaviors and thermal hyperalgesia in CCI rats might similarly be explained by its weak potency at inhibiting the NET in contrast to reboxetine (Table 1 and Table 2). The analgesic properties of the catecholamine uptake inhibitor nomifensine were investigated in the tail immersion, hot plate and formalin tests. Systemic administration of nomifensine produced analgesia only in the formalin test. The analgesia was not affected by the opioid receptor antagonist naltrexone suggesting that nomifensine analgesia appears to be DA-mediated but independent of opioid mechanisms [316]. Nevertheless, these data contrast with previous studies indicating that nomifensine did not affect nociceptive behavior [317] (or even produced hyperalgesic effects [318].
This poor literature contrasts with extensive data showing the role of DA and related receptors in pain [341]. For example, the systemic administration of the mixed dopamine agonist apomorphine in rodents induced a biphasic dose-response characterized by hyperalgesia at low doses and analgesia at higher doses [342]. Since the antinociceptive effects of higher doses of apomorphine were antagonized by central-acting sulpiride, but not by the peripheral D2-like receptor antagonist domperidone, it was proposed that the activation of central D2-like receptor was involved in the antinociceptive effects of apomorphine. The hyperalgesia due to low dose of apomorphine could stem from selective activation of high-affinity presynaptic autoreceptors, which would attenuate normal dopaminergic reactivity in response to stimulation, whereas higher doses would be necessary to bind postsynaptic targets adequately to affect analgesia [342]. Although the central region(s) mediating the effects of DA on nociception are not clearly identified, pharmacological experiments strongly suggested that the striatum, cerebral cortex and PAG play a major role. Indeed, microinjection of the D2-like receptor agonist quinpirole within the nucleus accumbens has been found to inhibit the persistent phase of formalin-induced nociception in a dose dependent fashion while the D1-like receptor agonist SKF-38393 was without effect [343]. Nevertheless, these results do not necessarily mean that the impact of D1-like receptor in the control of pain is insignifiant. Accordingly, microinjection of a D1-like receptor antagonist into the PAG likewise attenuated opiate-induced analgesia in the hot plate test in a dose-dependent manner, while D2-like receptor antagonism was without effect. These results demonstrated that the dopaminergic network of the PAG participates in supraspinal nociceptive responses after opiate administration through the involvement of D1-like receptors [344,345]. Finally, with respect to the role of DA an related receptors in the regulation of pain, its effect in the spinal cord should be considered. It was reported that intrathecal administration of dopamine induced thermal antinociceptive effects through D2-like receptors when assessed by the tail flick test [346,347]. Activation of spinal dopamine D2-like receptors also reduced pain-related behavior following the establishment of inflammatory pain in both the affected and contralateral limb, while D2-like receptor antagonism decreased pain thresholds [348]. Intrathecal administration of dopamine also increases the mechanical nociceptive threshold as does quinpirole, whereas D1-like receptor activation had no effect [349]. Interestingly, in a recent study combining the dual 5-HT/NE reuptake inhibitor duloxetine with selective D1- or D2-like receptor agonists, it was demonstrated in the rat formalin test, that combination of all pharmacological agents produced superior analgesic effect than that obtained with duloxetine alone [350]. This potentiation of duloxetine-mediated antinociception is interesting because it suggestes that antidepressant that can simultaneously enhance serotonergic, noradrenergic and dopaminergic neurotransmission within nociceptive pathways should provide a broader spectrum of antinociception than dual reuptake inhibitors.
3.5. Triple Reuptake Inhibitors TRIs
Taken together, these pharmacological studies strongly suggest that drugs simultaneously inhibiting the re-uptake of 5-HT, NA and DA may provide a broader spectrum of pain relief in animal models of experimental pain than single- or dual-acting agents. Precedent for the use of TRIs in the treatment of clinical pain exists with nefopam, a non-narcotic analgesic marketed in Europe [351]. DA plays a critical role in nefopam analgesia as indicated by the observation that rats treated with 6-OHDA plus desipramine, which selectively depletes brain dopamine, have a marked reduction in nefopam-induced analgesia [352]. A specific role for D2-like receptors in nefopam-induced analgesia was demonstrated from the observation that the D2-like receptor antagonist sulpiride inbibited the behovorial response of nefopam [353]. The potential interest of TRIs in the relief of pain has been corroborated by a recent publication characterizing the antinociceptive effects of the TRI bicifadine in acute, persistent and chronic models of pain [320] (Table 3). In this study, bicifadine potently suppressed pain responses in two models of acute inflammatory pain in both rats and mice. It also normalized the nociceptive threshold in the complete Freund's adjuvant model of persistent inflammatory pain and suppressed mechanical and thermal hyperalgesia and mechanical allodynia in the spinal nerve ligation model of chronic neuropathic pain. Mechanical hyperalgesia was also reduced by bicifadine in the STZ model of neuropathic pain [320]. The impact of bicifadine on 5-HT, NE and DA neurotransmissions was confirmed by in vitro binding assays and intracerebral in vivo microdialysis study in freely moving rats. In a second study, another TRI, NS7051, has shown comparable antinociceptive properties to tramadol confirming the interest of these antidepressants in the relief of pain [354]. The molecule has undergone several Phase II and III trials for the treatment of pain, including acute postsurgical pain and chronic low back pain [355,356], and is being evaluated for painful diabetic neuropathy [357]. However, bicifadine has failed to meet endpoints in a number of trials such as diabetic neuropathy [358] suggesting that TRIs may be used in specific pain. Other TRIs currently under development for depression should draw attention for future investigation in the field of pain and confirm whether or not they display any activity in diabetic neuropathy.
3.5. Bridging the gap between preclinical and clinical studies
In the perspective to develop analgesic drugs, it is really important to take into consideration that pain research still faces enormous challenges and there remain many obstacles in the treatment of clinical pain. Unfortunately, there are celebrated examples of failed “translation,” where efficacy in animal models predicted efficacy in human clinical trials, but no efficacy was found. The most definitive example of this failure to translate is the substance P (SP) neurokinin-1 (NK1) antagonist MK-869, which failed despite demonstrations of adequate exposure, penetration, and occupancy [359]. There are several criticisms on animal paradigms that can account for the poor relationship between clinical and preclinical studies. First of all, animal pain models only produce conditions of tissue damage but not necessarily all dimensions of clinical pain. Even if animal models do duplicate clinical pain experience like humans, there is a lack of effective assessment tools to detect different dimension of pain experiences in animals [360]. In particular, evidence suggests that there is a network of brain regions involved in sensory, emotional, cognitive and motor processing. Combined to varying extents, and dependent upon conditions, these regions interact to generate the unique forms of pain experienced by different individuals [20]. Consequently, behavioral measures, although very informative, reduce significantly these dimensions of pain suggesting that an integrative approach is necessary to predict a clinical condition or the effects of an analgesic drug [360,361]. Secondly, while the prevalent method of pain assessment in clinical research is a self-reporting system using the visual analog (or numerical) pain scale, animals cannot tell us how much pain they are suffering. Thus, the probability of misinterpreting the evidence obtained from experiments in species other than humans is always present [362]. To overcome this issue, a new index for measuring pain in mice based on the rodents' facial expressions has been recently developed [363]. This study provides a new preclinical approach for scoring the therapeutic potential of analgesic drugs in complement of conventional methods such as the tracking of animal position, locomotion and gross behavior (e.g., grooming, drinking, eating, social approach). This index of pain relies on changes to facial characteristics being identical to those observed in humans. Thirdly, despite this advance in translational research, simple reflex tests (licking an inflamed paw or flicking the tail in response to heat) are still extensively used to measure pain in animal. However, such responses to evoke pain do not closely match the experience of continuous spontaneous pain in humans [364], especially because a persistent pain almost never results from heat stimulation in the clinical setting. In addition, reflexes tend to rely on the spinal cord and brain stem, whereas learned responses involve the brain's cerebral cortex (and are thus closer to the human condition). In these conditions, it seems that brain imaging would offer interesting opportunities to better understand the neurobiology of pain and evaluate the therapeutic potential of analgesic drugs [65]. Functional imaging may be used as a language of translation to help overcome some of these deficits. Others criticisms have been raised to explain the bridge between basic and clinical research in the field of pain. For example, it has been proposed that the existing models using inflammatory mediators such as formalin, carrageenan and Freund's adjuvant, are too artificial, [365]. It has also been argued that design issues and reporting standards in animal experiments are greatly inferior to those currently prevailing in human clinical trials [366]. Specifically, details regarding blinding, randomization, and data dropouts are relatively reported in animal studies of pain, likely leading to high experimental bias. Despite all these limitations that may explain, at least in part, the fact that several drugs active in animals study failed to become analgesic drugs in human, animal models have obvious advantages with respect to standardization of genetic and environmental backgrounds. Novel integrative approaches may help improve preclinical approach to nociception and also to enable the building of bridges between scientists and clinicians.
4. Conclusions and Clinical Perspectives
This review emphasized the fact that pain and depression share common neurotransmitters pathways and that monoaminergic reuptake inhibitors may relieve chronic pain. If chronic pain may result in depression, pain is also a symptom frequently observed in depressed patients [367]. If it is well established that brain 5-HT and NE systems play an important role in the inhibitory descending pathway controlling pain sensitivity, growing evidence suggests that DA may also have a strong influence at the spinal and supraspinal levels [341]. This supports promising efficacity of triple reuptake inhibitors in the treatment of chronic pain, which simultaneously enhance 5-HT, NE and DA neurotransmissions. Although the efficacy of SSRIs in the chronic pain management indicate that SSRIs are more effective than placebo [368], there are indications that the role of NE is more important than that of 5-HT in the relief of pain in preclinical but also in clinical studies [369]. Indeed, in humans, various neuropathic pain syndromes respond to dual-acting agents but not to SSRIs [370]. For example, venlafaxine was shown to be effective in several chronic neuropathic pain syndromes, whereas SSRIs such as fluoxetine or citalopram, did not show any activity [371,372]. In an extensive review of the clinical data, Fishbain and colleagues determined that, overall, dual acting antidepressants were more active than NRIs, which were more active than SSRIs [373]. A pooled analysis of 31 double-blind studies comparing venlafaxine with SSRIs found that the SNRI was significantly more effective than SSRIs in treating somatic symptoms associated with depression [374]. In particular, the proportion of patients with full remission of their somatic symptoms was significantly greater with venlafaxine than with an SSRI [374,375]. Recently, the SNRI duloxetine was the first reuptake inhibitor approved for the treatment of diabetic peripheral neuropathic pain [370,371]. Efficacy of duloxetine, venlafaxine and milnacipran was studied only in fibromyalgia. Six trials including 2220 participants involved duloxetine. Three studies included participants with painful diabetic neuropathy and three treated participants with fibromyalgia. In an open-label trial, duloxetine has shown consistent efficacy in painful diabetic peripheral neuropathy [376], with effectiveness sustained for one year [373]. Duloxetine (60 mg daily) was effective in treating painful diabetic peripheral neuropathy in the short-term to 12 weeks, in fibromyalgia over 12 weeks and 28 weeks. There is moderately strong evidence that duloxetine (60 mg and 120 mg daily) is efficacious for treating pain in diabetic peripheral neuropathy and fibromyalgia. Direct comparisons of duloxetine with other antidepressants and with other drugs whose efficacy was already demonstrated in neuropathic pain would be appropriate [377]. Venlafaxine has shown efficacy in polyneuropathies of different origins, but not in post-herpetic neuropathy [376]. Two case reports have shown bupropion effectiveness in the treatment of non-depressed patients with chronic low back pain of undetermined origin [378]. Furthermore, an open-label trial of 22 patients suggested that bupropion might be an effective and well-tolerated treatment of the neuropathic pain [379]. A randomized, controlled, double blind, crossover study has shown relief of pain in a group of patients with mixed aetiology of neuropathies [380]. The authors concluded that bupropion may be useful for treatment of neuropathic pain, but large-scale studies are needed to confirm this expectation. Regarding triple reuptake inhibitors, bicifadine is being tested in the treatment of chronic diabetic pain (phase II) and chronic low back pain (phase III). The results of these clinical trials should be available in the next months and will likely help determine whether the addition of the dopaminergic component to SNRIs can improve their analgesic efficacy.
To date, when comparative studies are available, clinical trials for pain invariably indicate a superiority of the dual-acting agents for pain treatment compared to single reuptake inhibitors. With the development of milnacipran, duloxetine and now the triple reuptake inhibitors such as bicifadine focusing in their ability to relieve pain, more extensive comparative data should be forthcoming to make a more evidence-based judgement on the superiority of the SNRIs and TRIs in these indications. In this context, preclinical studies are useful to unveil which antidepressants may display the best therapeutic profile and may determine whether doses that produce antidepressant effects also affect pain and reciprocally.
One question that also should draw our attention is the management of side effects and particularly whether the simultaneous enhancement of serotonergic, noradrenergic and/or dopaminergic neurotransmission mitigate or accentuate these side effects. Despite the much improved side effect profile of newer antidepressants, all of them are still associated with adverse effects that vary between classes and within each class [381]. SSRIs have replaced tricyclics as the drugs of choice in the treatment of depression, mainly because of their improved tolerability and safety. Common side effects of all SSRIs include transient nausea, diarrhea or constipation, dry mouth, insomnia, anxiety, somnolence, weight gain and sexual dysfuntion [381,382]. The dual acting antidepressants (SNRIs) have tolerability profiles that are comparable to those of the SSRIs however with an elevation in diastolic blood pressure particularly for venlafaxine [381,382]. Regarding antidepressants that increase DA neurotransmission, bupropion causes dry mouth, constipation, headache, nausea, excessive sweating and tremor but few cardiovascular effects and little sedation. In additon, it has shown a favorable profile in terms of both weight gain and sexual dysfunction [381,382]. As expected from these observations, data from various studies suggested that sexual dysfunction complaints particularly with the SSRIs and SNRIs can be managed by adding of dopaminergic antidepressant such as bupropion [383-385]. On the contrary, if certain medications do have weight gain as a side effect, there is a logical risk of severe increased weight gain when two of these antidepressants are combined [386]. Interestingly, in an effort to manage most SSRI-induced weight gain, it was reported that clinicians opt to switch agents rather than add a specific medication to the exisiting SSRI [387].
Regarding TRIs, a concern with drugs that block DA transporters is their potential reinforcing property and abuse liability [388]. Nevertheless, drugs that block DAT do not necessarily lead to dependence. Indeed, Volkow and collaborators showed that DA-transporter-blocking drugs must induce more than 50% DAT blockade to produce reinforcing effects [388]. Hence, DA reuptake inhibitors have been classified into two groups: type 1 blockers, which produce addiction and euphoria, and type 2 blockers, which do not [389]. It is thus possible that the capacity of DA reuptake blockers to produce dependence may involve other mechanisms that should carefully be considered with multi-targets agents such as triple reuptake inhibitors. Several Phase 1 studies have been conducted to evaluate their adverse effects. In a dose-escalating, placebo-controlled, double blind, Phase 1a trial, no adverse effects were observed after doses of DOV216303 several times higher than the projected therapeutic doses [390]. In a Phase 1b, clinical trial, 10 subjects were given either placebo (n = 3) or drug (n = 7) at 3 doses (25 mg b.i.d., 25 mg t.i.d. and 50 mg b.i.d.) for 10 days. No severe side effects were noted, although diarrhea, vomiting and nausea were observed. These observations contrast with those reported in a recent study showing that more patients suffering from Parkinson's disease and treated with the triple reuptake inhibitor tesofensine (82%) than in the placebo group (74%) experienced adverse events such as a higher rate of nervous system disorders (dyskinesia and headache), gastrointestinal tract disorders (nausea and constipation), and psychiatric disorders (halluci- nations and insomnia) [391]. Finally, weight loss has been observed as an adverse event in studies with tesofensine [392], prompting further research for the indication of obesity.
In conclusion, tolerability profiles of the antidepressant or combination of antidepressants should be considered when making treatment choices. The decision to use such combinations must be tempered by and weighed in conjunction with the knowledge that specific side effects are expected. It seems that the lack of research studies on the role of adjunctive treatments in the management of antidepressant-induced side effects favors monotherapy over polytherapies [387]. In this prospect, further efforts to evaluate the safety, efficacy, and place for antidepressant medication combinations are called for.
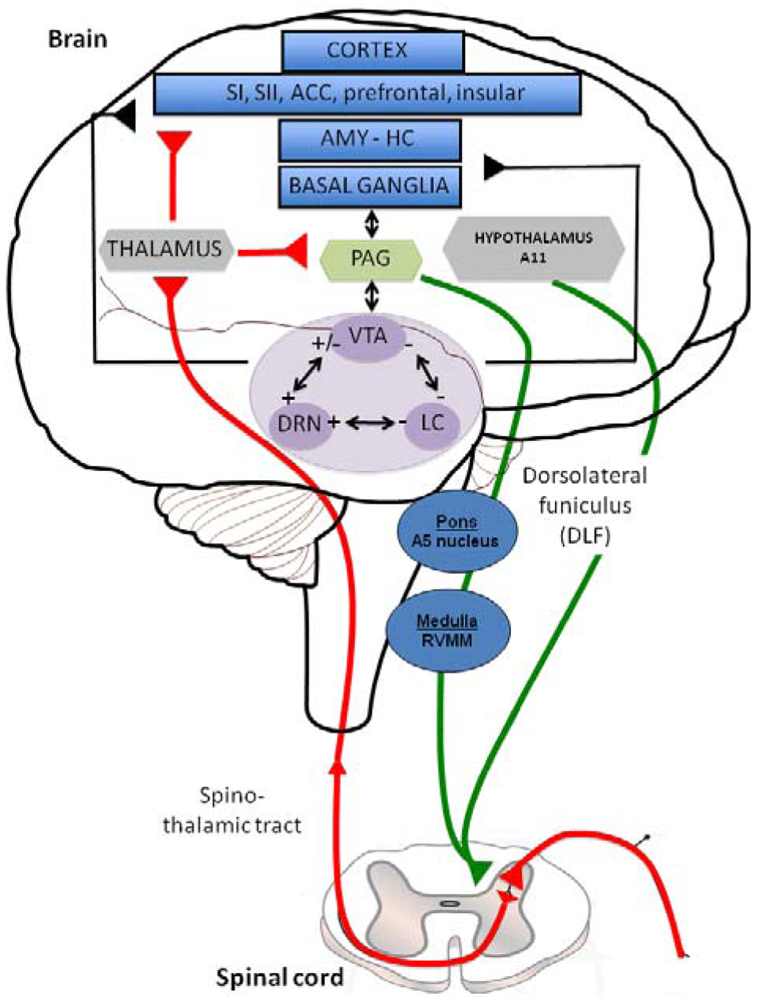

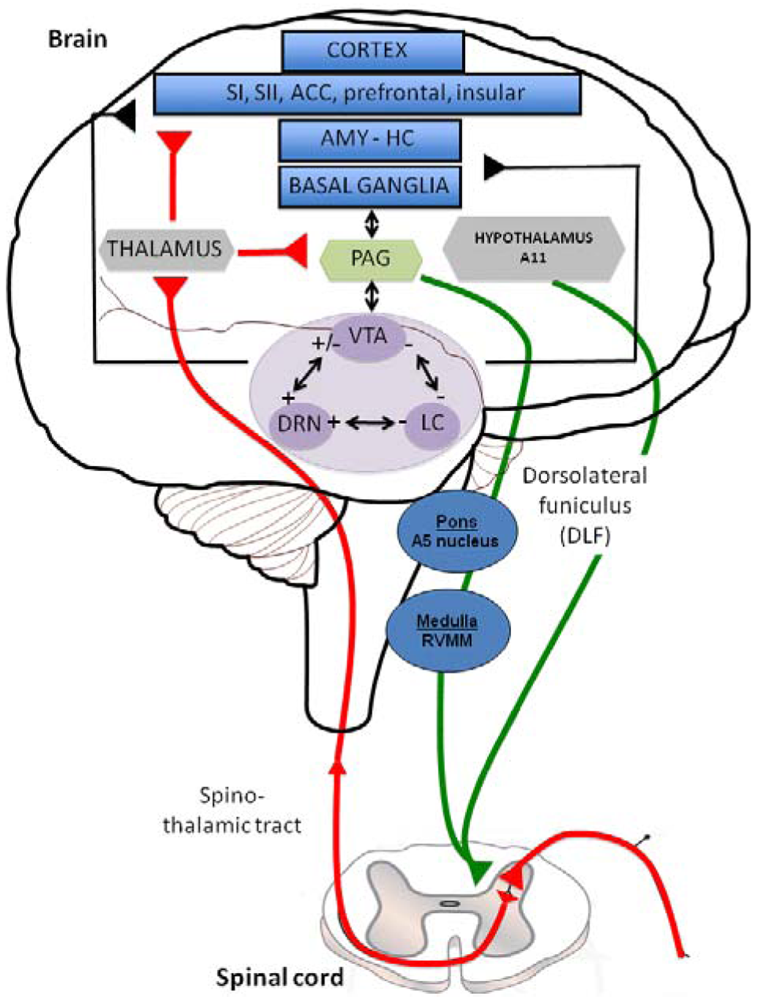{kind=link}
| Drug | Mechanism | In vitro functional activity – Uptake inhibition into rat brain synaptosomes (IC50, nM) | Ref | ||
|---|---|---|---|---|---|
| [3H]5-HT | [3H]NE | [3H]DA | |||
| Citalopram | SERT | ∼3.9 | ∼6100 | >10,000 | [140] |
| Escitalopram | ∼2 | ∼2500 | >10,000 | [140] | |
| Fluoxetine | ∼32 | ∼630 | ∼5170 | [141] | |
| Fluvoxamine | ∼10 | ∼1000 | >10,000 | [141] | |
| Paroxetine | ∼0.5 | ∼97 | ∼1170 | [141] | |
| Sertraline | ∼58 | ∼1200 | ∼1100 | [142] | |
| Atomoxetine | NET | ∼170 | ∼0.25 | ND | [143] |
| Desipramine | ∼380 | ∼1 | ∼3580 | [141] | |
| Reboxetine | ∼794 | ∼2 | >10,000 | [144] | |
| Duloxetine | SERT - NET | ∼1.5 | ∼40 | ND | [143] |
| Milnacipran | ∼28 | ∼29 | >10,000 | [141] | |
| Venlafaxine | ∼63 | ∼316 | ∼7940 | [144] | |
| Bupropion | NET - DAT | ∼19,900 | ∼1500 | ∼600 | [145] |
| Nomifensine | ∼830 | ∼6.6 | ∼48 | [145] | |
| DOV102677 | SERT - NET - DAT | ∼133 | ∼103 | ∼129 | [146] |
| DOV21947 | ∼12.3 | ∼22.8 | ∼96 | [147] | |
| JNJ-7925476 | ∼1* | ∼0.9* | ∼2.6* | [148] | |
| PRC200-SS | ∼2.1* | ∼1.5* | ∼61* | [149] | |
| Drug | Mechanism | In vivo functional activity – Inhibition of neuronal activity (IC50, μg/kg; iv) | Ref | ||
|---|---|---|---|---|---|
| DR 5-HT | LC NE | VTA DA | |||
| Citalopram | SERT | - - - (∼250) | 0 | - | [116] [133] [162] |
| Escitalopram | --- (∼60) | 0 | 0 | [116] [133] [163] | |
| Fluoxetine | --- (∼5000) | - | - | [162] [132] | |
| Fluvoxamine | --- (∼600) | ND | - | [164] | |
| Paroxetine | --- (∼240) | 0 | - | [164] [165] | |
| Sertraline | --- (∼180) | ND | - | [164] | |
| Atomoxetine | NET | ND | ND | ND | |
| Desipramine | 0 | --- (∼240) | ND | [166] [165] | |
| Reboxetine | 0 | --- (∼110–480) | 0 | [134] [144] [158] | |
| Duloxetine | SERT - NET | --- (∼10-700) | --- (∼480) | ND | [167] [168] |
| Milnacipran | --- (∼5700) | 0 | ND | [169] | |
| Venlafaxine | --- (∼110-230) | --- (∼700-1000) | 0 | [165] [144] | |
| Bupropion | NET - DAT | 0 | 0 | 0 | [170] [171] |
| Nomifensine | 0 | --- (∼40) | --- (∼450) | [172] | |
| SEP225289 | SERT - NET - DAT | - | - - | - | [173] |
| DOV216303 | - | - - | - | [173] | |
| Drug (Mechanism) | Animal | Treatment (Duration; dose; route) | Pain Model | Test | Effect | Ref |
|---|---|---|---|---|---|---|
| Citalopram (SERT) | Mice | A; 50 mg/kg; i.p. | Acute | Hot plate | + | [225] |
| Mice | A; 20 mg/kg; i.p | Acute | Tail flick | − | [246] | |
| Hot plate test | − | |||||
| Mice | A; 20 mg/kg; i.p. | Acute Inflammatory PBQ | Hot plate | ++ | [219] | |
| Abdominal writhes | + | |||||
| Rat | A; 5 to 20 mg/kg; i.p. | acute | Hot plate | 0 | [222] | |
| A; 20 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | |||
| Rat | A; 3–30 mg/kg; i.p. | Acute | Tail flick | 0 | [247] | |
| Hot plate | 0 | |||||
| A; 30 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | |||
| A; 3–30 mg/kg; i.p. | NP CCI | Von frey | 0 | |||
| Hind pressure | 0 | |||||
| A; 10-30 mg/kg; i.p. | NP CCI | Radiant Heat | ++ | |||
| Rat | A; 3–10 mg/kg; i.p. | Visceral | Colonic distension | 0 | [248] | |
| Rat | C; 3–10 mg/kg; i.p. | Visceral | Colonic distension | 0 | ||
| Rat | C; 10 mg/kg.j; i.p. | Comorbid anxiety | Hargreaves | + | [249] | |
| Rat | - ; -; i.t. | Inflammatory Acetic acid | Abdominal writhing | 0 | [223] | |
| Rat | - ; - ; i.c.v | Inflammatory Acetic acid | Abdominal writhing | ++ | ||
| Rat | A; 10–25 mg/kg; i.p. | Acute | Tail flick | 0 | [224] | |
| Tail withdrawal | ++ | |||||
| C; 10–25 mg/kg; i.p. | Acute | Tail flick | 0 | |||
| Tail withdrawal | 0 | |||||
| A; 5–10 mg/kg; po | Inflammatory Acetic acid | Abdominal writhing | 0 | |||
| C; 25 mg/kg; po | Inflammatory Acetic acid | Abdominal writhing | 0 | |||
| A; 10–25 mg/kg; i.p. | Electrical | Flinch-jump test | 0 | |||
| C; 10–25 mg/kg; i.p. | Electrical | Flinch-jump test | 0 | |||
| Escitalopram (SERT) | Mice | A; 0,5 to 50 mg/kg; i.p. | Acute | Hot plate | 0 | [225] |
| Fluoxetine (SERT) | Mice | A; 10–30 mg/kg; Po | NP CCI | Von Frey | ++ | [251] |
| Mice | A; 10–40 mg/kg; Po | Acute | Hot plate | ++ | [252] | |
| C; 10 mg/kg; Po | Acute | Hot plate | ++ | |||
| A; 30 mg/kg; po | Inflammatory Formalin | Formalin test | ++ | |||
| Mice | A; 20 mg/kg; i.p. | Acute | Tail flick | ++ | [253] | |
| Inflammatory Formalin | Formalin test | ++ | ||||
| Inflammatory Carrageenan | Von Frey | + | ||||
| Mice | A; 15 μg; i.c.v. | NP CCI | Von Frey | 0 | [234] | |
| Tail Pinch | 0 | |||||
| A; 5–10 mg/kg; i.t. | NP CCI | Von Frey | 0 | |||
| Tail Pinch | 0 | |||||
| A; 15 μg; i.c.v. | NP STZ | Von Frey | 0 | |||
| Tail Pinch | 0 | |||||
| A; 5–10 mg/kg; i.t. | NP STZ | Von Frey | 0 | |||
| Tail Pinch | 0 | |||||
| Mice | A; 5–10–20 mg/kg; i.p. | acute | Hot plate | + | [226] | |
| Mice | A; 25 mg/kg; i.p. | Acute | Hot plate | + | [225] | |
| Mice | A; 10–20 mg/kg; i.p. | NP STZ | Tail Immersion | + | [254] | |
| Hot Plate | + | |||||
| Mice | -; -; - | Acute | Hot plate | + | [255] | |
| NP STZ | Hot plate | 0 | ||||
| Mice | A; 30 mg/kg; s.c. | Inflammatory PPQ | Abdominal writhes | + | [256] | |
| Rat | A; 56 mg/kg; s.c. | NP SNL | Von Frey | + | ||
| Rat | -; -; - | Acute | Tail flick | 0 | [238] | |
| Hot plate | 0 | |||||
| Paw pressure | 0 | |||||
| Rat | SC; 20 mg/kg.j; i.p. | NP STZ | Paw pressure | + | [257] | |
| Rat | -; -; s.c. | NP CCI | Radiant heat | 0 | [258] | |
| Rat | A; 3–10–30 mg/kg; s.c. | NP SNL | Von Frey | + | [259] | |
| Rat | A; 10–60 mg/kg; i.p. | Inflammatory Carrageenan | Paw oedema | ++ | [260] | |
| C; 20 mg/kg.j; i.p. | Inflammatory Carrageenan | Paw oedema | ++ | |||
| Rat | A; 10 mg/kg; i.p. | Inflammatory Carrageenan | Paw oedema | + | [261] | |
| Rat | C; 0,8 mg/kg; Po | Inflammatory Formalin | Formalin test | ++ | [233] | |
| A; 0.32 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | |||
| C; 0.16 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | |||
| A; 10 μg; i.t. | Inflammatory Formalin | Formalin test | ++ | |||
| Rat | A; 20 mg/kg; i.p. | Acute | Tail flick | ++ | [227] | |
| Hot plate | ++ | |||||
| A; 5–20 mg/kg; i.p. | Inflammatory Acetic acid | Abdominal writhing | ++ | |||
| A; 1 μg; i.c.v. | Inflammatory Acetic acid | Abdominal writhing | ++ | |||
| Rat | A; 100–300 nmol; i.pl. | Inflammatory Formalin | Formalin 2nd phase | + | [262] | |
| NP SNL | Thermal hyperalgesia | 0 | ||||
| Rat | A; 0.5–1–2–4 mg/kg; i.v. | Acute | Hot plate | 0 | [263] | |
| Tail flick | 0 | |||||
| A; 0.25 mg/kg; i.v. | Electrical | NIWR | - | |||
| Rat | A; 3–30 mg/kg; s.c. | Inflammatory Formalin | Formalin test | 0 | [264] | |
| NP SNL | Von Frey | 0 | ||||
| Pin Prick | 0 | |||||
| Rat | A; 3–30 mg/kg; i.p. | Acute | Tail flick | + | [265] | |
| A; 30 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | |||
| A; 3–30 mg/kg; i.p. | NP CCI | Von Frey | ++ | |||
| Radiant heat | 0 | |||||
| NP SNL | Von Frey | 0 | ||||
| Radiant Heat | 0 | |||||
| Rat | A; 5–10 mg/kg; - | acute | Tail jerk | 0 | [239] | |
| Fluvoxamine (SERT) | Mice | A; 2–6–12 μg; i.c.v. | Inflammatory Formalin | Formalin test | ++ | [266] |
| NP SNL | Radiant heat | ++ | ||||
| Mice | A; 10 mg/kg; s.c. | NP SNL | Von Frey | + | [267] | |
| Hargreaves | 0 | |||||
| Mice | A; 10–30 mg/kg; i.p. | Acute | Paw pressure | ++ | [268] | |
| A; 10–30–100 μg; i.t. | NP SNL | Von Frey | ++ | |||
| A; 10–30–100 μg; i.c.v. | NP SNL | Von Frey | 0 | |||
| Mice | A; 1–70 mg/kg;i.p. | acute | Hot plate | +++ | [225] | |
| Mice | A; 30 mg/kg; i.p. | acute | Hot plate | +++ | [269] | |
| A; 2.6–10 μg; i.t. | acute | Hot plate | ++ | |||
| A; 0.6–2.7 μg; i.c.v. | acute | Hot plate | ++ | |||
| Mice | A; 20 mg/kg; i.p. | Acute | Hot plate | ++ | [219] | |
| Inflammatory PBQ | Abdominal writhes | + | ||||
| Rat | C; 20 mg/kg.j; i.p. | Arthritis | Paw pressure | 0 | [236] | |
| Rat | A; 20 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | [228] | |
| Rat | A; 0.5–1–2–4 mg/kg; i.v. | Acute | Hot plate | 0 | [263] | |
| Tail flick | 0 | |||||
| A; 0.25 mg/kg; i.v. | Electrical | NIWR | − | |||
| Rat | A; 5 to 20 mg/kg; i.p. | acute | Hot plate | 0 | [222] | |
| A; 20 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | |||
| Rat | A; 1 mol to 1 μmol; i.t. | NP CCI | Von Frey | 0 | [241] | |
| NP STZ | Von Frey | 0 | ||||
| Paroxetine (SERT) | Mice | A; 4 mg/kg; s.c. | NP SNL | Von Frey | ++ | [102] |
| Radiant Heat | ++ | |||||
| SC; 5 mmol/j; i.c.v. | NP SNL | Von Frey | 0 | |||
| Radiant Heat | 0 | |||||
| Mice | A; 5–10 mg/kg; i.p. | Acute | Hot plate test | ++ | [231] | |
| Mice | -; -; - | Acute | Hot plate test | ++ | [255] | |
| NP STZ | Hot plate test | 0 | ||||
| Mice | A; 5–10–20 mg/kg; i.p. | Inflammatory Acetic acid | Abdominal writhing | ++ | [235] | |
| Mice | A; 5 mg/kg; i.p. | Acute | Hot plate test | ++ | [230] | |
| Mice | A; 3.8 mg/kg (ED50); s.c. | Inflammatory Acetic acid | Abdominal writhing | ++ | [270] | |
| Mice | A; 30 mg/kg; s.c. | Inflammatory PPQ | Abdominal writhes | + | [256] | |
| Rat | A; 10–30 mg/kg; s.c. | NP SNL | Von Frey | ++ | ||
| Rat | A; 0.1–100 nmol; i.t. | NP STZ | Von Frey | + | [241] | |
| NP CCI | Von Frey | 0 | ||||
| Rat | A; 10–100 μg; i.t. | NP SNL | Von Frey | 0 | [271] | |
| Sertraline (SERT) | Mice | A; 5 mg/kg; i.p. | acute | Hot plate | ++ | [232] |
| C; 5 mg/kg; i.p. | acute | Hot plate | ++ | |||
| Rat | C; 30 mg/kg; Po | NP STZ | Hot plate | ++ | [229] | |
| Rat | A; 5–20 mg/kg; i.p. | acute | Hot plate | 0 | [222] | |
| A; 20 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | |||
| Rat | A; 3.6 to 28.8 mg/kg; i.p. | Inflammatory Carrageenan | Paw oedema | ++ | [261] | |
| Rat | A; 30 μg; i.t. | Acute | Tail flick | 0 | [242] | |
| Desipramine (NET) | Mice | C; 10 mg/kg.j; i.p. | NP SNL | Von Frey | ++ | [272] |
| Mice | A; 20 mg/kg; i.p. | acute | Tail flick | ++ | [273] | |
| A; 2.5 to 20mg/kg; i.p. | acute | Hot Plate | 0 | |||
| A; 2.5–20 mg/kg; i.p. | Inflammatory Acetic acid | Abdominal writhes | ++ | |||
| A; 2.5–20 mg/kg; i.p. | Inflammatory Formalin | Formalin | ++ | |||
| Mice | A; 20 mg/kg; i.p. | acute | Hot plate | + | [219] | |
| Inflammatory PBQ | Abdominal writhes | ++ | ||||
| Mice | A; 30 mg/kg; s.c. | Inflammatory PPQ | Abdominal writhes | ++ | [256] | |
| Rat | A; 100mg/kg; s.c. | NP SNL | Von Frey | ++ | ||
| Rat | C; 10 mg/kg.j; i.p. | NP SNL | Von Frey | ++ | [274] | |
| Rat | A; 3–30 μg ; i.p. | Inflammatory Formalin | Formalin test | ++ | [275] | |
| A; 60–100 μg; i.t. | Inflammatory Formalin | Formalin test | ++ | |||
| Rat | A; 20 mg/kg; i.p. | acute | Hot plate | ++ | [222] | |
| Inflammatory Formalin | Formalin test | +++ | ||||
| Rat | A; 10–30–60–100 μg; it. | Inflammatory Carrageenan | Radiant heat | ++ | [276] | |
| Rat | A; 100–300 mmol; i.pl | Inflammatory Formalin | Formalin test | + | [262] | |
| NP SNL | Thermal hyperalgesia | ++ | ||||
| Rat | A; 3μg; i.t. | Acute | Tail flick | ++ | [277] | |
| Rat | A; 25 mg/kg; - | Acute | Hot plate | 0 | [278] | |
| Rat | A; 3–10–30–100 mg/kg; s.c. | Inflammatory Formalin | Formalin test | ++ | [264] | |
| A; 10–100 mg/kg; s.c. | NP SNL | Von Frey | 0 | |||
| Pin Prick | ++ | |||||
| Rat | A; 2 mg/kg; i.v. | NP SNL | Paw pressure | ++ | [279] | |
| «Pain related behaviour» | ++ | |||||
| Rat | A; 25 mg/kg; Po | Acute | Tail flick | + | [280] [281] | |
| Rat | C; - ; Po | Acute | Tail flick | + | [282] | |
| Hot Plate | + | |||||
| Reboxetine (NET) | Mice | A; 10 mg/kg; i.p. | Acute | Hot plate | + | [283] |
| Mice | C; 1.6 mg/kg.j; i.p. | NP SNL | Von Frey | ++ | [272] | |
| Mice | A; 3–10–30 mg/kg; s.c. | Inflammatory PPQ | Abdominal writhes | +++ | [256] | |
| Rat | A; 100 mg/kg; s.c. | NP SNL | Von Frey | ++ | ||
| Rat | A; 0.5–5 μg; i.t. | Paw incisional injury | + | [284] | ||
| Rat | A; 3–30 mg/kg; i.p. | Acute | Tail flick | + | [265] | |
| Inflammatory Formalin | Formalin test | ++ | ||||
| NP CCI | Von Frey | 0 | ||||
| Radiant heat | ++ | |||||
| NP SNL | Von Frey | 0 | ||||
| Radiant Heat | ++ | |||||
| Duloxetine (SERT – NET) | Mice | A; 30 mg/kg; i.p. | Acute | Tail flick | + | [253] |
| Inflammatory Formalin | Formalin test | ++ | ||||
| Inflammatory Carrageenan | Von Frey | +++ | ||||
| Mice | A; 5–10–20 mg/kg; i.p. | NP STZ | Tail immersion | ++ | [285] | |
| Hot plate | ++ | |||||
| Mice | A; 30–100 mg/kg; po | NP SNL | Von Frey | + | [286] | |
| Mice | A; 3–30 mg/kg; i.p. | Chronic pelvic-perineal | “pain behaviour” | + | [287] | |
| Mice | A; 1–100 mg/kg; Po | Inflammatory Acetic acid | Abdominal writhing | ++ | [288] | |
| Inflammatory Carrageenan | Von Frey | ++ | ||||
| Hargreaves | ++ | |||||
| Mice | A; 1–30 mg/kg; i.p. | Acute | Tail Flick | 0 | [289] | |
| Hot Plate | ++ | |||||
| A; 1 to 30 mg/kg; Po | Inflammatory Acetic acid | Abdominal writhing | + | |||
| Rat | A; 1–30 mg/kg; i.p | Inflammatory Carrageenan | Von Frey | ++ | ||
| Tail flick | ++ | |||||
| A; 30 mg/kg; i.p. | Inflammatory Capsaicin | Von Frey | ++ | |||
| Rat | A; 0.16–40 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | [290] | |
| A; 5–10–20–40 mg/kg; i.p. | Stress induced | USV | ++ | |||
| Rat | A; 30–90–150 μg/kg; Po | Inflammatory Osteoathritis | Grip force | ++ | [291] | |
| Rat | A; 3-60 mg/kg; s.c. | Inflammatory Formalin | Formalin test | ++ | [292] | |
| Rat | A; 3–30 mg/kg; i.p. | Acute | Tail flick | 0 | [247] | |
| Hot plate | + | |||||
| A; 30 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | |||
| NP CCI | Von frey | 0 | ||||
| Hind pressure | ++ | |||||
| Radiant Heat | ++ | |||||
| Rat | A; 3–30 mg/kg; po | Acute | Tail flick | ++ | [293] | |
| A; 3–15 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | |||
| A; 5–30 mg/kg; po | NP SNL | Von Frey | + | |||
| Rat | A; 30 mg/kg; i.p. | EAE | Paint-Brush test | 0 | [294] | |
| Pinch test | 0 | |||||
| Tail immersion | + | |||||
| Cold plate | + | |||||
| Rat | A; 0.4–20 mg/kg; i.p. | TASM ligation | Von Frey | ++ | [295] | |
| Milnacipran (SERT – NET) | Mice | A; 210 ng–21 μg ; i.c.v. | NP SNL | Von frey | ++ | [296] |
| Radiant heat | ++ | |||||
| A; 210 ng–21 μg ; i.t. | NP SNL | Von frey | ++ | |||
| Radiant heat | ++ | |||||
| A; 210 ng–21 μg ; local | NP SNL | Von frey | ++ | |||
| Radiant heat | ++ | |||||
| A; 30–120 mg/kg; po | NP SNL | Von frey | ++ | |||
| Radiant heat | ++ | |||||
| Mice | A; 2.5-20 mg/kg; i.p. | Inflammatory Acetic acid | Abdominal writhing | ++ | [297] | |
| Mice | A; 10 mg/kg; s.c. | NP SNL | Von Frey | ++ | [102] | |
| Radiant Heat | ++ | |||||
| Rat | A; 1–30 μg; i.t. | Post operative | Von Frey | ++ | [298] | |
| Rat | A; 0.16–60 mg/kg; i.p | Inflammatory Formalin | Formalin test | + | [290] | |
| A; 2.5-20 mg/kg; i.p. | Stress induced | USV | ++ | |||
| Rat | C; 10 mg/kg.j; s.c. | NP CCI | Von Frey | ++ | [299] | |
| Rat | A; 60 mg/kg; i.p. | Acute | Analgesimeter | 0 | [300] | |
| Rat | A; 1μmol to 10mmol; i.t. | NP CCI | Von Frey | + | [241] | |
| NP STZ | Von Frey | + | ||||
| Rat | A; 60 mg/kg; i.p. | NP CCI | Analgesimeter | ++ | [301] | |
| Rat | A; 3–100 μg; i.t. | NP SNL | Von Frey | ++ | [271] | |
| Rat | A; 10 mg/kg; i.v. | Visceral | Rectal distension | 0 | [302] | |
| A; 1 to 10 μg; i.t. | Visceral | Rectal distension | 0 | |||
| Rat | A; 10–30 mg/kg; i.p. | Inflammatory Formalin | Formalin test | + | [293] | |
| A; 300 mg/kg; po | NP SNL | Von Fre | + | |||
| Rat | A; 5 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | [228] | |
| Venlafaxine (SERT –NET) | Mice | C; 10 mg/kg.j; i.p. | NP SNL | Von Frey | ++ | [272] |
| Mice | -; -; - | Acute | Hot plate | + | [303] | |
| Mice | A; 70 mg/kg; i.p. | Acute | Hot plate | ++ | [304] | |
| Mice | A;l 1 to 30 mg/kg; i.p. | Acute | Hot plate | + | [305] | |
| Mice | A; 1–30 mg/kg; i.p. | Acute | Hot plate | ++ | [306] | |
| Rat | A; 10–30 mg/kg; i.p. | Inflammatory Formalin | Formalin test | ++ | [293] | |
| A; 300 mg/kg; po | NP SNL | Von Frey | + | |||
| Rat | A; 50–100 mg/kg; i.p. | Inflammatory Carrageenan | Von Frey | ++ | [307] | |
| Rat | A; 2.5–5–10 mg/kg; s.c. | NP STZ | Paw pressure | ++ | [272] | |
| Rat | A; 10–40 mg/kg; s.c. | NP Vincristin | Paw pressure | ++ | [308] | |
| Rat | A; 7.5 mg/kg; s.c. | NP Oxaliplatine | Tail-immersion test | ++ | [309] | |
| Rat | A; 22 mg/kg; i.p. | NP | Paw pressure | 0 | [310] | |
| Rat | A; 25 mg/kg; i.p. | Acute | Hot plate test | +++ | [311] | |
| Rat | A; 10–25–50 mg/kg; s.c. | NP SNL | Von Frey | 0 | [312] | |
| Formalin test | ++ | |||||
| Rat | A; 2.5 mg/kg; s.c. | Acute | Paw pressure | 0 | [313] | |
| Inflammatory Formalin | Formalin test 2nd phase | ++ | ||||
| Rat | - ; - ; - | NP SNL | Paw pressure | ++ | [314] | |
| Rat | SC; 22 mg/kg ; Po | NP CCI | Radiant Heat | ++ | [315] | |
| Rat | A; 3–100 mg/kg; i.p. | Acute | Tail flick | + | [265] | |
| Inflammatory Formalin | Formalin test 2nd phase | + | ||||
| NP CCI | Von Frey | 0 | ||||
| Radiant heat | + | |||||
| NP SNL | Von Frey | 0 | ||||
| Radiant Heat | 0 | |||||
| Bupropion (NET – DAT) | Mice | A; 10–30 mg/kg; Po | NP CCI | Von Frey | ++ | [251] |
| Rat | A; 3–30 mg/kg; i.p. | Acute | Tail flick | + | [265] | |
| Inflammatory Formalin | Formalin test | 0 | ||||
| NP CCI | Von Frey | ++ | ||||
| Radiant heat | + | |||||
| NP SNL | Von Frey | ++ | ||||
| Radiant Heat | 0 | |||||
| Nomifensine (NET – DAT) | Rat | A; 0.625–5 mg/kg; s.c. | Inflammatory Formalin | Formalin test | ++ | [316] |
| Rat | A; 25 mg/kg; i.p. | Acute | Charpentier pain mode | 0 | [317] | |
| Rat | A; 0.1–10 mg/kg; i.p. | Acute | Tail immersion | - | [318] | |
| Rat | A; 10–40 mg/kg; s.c. | Acute | Hot plate | 0 | [319] | |
| A; 2.5–5–10 mg/kg; s.c. | Tail immersion | - | ||||
| Bicifadine (SERT – NET – DAT) | Rat | A; 4-8-12 mg/kg; po | Acute inflamatory | Randall Selito test | ++ | [320] |
| A; 50 mg/kg; po | Kaolin-induced arthritis test | ++ | ||||
| A; 25–50–75–100 mg/kg; po | Acute | Tail flick | ++ | |||
| Hot plate | 0 | |||||
| A; 10–30 mg/kg; po | Inflammatory PPQ | Abdominal writhing | ++ | |||
| A; 5–10–20–40 mg/kg; po | Visceral | Colonic distension | ++ | |||
| A; 40–60 mg/kg; po | Inflammatory | Freund adjuvant | ++ | |||
| A; 10–20–30 mg/kg; po | Inflammatory Formalin | Formalin test | ++ | |||
| A; 40 mg/kg; po | NP SNL | Paw pressure | ++ | |||
| A; 40 mg/kg; po | Von Frey | ++ | ||||
| A; 12.5–25–100 mg/kg; po | Radiant heat | ++ | ||||
| A; 12.5–25 mg/kg; po | NP STZ | Paw pressure | ++ |
Treatment A: acute, SC: Subchronic from 2 to 5 consecutive days, C: Chronic more than 5 consecutive days. i.p.: intreperitoneal; i.c.v: intracerebroventricular; i.t.: intrathecal; i.v.intravenous; po: per os; s.c.: subcutaneous; Pain model NP: neuropathic pain; SNL: sciatic nerve ligation; CCI: chronic constriction injury; PH: post herpetic; EAE: experimental autoimmune encephalomyelitis; TASM: tendon of the anterior superficial part of the rat masseter muscle; NIWR: noxious-induced withdrawal reflexes; PBQ: para-benzoquinone; PPQ: para-phenylquinone. Test USV: Ultrasound vocalization. Effect (−) pronociceptive effect; (0) No effect; (++) antinociceptive effect; (+) when authors specifie “weak”, “little” or “mild” antinociceptive effect; (+++) when authors specifie “strong” or “important” antinociceptive effect.
References
- Price, D.D. Psychological and neural mechanisms of the affective dimension of pain. Science 2000, 288, 1769–1772. [Google Scholar]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar]
- Silveira, J.W.; Dias, Q.M.; Del Bel, E.A.; Prado, W.A. Serotonin receptors are involved in the spinal mediation of descending facilitation of surgical incision-induced increase of Fos-like immunoreactivity in rats. Mol. Pain 2010, 6, 17. [Google Scholar]
- Rahman, W.; D'Mello, R.; Dickenson, A.H. Peripheral nerve injury-induced changes in spinal alpha(2)-adrenoceptor-mediated modulation of mechanically evoked dorsal horn neuronal responses. J. Pain 2008, 9, 350–359. [Google Scholar]
- Willis, W.D., Jr. The somatosensory system, with emphasis on structures important for pain. Brain Res. Rev. 2007, 55, 297–313. [Google Scholar]
- Jeon, D.; Kim, S.; Chetana, M.; Jo, D.; Ruley, H.E.; Lin, S.Y.; Rabah, D.; Kinet, J.P.; Shin, H.S. Observational fear learning involves affective pain system and Cav1.2 Ca2+ channels in ACC. Nat. Neurosci. 2010, 13, 482–488. [Google Scholar]
- Xie, Y.F.; Huo, F.Q.; Tang, J.S. Cerebral cortex modulation of pain. Acta Pharmacol. Sin. 2009, 30, 31–41. [Google Scholar]
- Chen, T.L.; Babiloni, C.; Ferretti, A.; Perrucci, M.G.; Romani, G.L.; Rossini, P.M.; Tartaro, A.; Del Gratta, C. Human secondary somatosensory cortex is involved in the processing of somatosensory rare stimuli: an fMRI study. Neuroimage 2008, 40, 1765–1771. [Google Scholar]
- Ji, G.; Sun, H.; Fu, Y.; Li, Z.; Pais-Vieira, M.; Galhardo, V.; Neugebauer, V. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J. Neurosci. 2010, 30, 5451–5464. [Google Scholar]
- Liu, M.G.; Chen, J. Roles of the hippocampal formation in pain information processing. Neurosci. Bull. 2009, 25, 237–266. [Google Scholar]
- Verdu, B.; Decosterd, I.; Buclin, T.; Stiefel, F.; Berney, A. Antidepressants for the treatment of chronic pain. Drugs 2008, 68, 2611–2632. [Google Scholar]
- Furst, S. Transmitters involved in antinociception in the spinal cord. Brain Res. Bull. 1999, 48, 129–141. [Google Scholar]
- Benarroch, E.E. Descending monoaminergic pain modulation: bidirectional control and clinical relevance. Neurology 2008, 71, 217–221. [Google Scholar]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar]
- Guiard, B.P.; El Mansari, M.; Blier, P. Cross-talk between dopaminergic and noradrenergic systems in the rat ventral tegmental area, locus ceruleus, and dorsal hippocampus. Mol. Pharmacol. 2008, 74, 1463–1475. [Google Scholar]
- Bymaster, F.P.; Zhang, W.; Carter, P.A.; Shaw, J.; Chernet, E.; Phebus, L.; Wong, D.T.; Perry, K.W. Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology (Berl) 2002, 160, 353–361. [Google Scholar]
- Moron, J.A.; Brockington, A.; Wise, R.A.; Rocha, B.A.; Hope, B.T. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J. Neurosci. 2002, 22, 389–395. [Google Scholar]
- Carboni, E.; Silvagni, A. Dopamine reuptake by norepinephrine neurons: exception or rule? Crit. Rev. Neurobiol. 2004, 16, 121–128. [Google Scholar]
- Chapman, C.R.; Tuckett, R.P.; Song, C.W. Pain and stress in a systems perspective: reciprocal neural, endocrine, and immune interactions. J. Pain 2008, 9, 122–145. [Google Scholar]
- Tracey, I.; Mantyh, P.W. The cerebral signature for pain perception and its modulation. Neuron 2007, 55, 377–391. [Google Scholar]
- Heinricher, M.M.; Tavares, I.; Leith, J.L.; Lumb, B.M. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res. Rev. 2009, 60, 214–225. [Google Scholar]
- Jensen, T.S.; Yaksh, T.L. Comparison of the antinociceptive effect of morphine and glutamate at coincidental sites in the periaqueductal gray and medial medulla in rats. Brain Res. 1989, 476, 1–9. [Google Scholar]
- Jacquet, Y.F.; Squires, R.F. Excitatory amino acids: role in morphine excitation in rat periaqueductal gray. Behav. Brain Res. 1988, 31, 85–88. [Google Scholar]
- Deakin, J.F.; Dostrovsky, J.O. Involvement of the periaqueductal grey matter and spinal 5-hydroxytryptaminergic pathways in morphine analgesia: effcts of lesions and 5-hydroxytryptamine depletion. Br. J. Pharmacol. 1978, 63, 159–165. [Google Scholar]
- Jansen, A.S.; Farkas, E.; Mac Sams, J.; Loewy, A.D. Local connections between the columns of the periaqueductal gray matter: a case for intrinsic neuromodulation. Brain Res. 1998, 784, 329–336. [Google Scholar]
- Stezhka, V.V.; Lovick, T.A. Inhibitory and excitatory projections from the dorsal raphe nucleus to neurons in the dorsolateral periaqueductal gray matter in slices of midbrain maintained in vitro. Neuroscience 1994, 62, 177–187. [Google Scholar]
- Wang, Q.P.; Nakai, Y. The dorsal raphe: an important nucleus in pain modulation. Brain Res. Bull. 1994, 34, 575–585. [Google Scholar]
- Beitz, A.J.; Clements, J.R.; Mullett, M.A.; Ecklund, L.J. Differential origin of brainstem serotoninergic projections to the midbrain periaqueductal gray and superior colliculus of the rat. J. Comp. Neurol. 1986, 250, 498–509. [Google Scholar]
- Ghazni, N.F.; Cahill, C.M.; Stroman, P.W. Tactile sensory and pain networks in the human spinal cord and brain stem mapped by means of functional MR imaging. AJNR Am. J. Neuroradiol. 2010, 31, 661–667. [Google Scholar]
- Commons, K.G.; Valentino, R.J. Cellular basis for the effects of substance P in the periaqueductal gray and dorsal raphe nucleus. J. Comp. Neurol. 2002, 447, 82–97. [Google Scholar]
- Mohrland, J.S.; Gebhart, G.F. Substance P-induced analgesia in the rat. Brain Res. 1979, 171, 556–559. [Google Scholar]
- Frederickson, R.C.; Burgis, V.; Harrell, C.E.; Edwards, J.D. Dual actions of substance P on nociception: possible role of endogenous opioids. Science 1978, 199, 1359–1362. [Google Scholar]
- Malick, J.B.; Goldstein, J.M. Analgesic activity of substance P following intracerebral administration in rats. Life Sci. 1978, 23, 835–844. [Google Scholar]
- Stewart, J.M.; Getto, C.J.; Neldner, K.; Reeve, E.B.; Krivoy, W.A.; Zimmermann, E. Substance P and analgesia. Nature 1976, 262, 784–785. [Google Scholar]
- Naranjo, J.R.; Sanchez-Franco, F.; Garzon, J.; del Rio, J. Analgesic activity of substance P in rats: apparent mediation by met-enkephalin release. Life Sci. 1982, 30, 441–446. [Google Scholar]
- Guiard, B.P.; Guilloux, J.P.; Reperant, C.; Hunt, S.P.; Toth, M.; Gardier, A.M. Substance P neurokinin 1 receptor activation within the dorsal raphe nucleus controls serotonin release in the mouse frontal cortex. Mol. Pharmacol. 2007, 72, 1411–1418. [Google Scholar]
- Liu, R.; Ding, Y.; Aghajanian, G.K. Neurokinins activate local glutamatergic inputs to serotonergic neurons of the dorsal raphe nucleus. Neuropsychopharmacology 2002, 27, 329–340. [Google Scholar]
- Van Bockstaele, E.J.; Bajic, D.; Proudfit, H.; Valentino, R.J. Topographic architecture of stress-related pathways targeting the noradrenergic locus coeruleus. Physiol. Behav. 2001, 73, 273–283. [Google Scholar]
- Bajic, D.; Proudfit, H.K. Projections of neurons in the periaqueductal gray to pontine and medullary catecholamine cell groups involved in the modulation of nociception. J. Comp. Neurol. 1999, 405, 359–379. [Google Scholar]
- Ennis, M.; Behbehani, M.; Shipley, M.T.; Van Bockstaele, E.J.; Aston-Jones, G. Projections from the periaqueductal gray to the rostromedial pericoerulear region and nucleus locus coeruleus: anatomic and physiologic studies. J. Comp. Neurol. 1991, 306, 480–494. [Google Scholar]
- Van Bockstaele, E.J.; Menko, A.S.; Drolet, G. Neuroadaptive responses in brainstem noradrenergic nuclei following chronic morphine exposure. Mol. Neurobiol. 2001, 23, 155–171. [Google Scholar]
- Valentino, R.J.; Foote, S.L. Corticotropin-releasing hormone increases tonic but not sensory-evoked activity of noradrenergic locus coeruleus neurons in unanesthetized rats. J. Neurosci. 1988, 8, 1016–1025. [Google Scholar]
- Williams, J.T.; North, R.A.; Tokimasa, T. Inward rectification of resting and opiate-activated potassium currents in rat locus coeruleus neurons. J. Neurosci. 1988, 8, 4299–4306. [Google Scholar]
- Manning, B.H.; Morgan, M.J.; Franklin, K.B. Morphine analgesia in the formalin test: evidence for forebrain and midbrain sites of action. Neuroscience 1994, 63, 289–294. [Google Scholar]
- Yaksh, T.L.; DuChateau, J.C.; Rudy, T.A. Antagonism by methysergide and cinanserin of the antinociceptive action of morphine administered into the periaqueductal gray. Brain Res. 1976, 104, 367–372. [Google Scholar]
- Pert, A.; Yaksh, T. Localization of the antinociceptive action of morphine in primate brain. Pharmacol. Biochem. Behav. 1975, 3, 133–138. [Google Scholar]
- Flores, J.A.; El Banoua, F.; Galan-Rodriguez, B.; Fernandez-Espejo, E. Opiate anti-nociception is attenuated following lesion of large dopamine neurons of the periaqueductal grey: critical role for D1 (not D2) dopamine receptors. Pain 2004, 110, 205–214. [Google Scholar]
- Dong, H.W.; Swanson, L.W. Projections from bed nuclei of the stria terminalis, anteromedial area: cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J. Comp. Neurol. 2006, 494, 142–178. [Google Scholar]
- Hasue, R.H.; Shammah-Lagnado, S.J. Origin of the dopaminergic innervation of the central extended amygdala and accumbens shell: a combined retrograde tracing and immunohistochemical study in the rat. J. Comp. Neurol. 2002, 454, 15–33. [Google Scholar]
- Nestler, E.J. Under siege: The brain on opiates. Neuron 1996, 16, 897–900. [Google Scholar]
- Wood, P.L. Opioid regulation of CNS dopaminergic pathways: a review of methodology, receptor types, regional variations and species differences. Peptides 1983, 4, 595–601. [Google Scholar]
- Wood, P.L.; Stotland, M.; Richard, J.W.; Rackham, A. Actions of mu, kappa, sigma, delta and agonist/antagonist opiates on striatal dopaminergic function. J. Pharmacol. Exp. Ther. 1980, 215, 697–703. [Google Scholar]
- Meyer, P.J.; Morgan, M.M.; Kozell, L.B.; Ingram, S.L. Contribution of dopamine receptors to periaqueductal gray-mediated antinociception. Psychopharmacology (Berl) 2009, 204, 531–540. [Google Scholar]
- Mark, V.H.; Ervin, F.R. Role of thalamotomy in treatment of chronic severe pain. Postgrad. Med. 1965, 37, 563–571. [Google Scholar]
- Andersen, E.; Dafny, N. An ascending serotonergic pain modulation pathway from the dorsal raphe nucleus to the parafascicularis nucleus of the thalamus. Brain Res. 1983, 269, 57–67. [Google Scholar]
- Reyes-Vazquez, C.; Qiao, J.T.; Dafny, N. Nociceptive responses in nucleus parafascicularis thalami are modulated by dorsal raphe stimulation and microiontophoretic application of morphine and serotonin. Brain Res. Bull. 1989, 23, 405–411. [Google Scholar]
- Garcia-Cabezas, M.A.; Rico, B.; Sanchez-Gonzalez, M.A.; Cavada, C. Distribution of the dopamine innervation in the macaque and human thalamus. Neuroimage 2007, 34, 965–984. [Google Scholar]
- Condes, L.M.; Leon, O.M.; Sanchez, A.M.; Omana, Z.I. Mesencephalic projections to the thalamic centralis lateralis and medial prefrontal cortex: a WGA-HRP study. Brain Res. 1990, 509, 321–324. [Google Scholar]
- Groenewegen, H.J. Organization of the afferent connections of the mediodorsal thalamic nucleus in the rat, related to the mediodorsal-prefrontal topography. Neuroscience 1988, 24, 379–431. [Google Scholar]
- Voisin, D.L.; Guy, N.; Chalus, M.; Dallel, R. Nociceptive stimulation activates locus coeruleus neurones projecting to the somatosensory thalamus in the rat. J. Physiol. 2005, 566, 929–937. [Google Scholar]
- Baulmann, J.; Spitznagel, H.; Herdegen, T.; Unger, T.; Culman, J. Tachykinin receptor inhibition and c-Fos expression in the rat brain following formalin-induced pain. Neuroscience 2000, 95, 813–820. [Google Scholar]
- Gao, H.R.; Shi, T.F.; Yang, C.X.; Zhang, D.; Zhang, G.W.; Zhang, Y.; Jiao, R.S.; Zhang, H.; Xu, M.Y. The effect of dopamine on pain-related neurons in the parafascicular nucleus of rats. J. Neural. Transm. 2010, 117, 585–591. [Google Scholar]
- Govindaiah, G.; Wang, Y.; Cox, C.L. Dopamine enhances the excitability of somatosensory thalamocortical neurons. Neuroscience 2010, 170, 981–991. [Google Scholar]
- Chudler, E.H.; Dong, W.K. The role of the basal ganglia in nociception and pain. Pain 1995, 60, 3–38. [Google Scholar]
- Borsook, D.; Becerra, L. CNS animal fMRI in pain and analgesia. Neurosci. Biobehav. Rev. 2010. [Google Scholar] [CrossRef]
- Russmann, H.; Vingerhoets, F.; Ghika, J.; Maeder, P.; Bogousslavsky, J. Acute infarction limited to the lenticular nucleus: clinical, etiologic, and topographic features. Arch. Neurol. 2003, 60, 351–355. [Google Scholar]
- Loher, T.J.; Burgunder, J.M.; Weber, S.; Sommerhalder, R.; Krauss, J.K. Effect of chronic pallidal deep brain stimulation on off period dystonia and sensory symptoms in advanced Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 2002, 73, 395–399. [Google Scholar]
- Tork, I. Anatomy of the serotonergic system. Ann. NY Acad. Sci. 1990, 600, 9–34, discussion 34-35. [Google Scholar]
- Bloom, F.E.; Rossier, J.; Battenberg, E.L.; Bayon, A.; French, E.; Henriksen, S.J.; Siggins, G.R.; Segal, D.; Browne, R.; Ling, N.; Guillemin, R. beta-endorphin: cellular localization, electrophysiological and behavioral effects. Adv. Biochem. Psychopharmacol. 1978, 18, 89–109. [Google Scholar]
- Tseng, L.F.; Wang, Q. Forebrain sites differentially sensitive to beta-endorphin and morphine for analgesia and release of Met-enkephalin in the pentobarbital-anesthesized rat. J. Pharmacol. Exp. Ther. 1992, 261, 1028–1036. [Google Scholar]
- Foley, K.M.; Kourides, I.A.; Inturrisi, C.E.; Kaiko, R.F.; Zaroulis, C.G.; Posner, J.B.; Houde, R.W.; Li, C.H. beta-Endorphin: analgesic and hormonal effects in humans. Proc. Natl. Acad. Sci. USA 1979, 76, 5377–5381. [Google Scholar]
- Bloom, F.; Segal, D.; Ling, N.; Guillemin, R. Endorphins: profound behavioral effects in rats suggest new etiological factors in mental illness. Science 1976, 194, 630–632. [Google Scholar]
- Loh, H.H.; Tseng, L.F.; Wei, E.; Li, C.H. beta-endorphin is a potent analgesic agent. Proc. Natl. Acad. Sci. USA 1976, 73, 2895–2898. [Google Scholar]
- Brown, R.S.; Bottomley, W.K. Utilization and mechanism of action of tricyclic antidepressants in the treatment of chronic facial pain: a review of the literature. Anesth. Prog. 1990, 37, 223–229. [Google Scholar]
- Sacerdote, P.; Brini, A.; Mantegazza, P.; Panerai, A.E. A role for serotonin and beta-endorphin in the analgesia induced by some tricyclic antidepressant drugs. Pharmacol. Biochem. Behav. 1987, 26, 153–158. [Google Scholar]
- Zangen, A.; Nakash, R.; Yadid, G. Serotonin-mediated increases in the extracellular levels of beta-endorphin in the arcuate nucleus and nucleus accumbens: a microdialysis study. J. Neurochem. 1999, 73, 2569–2574. [Google Scholar]
- Han, J.S.; Xuan, Y.T. A mesolimbic neuronal loop of analgesia: I. Activation by morphine of a serotonergic pathway from periaqueductal gray to nucleus accumbens. Int. J. Neurosci. 1986, 29, 109–117. [Google Scholar]
- Moss, M.S.; Glazer, E.J.; Basbaum, A.I. Enkephalin-immunoreactive perikarya in the cat raphe dorsalis. Neurosci. Lett. 1981, 21, 33–37. [Google Scholar]
- Park, M.R.; Gonzales-Vegas, J.A.; Kitai, S.T. Serotonergic excitation from dorsal raphe stimulation recorded intracellularly from rat caudate-putamen. Brain Res. 1982, 243, 49–58. [Google Scholar]
- Zhang, W.; Ordway, G.A. The alpha2C-adrenoceptor modulates GABA release in mouse striatum. Brain Res. Mol. Brain Res. 2003, 112, 24–32. [Google Scholar]
- Dawson, L.A.; Nguyen, H.Q.; Li, P. In vivo effects of the 5-HT(6) antagonist SB-271046 on striatal and frontal cortex extracellular concentrations of noradrenaline, dopamine, 5-HT, glutamate and aspartate. Br. J. Pharmacol. 2000, 130, 23–26. [Google Scholar]
- Hagelberg, N.; Jaaskelainen, S.K.; Martikainen, I.K.; Mansikka, H.; Forssell, H.; Scheinin, H.; Hietala, J.; Pertovaara, A. Striatal dopamine D2 receptors in modulation of pain in humans: a review. Eur. J. Pharmacol. 2004, 500, 187–192. [Google Scholar]
- Wu, G.J.; Chen, Z.Q.; Shi, H. Roles of entopeduncular nucleus in acupuncture analgesia and caudate-putamen nucleus stimulation-induced analgesia. Sheng Li Xue Bao 2002, 54, 55–59. [Google Scholar]
- Zhang, G.W.; Yang, C.X.; Gao, H.R.; Zhang, D.; Zhang, Y.; Jiao, R.S.; Zhang, H.; Liang, Y.; Xu, M.Y. Microinjection of different doses of norepinephrine into the caudate putamen produces opposing effects in rats. Neurosci. Lett. 2010, 471, 125–128. [Google Scholar]
- Zhang, G.W.; Yang, C.X.; Zhang, D.; Gao, H.E.; Zhang, Y.; Jiao, R.S.; Zhang, H.; Liang, Y.; Xu, M.Y. Noradrenergic mechanism involved in the nociceptive modulation of nociceptive-related neurons in the caudate putamen. Neurosci. Lett. 2010, 480, 59–63. [Google Scholar]
- Fallon, J.H.; Moore, R.Y. Catecholamine innervation of the basal forebrain. IV. Topography of the dopamine projection to the basal forebrain and neostriatum. J. Comp. Neurol. 1978, 180, 545–580. [Google Scholar]
- Chudler, E.H.; Lu, Y. Nociceptive behavioral responses to chemical, thermal and mechanical stimulation after unilateral, intrastriatal administration of 6-hydroxydopamine. Brain Res. 2008, 1213, 41–47. [Google Scholar]
- Tassorelli, C.; Armentero, M.T.; Greco, R.; Fancellu, R.; Sandrini, G.; Nappi, G.; Blandini, F. Behavioral responses and Fos activation following painful stimuli in a rodent model of Parkinson's disease. Brain Res. 2007, 1176, 53–61. [Google Scholar]
- Altier, N.; Stewart, J. The role of dopamine in the nucleus accumbens in analgesia. Life Sci. 1999, 65, 2269–2287. [Google Scholar]
- Takeda, R.; Ikeda, T.; Tsuda, F.; Abe, H.; Hashiguchi, H.; Ishida, Y.; Nishimori, T. Unilateral lesions of mesostriatal dopaminergic pathway alters the withdrawal response of the rat hindpaw to mechanical stimulation. Neurosci. Res. 2005, 52, 31–36. [Google Scholar]
- Saade, N.E.; Atweh, S.F.; Bahuth, N.B.; Jabbur, S.J. Augmentation of nociceptive reflexes and chronic deafferentation pain by chemical lesions of either dopaminergic terminals or midbrain dopaminergic neurons. Brain Res. 1997, 751, 1–12. [Google Scholar]
- Carey, R.J. Acute ipsilateral hyperalgesia and chronic contralateral hypoalgesia after unilateral 6-hydroxydopamine lesions of the substantia nigra. Exp. Neurol. 1986, 91, 277–284. [Google Scholar]
- Robinson, M.J.; Edwards, S.E.; Iyengar, S.; Bymaster, F.; Clark, M.; Katon, W. Depression and pain. Front. BioSci. 2009, 14, 5031–5051. [Google Scholar]
- Oshiro, Y.; Quevedo, A.S.; McHaffie, J.G.; Kraft, R.A.; Coghill, R.C. Brain mechanisms supporting spatial discrimination of pain. J. Neurosci. 2007, 27, 3388–3394. [Google Scholar]
- Salomons, T.V.; Johnstone, T.; Backonja, M.M.; Shackman, A.J.; Davidson, R.J. Individual differences in the effects of perceived controllability on pain perception: critical role of the prefrontal cortex. J. Cogn. Neurosci. 2007, 19, 993–1003. [Google Scholar]
- Seifert, F.; Maihofner, C. Representation of cold allodynia in the human brain--a functional MRI study. Neuroimage 2007, 35, 1168–1180. [Google Scholar]
- Baliki, M.N.; Chialvo, D.R.; Geha, P.Y.; Levy, R.M.; Harden, R.N.; Parrish, T.B.; Apkarian, A.V. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J. Neurosci. 2006, 26, 12165–12173. [Google Scholar]
- Frot, M.; Mauguiere, F. Dual representation of pain in the operculo-insular cortex in humans. Brain 2003, 126, 438–450. [Google Scholar]
- Donahue, R.R.; LaGraize, S.C.; Fuchs, P.N. Electrolytic lesion of the anterior cingulate cortex decreases inflammatory, but not neuropathic nociceptive behavior in rats. Brain Res. 2001, 897, 131–138. [Google Scholar]
- Lopez-Avila, A.; Coffeen, U.; Ortega-Legaspi, J.M.; del Angel, R.; Pellicer, F. Dopamine and NMDA systems modulate long-term nociception in the rat anterior cingulate cortex. Pain 2004, 111, 136–143. [Google Scholar]
- Ohara, P.T.; Vit, J.P.; Jasmin, L. Cortical modulation of pain. Cell. Mol. Life Sci. 2005, 62, 44–52. [Google Scholar]
- Matsuzawa-Yanagida, K.; Narita, M.; Nakajima, M.; Kuzumaki, N.; Niikura, K.; Nozaki, H.; Takagi, T.; Tamai, E.; Hareyama, N.; Terada, M.; Yamazaki, M.; Suzuki, T. Usefulness of antidepressants for improving the neuropathic pain-like state and pain-induced anxiety through actions at different brain sites. Neuropsychopharmacology 2008, 33, 1952–1965. [Google Scholar]
- Kurth, F.; Eickhoff, S.B.; Schleicher, A.; Hoemke, L.; Zilles, K.; Amunts, K. Cytoarchitecture and probabilistic maps of the human posterior insular cortex. Cereb Cortex 2010, 20, 1448–1461. [Google Scholar]
- Nemeroff, C. Part IV: Mood Disorders. In Neurobiology of Mental Illness; Charney, D.S., Nestler, E.J., Eds.; Oxford University Press: New York, USA, 2004. [Google Scholar]
- Kenneth, C.; Tri, T. Cortical mechanisms mediating acute and chronic pain in humans. In Handbook of Clin Neurology; Cervero, F., Jensen, T.S., Eds.; Elsevier, Massachusetts: Boston, USA, 2006. [Google Scholar]
- Neugebauer, V.; Li, W.; Bird, G.C.; Han, J.S. The amygdala and persistent pain. Neuroscientist 2004, 10, 221–234. [Google Scholar]
- Geha, P.Y.; Baliki, M.N.; Chialvo, D.R.; Harden, R.N.; Paice, J.A.; Apkarian, A.V. Brain activity for spontaneous pain of postherpetic neuralgia and its modulation by lidocaine patch therapy. Pain 2007, 128, 88–100. [Google Scholar]
- Bingel, U.; Quante, M.; Knab, R.; Bromm, B.; Weiller, C.; Buchel, C. Subcortical structures involved in pain processing: evidence from single-trial fMRI. Pain 2002, 99, 313–321. [Google Scholar]
- Duric, V.; McCarson, K.E. Persistent pain produces stress-like alterations in hippocampal neurogenesis and gene expression. J. Pain 2006, 7, 544–555. [Google Scholar]
- Ploghaus, A.; Narain, C.; Beckmann, C.F.; Clare, S.; Bantick, S.; Wise, R.; Matthews, P.M.; Rawlins, J.N.; Tracey, I. Exacerbation of pain by anxiety is associated with activity in a hippocampal network. J. Neurosci. 2001, 21, 9896–9903. [Google Scholar]
- Mori, M.; Jefferson, J.J.; Hummel, M.; Garbe, D.S. CNTF: a putative link between dopamine D2 receptors and neurogenesis. J. Neurosci. 2008, 28, 5867–5869. [Google Scholar]
- Yang, P.; Arnold, S.A.; Habas, A.; Hetman, M.; Hagg, T. Ciliary neurotrophic factor mediates dopamine D2 receptor-induced CNS neurogenesis in adult mice. J. Neurosci. 2008, 28, 2231–2241. [Google Scholar]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar]
- Terada, M.; Kuzumaki, N.; Hareyama, N.; Imai, S.; Niikura, K.; Narita, M.; Yamazaki, M.; Suzuki, T. Suppression of enriched environment-induced neurogenesis in a rodent model of neuropathic pain. Neurosci. Lett. 2008, 440, 314–318. [Google Scholar]
- Guiard, B.P.; El Mansari, M.; Blier, P. Prospect of a dopamine contribution in the next generation of antidepressant drugs: the triple reuptake inhibitors. Curr. Drug Targets 2009, 10, 1069–1084. [Google Scholar]
- El Mansari, M.; Sanchez, C.; Chouvet, G.; Renaud, B.; Haddjeri, N. Effects of acute and long-term administration of escitalopram and citalopram on serotonin neurotransmission: an in vivo electrophysiological study in rat brain. Neuropsychopharmacology 2005, 30, 1269–1277. [Google Scholar]
- Sanchez, C.; Hyttel, J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell. Mol. Neurobiol. 1999, 19, 467–489. [Google Scholar]
- Tremblay, P.; Blier, P. Catecholaminergic strategies for the treatment of major depression. Curr. Drug Targets 2006, 7, 149–158. [Google Scholar]
- Guiard, B.P.; Lanfumey, L.; Gardier, A.M. Microdialysis approach to study serotonin outflow in mice following selective serotonin reuptake inhibitors and substance P (neurokinin 1) receptor antagonist administration: a review. Curr. Drug Targets 2006, 7, 187–201. [Google Scholar]
- Aronson, S.; Delgado, P. Escitalopram. Drugs Today (Barc) 2004, 40, 121–131. [Google Scholar]
- Kobayashi, T.; Hayashi, E.; Shimamura, M.; Kinoshita, M.; Murphy, N.P. Neurochemical responses to antidepressants in the prefrontal cortex of mice and their efficacy in preclinical models of anxiety-like and depression-like behavior: a comparative and correlational study. Psychopharmacology (Berl) 2008, 197, 567–580. [Google Scholar]
- David, D.J.; Bourin, M.; Jego, G.; Przybylski, C.; Jolliet, P.; Gardier, A.M. Effects of acute treatment with paroxetine, citalopram and venlafaxine in vivo on noradrenaline and serotonin outflow: a microdialysis study in Swiss mice. Br. J. Pharmacol. 2003, 140, 1128–1136. [Google Scholar]
- Beyer, C.E.; Boikess, S.; Luo, B.; Dawson, L.A. Comparison of the effects of antidepressants on norepinephrine and serotonin concentrations in the rat frontal cortex: an in-vivo microdialysis study. J. Psychopharmacol. 2002, 16, 297–304. [Google Scholar]
- Koch, S.; Hemrick-Luecke, S.K.; Thompson, L.K.; Evans, D.C.; Threlkeld, P.G.; Nelson, D.L.; Perry, K.W.; Bymaster, F.P. Comparison of effects of dual transporter inhibitors on monoamine transporters and extracellular levels in rats. Neuropharmacology 2003, 45, 935–944. [Google Scholar]
- Hajos-Korcsok, E.; McTavish, S.F.; Sharp, T. Effect of a selective 5-hydroxytryptamine reuptake inhibitor on brain extracellular noradrenaline: microdialysis studies using paroxetine. Eur. J. Pharmacol. 2000, 407, 101–107. [Google Scholar]
- Thomas, D.N.; Nutt, D.J.; Holman, R.B. Sertraline, a selective serotonin reuptake inhibitor modulates extracellular noradrenaline in the rat frontal cortex. J. Psychopharmacol. 1998, 12, 366–370. [Google Scholar]
- Shachar, D.; Klein, E.; Tabak, A.; Finberg, J.P. Effect of single and repeated administration of fluvoxamine on noradrenaline release in rat brain. Eur. J. Pharmacol. 1997, 332, 237–243. [Google Scholar]
- Matsumoto, M.; Yoshioka, M.; Togashi, H.; Tochihara, M.; Ikeda, T.; Saito, H. Modulation of norepinephrine release by serotonergic receptors in the rat hippocampus as measured by in vivo microdialysis. J. Pharmacol. Exp. Ther. 1995, 272, 1044–1051. [Google Scholar]
- Jordan, S.; Kramer, G.L.; Zukas, P.K.; Moeller, M.; Petty, F. In vivo biogenic amine efflux in medial prefrontal cortex with imipramine, fluoxetine, and fluvoxamine. Synapse 1994, 18, 294–297. [Google Scholar]
- Hajos-Korcsok, E.; McQuade, R.; Sharp, T. Influence of 5-HT1A receptors on central noradrenergic activity: microdialysis studies using (+/−)-MDL 73005EF and its enantiomers. Neuropharmacology 1999, 38, 299–306. [Google Scholar]
- Mongeau, R.; De Montigny, C.; Blier, P. Activation of 5-HT3 receptors enhances the electrically evoked release of [3H]noradrenaline in rat brain limbic structures. Eur. J. Pharmacol. 1994, 256, 269–279. [Google Scholar]
- Miguelez, C.; Fernandez-Aedo, I.; Torrecilla, M.; Grandoso, L.; Ugedo, L. alpha(2)-Adrenoceptors mediate the acute inhibitory effect of fluoxetine on locus coeruleus noradrenergic neurons. Neuropharmacology 2009, 56, 1068–1073. [Google Scholar]
- Dremencov, E.; El Mansari, M.; Blier, P. Noradrenergic augmentation of escitalopram response by risperidone: electrophysiologic studies in the rat brain. Biol. Psychiatry 2007, 61, 671–678. [Google Scholar]
- Szabo, S.T.; Blier, P. Effect of the selective noradrenergic reuptake inhibitor reboxetine on the firing activity of noradrenaline and serotonin neurons. Eur. J. Neurosci. 2001, 13, 2077–2087. [Google Scholar]
- Gilmor, M.L.; Owens, M.J.; Nemeroff, C.B. Inhibition of norepinephrine uptake in patients with major depression treated with paroxetine. Am. J. Psychiatry 2002, 159, 1702–1710. [Google Scholar]
- Owens, M.J.; Knight, D.L.; Nemeroff, C.B. Paroxetine binding to the rat norepinephrine transporter in vivo. Biol. Psychiatry 2000, 47, 842–845. [Google Scholar]
- Vizi, E.S.; Zsilla, G.; Caron, M.G.; Kiss, J.P. Uptake and release of norepinephrine by serotonergic terminals in norepinephrine transporter knock-out mice: implications for the action of selective serotonin reuptake inhibitors. J. Neurosci. 2004, 24, 7888–7894. [Google Scholar]
- Ni, Y.G.; Miledi, R. Blockage of 5HT2C serotonin receptors by fluoxetine (Prozac). Proc. Natl. Acad. Sci. USA 1997, 94, 2036–2040. [Google Scholar]
- Szabo, S.T.; Blier, P. Effects of serotonin (5-hydroxytryptamine, 5-HT) reuptake inhibition plus 5-HT(2A) receptor antagonism on the firing activity of norepinephrine neurons. J. Pharmacol. Exp. Ther. 2002, 302, 983–991. [Google Scholar]
- Sanchez, C.; Bergqvist, P.B.; Brennum, L.T.; Gupta, S.; Hogg, S.; Larsen, A.; Wiborg, O. Escitalopram, the S-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive of antidepressant and anxiolytic activities. Psychopharmacology (Berl) 2003, 167, 353–362. [Google Scholar]
- Mochizuki, D.; Tsujita, R.; Yamada, S.; Kawasaki, K.; Otsuka, Y.; Hashimoto, S.; Hattori, T.; Kitamura, Y.; Miki, N. Neurochemical and behavioural characterization of milnacipran, a serotonin and noradrenaline reuptake inhibitor in rats. Psychopharmacology (Berl) 2002, 162, 323–332. [Google Scholar]
- Koe, B.K. Preclinical pharmacology of sertraline: a potent and specific inhibitor of serotonin reuptake. J. Clin. Psychiatry 1990, 51 Suppl. B, 13–17. [Google Scholar]
- Mantovani, M.; Dooley, D.J.; Weyerbrock, A.; Jackisch, R.; Feuerstein, T.J. Differential inhibitory effects of drugs acting at the noradrenaline and 5-hydroxytryptamine transporters in rat and human neocortical synaptosomes. Br. J. Pharmacol. 2009, 158, 1848–1856. [Google Scholar]
- Millan, M.J.; Gobert, A.; Lejeune, F.; Newman-Tancredi, A.; Rivet, J.M.; Auclair, A.; Peglion, J.L. S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J. Pharmacol. Exp. Ther. 2001, 298, 565–580. [Google Scholar]
- Arnt, J.; Christensen, A.V.; Hyttel, J. Pharmacology in vivo of the phenylindan derivative, Lu 19-005, a new potent inhibitor of dopamine, noradrenaline and 5-hydroxytryptamine uptake in rat brain. Naunyn Schmiedebergs Arch. Pharmacol. 1985, 329, 101–107. [Google Scholar]
- Popik, P.; Krawczyk, M.; Golembiowska, K.; Nowak, G.; Janowsky, A.; Skolnick, P.; Lippa, A.; Basile, A.S. Pharmacological profile of the “triple” monoamine neurotransmitter uptake inhibitor, DOV 102,677. Cell. Mol. Neurobiol. 2006, 26, 857–873. [Google Scholar]
- Skolnick, P.; Popik, P.; Janowsky, A.; Beer, B.; Lippa, A.S. Antidepressant-like actions of DOV 21,947: a “triple” reuptake inhibitor. Eur. J. Pharmacol. 2003, 461, 99–104. [Google Scholar]
- Maryanoff, B.E.; McComsey, D.F.; Gardocki, J.F.; Shank, R.P.; Costanzo, M.J.; Nortey, S.O.; Schneider, C.R.; Setler, P.E. Pyrroloisoquinoline antidepressants. 2. In-depth exploration of structure-activity relationships. J. Med. Chem. 1987, 30, 1433–1454. [Google Scholar]
- Liang, Y.; Shaw, A.M.; Boules, M.; Briody, S.; Robinson, J.; Oliveros, A.; Blazar, E.; Williams, K.; Zhang, Y.; Carlier, P.R.; Richelson, E. Antidepressant-like pharmacological profile of a novel triple reuptake inhibitor, (1S,2S)-3-(methylamino)-2-(naphthalen-2-yl)-1-phenylpropan-1-ol (PRC200-SS). J. Pharmacol. Exp. Ther. 2008, 327, 573–583. [Google Scholar]
- Hyttel, J. Citalopram--pharmacological profile of a specific serotonin uptake inhibitor with antidepressant activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 1982, 6, 277–295. [Google Scholar]
- Ortega, J.E.; Fernandez-Pastor, B.; Callado, L.F.; Meana, J.J. In vivo potentiation of reboxetine and citalopram effect on extracellular noradrenaline in rat brain by alpha(2)-adrenoceptor antagonism. Eur. Neuropsychopharmacol. 2010, 20, 813–822. [Google Scholar]
- Parini, S.; Renoldi, G.; Battaglia, A.; Invernizzi, R.W. Chronic reboxetine desensitizes terminal but not somatodendritic alpha2-adrenoceptors controlling noradrenaline release in the rat dorsal hippocampus. Neuropsychopharmacology 2005, 30, 1048–1055. [Google Scholar]
- Invernizzi, R.W.; Garattini, S. Role of presynaptic alpha2-adrenoceptors in antidepressant action: recent findings from microdialysis studies. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 819–827. [Google Scholar]
- Owen, J.C.; Whitton, P.S. Reboxetine modulates norepinephrine efflux in the frontal cortex of the freely moving rat: the involvement of alpha 2 and 5-HT1A receptors. Neurosci. Lett. 2003, 348, 171–174. [Google Scholar]
- Mateo, Y.; Fernandez-Pastor, B.; Meana, J.J. Acute and chronic effects of desipramine and clorgyline on alpha(2)-adrenoceptors regulating noradrenergic transmission in the rat brain: a dual-probe microdialysis study. Br. J. Pharmacol. 2001, 133, 1362–1370. [Google Scholar]
- Sacchetti, G.; Bernini, M.; Bianchetti, A.; Parini, S.; Invernizzi, R.W.; Samanin, R. Studies on the acute and chronic effects of reboxetine on extracellular noradrenaline and other monoamines in the rat brain. Br. J. Pharmacol. 1999, 128, 1332–1338. [Google Scholar]
- Miguelez, C.; Grandoso, L.; Ugedo, L. Locus coeruleus and dorsal raphe neuron activity and response to acute antidepressant administration in a rat model of Parkinson's disease. Int. J. Neuropsychopharmacol. 2010, 1–14. [Google Scholar]
- Linner, L.; Wiker, C.; Arborelius, L.; Schalling, M.; Svensson, T.H. Selective noradrenaline reuptake inhibition enhances serotonergic neuronal activity and transmitter release in the rat forebrain. J. Neural. Transm. 2004, 111, 127–139. [Google Scholar]
- Page, M.E.; Lucki, I. Effects of acute and chronic reboxetine treatment on stress-induced monoamine efflux in the rat frontal cortex. Neuropsychopharmacology 2002, 27, 237–247. [Google Scholar]
- Pudovkina, O.L.; Cremers, T.I.; Westerink, B.H. Regulation of the release of serotonin in the dorsal raphe nucleus by alpha1 and alpha2 adrenoceptors. Synapse 2003, 50, 77–82. [Google Scholar]
- Mongeau, R.; Blier, P.; de Montigny, C. The serotonergic and noradrenergic systems of the hippocampus: their interactions and the effects of antidepressant treatments. Brain Res. Brain Res. Rev. 1997, 23, 145–195. [Google Scholar]
- Prisco, S.; Esposito, E. Differential effects of acute and chronic fluoxetine administration on the spontaneous activity of dopaminergic neurones in the ventral tegmental area. Br. J. Pharmacol. 1995, 116, 1923–1931. [Google Scholar]
- Dremencov, E.; El Mansari, M.; Blier, P. Effects of sustained serotonin reuptake inhibition on the firing of dopamine neurons in the rat ventral tegmental area. J. Psychiatry Neurosci. 2009, 34, 223–229. [Google Scholar]
- Di Mascio, M.; Di Giovanni, G.; Di Matteo, V.; Prisco, S.; Esposito, E. Selective serotonin reuptake inhibitors reduce the spontaneous activity of dopaminergic neurons in the ventral tegmental area. Brain Res. Bull. 1998, 46, 547–554. [Google Scholar]
- Beique, J.C.; de Montigny, C.; Blier, P.; Debonnel, G. Venlafaxine: discrepancy between in vivo 5-HT and NE reuptake blockade and affinity for reuptake sites. Synapse 1999, 32, 198–211. [Google Scholar]
- Cunningham, K.A.; Lakoski, J.M. The interaction of cocaine with serotonin dorsal raphe neurons. Single-unit extracellular recording studies. Neuropsychopharmacology 1990, 3, 41–50. [Google Scholar]
- Smith, J.E.; Lakoski, J.M. Electrophysiological effects of fluoxetine and duloxetine in the dorsal raphe nucleus and hippocampus. Eur. J. Pharmacol. 1997, 323, 69–73. [Google Scholar]
- Kasamo, K.; Blier, P.; De Montigny, C. Blockade of the serotonin and norepinephrine uptake processes by duloxetine: in vitro and in vivo studies in the rat brain. J. Pharmacol. Exp. Ther. 1996, 277, 278–286. [Google Scholar]
- Mongeau, R.; Weiss, M.; de Montigny, C.; Blier, P. Effect of acute, short- and long-term milnacipran administration on rat locus coeruleus noradrenergic and dorsal raphe serotonergic neurons. Neuropharmacology 1998, 37, 905–918. [Google Scholar]
- Ghanbari, R.; El Mansari, M.; Blier, P. Electrophysiological effects of the co-administration of escitalopram and bupropion on rat serotonin and norepinephrine neurons. J. Psychopharmacol. 2010, 24, 39–50. [Google Scholar]
- Dong, J.; Blier, P. Modification of norepinephrine and serotonin, but not dopamine, neuron firing by sustained bupropion treatment. Psychopharmacology (Berl) 2001, 155, 52–57. [Google Scholar]
- Katz, N.S.; Guiard, B.P.; El Mansari, M.; Blier, P. Effects of acute and sustained administration of the catecholamine reuptake inhibitor nomifensine on the firing activity of monoaminergic neurons. J. Psychopharmacol. 2010, 24, 1223–1235. [Google Scholar]
- Guiard, B.P.; Chenu, F.; Mansari, M.E.; Blier, P. Characterization of the electrophysiological properties of triple reuptake inhibitors on monoaminergic neurons. Int. J. Neuropsychopharmacol. 2010, 1–13. [Google Scholar]
- Cipriani, A.; Santilli, C.; Furukawa, T.A.; Signoretti, A.; Nakagawa, A.; McGuire, H.; Churchill, R.; Barbui, C. Escitalopram versus other antidepressive agents for depression. Cochrane Database Syst. Rev. 2009, CD006532. [Google Scholar]
- Thase, M.E. Are SNRIs more effective than SSRIs? A review of the current state of the controversy. Psychopharmacol. Bull. 2008, 41, 58–85. [Google Scholar]
- Gobert, A.; Rivet, J.M.; Cistarelli, L.; Millan, M.J. Potentiation of the fluoxetine-induced increase in dialysate levels of serotonin (5-HT) in the frontal cortex of freely moving rats by combined blockade of 5-HT1A and 5-HT1B receptors with WAY 100,635 and GR 127,935. J. Neurochem. 1997, 68, 1159–1163. [Google Scholar]
- Engleman, E.A.; Perry, K.W.; Mayle, D.A.; Wong, D.T. Simultaneous increases of extracellular monoamines in microdialysates from hypothalamus of conscious rats by duloxetine, a dual serotonin and norepinephrine uptake inhibitor. Neuropsychopharmacology 1995, 12, 287–295. [Google Scholar]
- Kihara, T.; Ikeda, M. Effects of duloxetine, a new serotonin and norepinephrine uptake inhibitor, on extracellular monoamine levels in rat frontal cortex. J. Pharmacol. Exp. Ther. 1995, 272, 177–183. [Google Scholar]
- Dawson, L.A.; Nguyen, H.Q.; Geiger, A. Effects of venlafaxine on extracellular concentrations of 5-HT and noradrenaline in the rat frontal cortex: augmentation via 5-HT1A receptor antagonism. Neuropharmacology 1999, 38, 1153–1163. [Google Scholar]
- Wikell, C.; Bergqvist, P.B.; Hjorth, S.; Apelqvist, G.; Bjork, H.; Bengtsson, F. Brain monoamine output alterations after a single venlafaxine challenge in experimental hepatic encephalopathy. Clin. Neuropharmacol. 1998, 21, 296–306. [Google Scholar]
- Weikop, P.; Kehr, J.; Scheel-Kruger, J. The role of alpha1- and alpha2-adrenoreceptors on venlafaxine-induced elevation of extracellular serotonin, noradrenaline and dopamine levels in the rat prefrontal cortex and hippocampus. J. Psychopharmacol. 2004, 18, 395–403. [Google Scholar]
- Piacentini, M.F.; Clinckers, R.; Meeusen, R.; Sarre, S.; Ebinger, G.; Michotte, Y. Effects of venlafaxine on extracellular 5-HT, dopamine and noradrenaline in the hippocampus and on peripheral hormone concentrations in the rat in vivo. Life Sci. 2003, 73, 2433–2442. [Google Scholar]
- Moret, C.; Briley, M. Effects of milnacipran and pindolol on extracellular noradrenaline and serotonin levels in guinea pig hypothalamus. J. Neurochem. 1997, 69, 815–822. [Google Scholar]
- Stahl, S.M.; Grady, M.M.; Moret, C.; Briley, M. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005, 10, 732–747. [Google Scholar]
- Kitaichi, Y.; Inoue, T.; Nakagawa, S.; Izumi, T.; Koyama, T. Effect of milnacipran on extracellular monoamine concentrations in the medial prefrontal cortex of rats pre-treated with lithium. Eur. J. Pharmacol. 2005, 516, 219–226. [Google Scholar]
- Wong, D.T. Duloxetine (LY 248686): an inhibitor of serotonin and noradrenaline uptake and an antidepressant drug candidate. Expert. Opin. Investig. Drugs 1998, 7, 1691–1699. [Google Scholar]
- Vaishnavi, S.N.; Nemeroff, C.B.; Plott, S.J.; Rao, S.G.; Kranzler, J.; Owens, M.J. Milnacipran: a comparative analysis of human monoamine uptake and transporter binding affinity. Biol. Psychiatry 2004, 55, 320–322. [Google Scholar]
- Cassano, P.; Lattanzi, L.; Soldani, F.; Navari, S.; Battistini, G.; Gemignani, A.; Cassano, G.B. Pramipexole in treatment-resistant depression: an extended follow-up. Depress. Anxiety 2004, 20, 131–138. [Google Scholar]
- Santamaria, A.; Arias, H.R. Neurochemical and behavioral effects elicited by bupropion and diethylpropion in rats. Behav. Brain Res. 2010, 211, 132–139. [Google Scholar]
- Zocchi, A.; Varnier, G.; Arban, R.; Griffante, C.; Zanetti, L.; Bettelini, L.; Marchi, M.; Gerrard, P. A.; Corsi, M. Effects of antidepressant drugs and GR 205171, an neurokinin-1 (NK1) receptor antagonist, on the response in the forced swim test and on monoamine extracellular levels in the frontal cortex of the mouse. Neurosci. Lett. 2003, 345, 73–76. [Google Scholar]
- Nomikos, G.G.; Damsma, G.; Wenkstern, D.; Fibiger, H.C. In vivo characterization of locally applied dopamine uptake inhibitors by striatal microdialysis. Synapse 1990, 6, 106–112. [Google Scholar]
- Nomikos, G.G.; Damsma, G.; Wenkstern, D.; Fibiger, H.C. Acute effects of bupropion on extracellular dopamine concentrations in rat striatum and nucleus accumbens studied by in vivo microdialysis. Neuropsychopharmacology 1989, 2, 273–279. [Google Scholar]
- Meyer, J.H.; Goulding, V.S.; Wilson, A.A.; Hussey, D.; Christensen, B.K.; Houle, S. Bupropion occupancy of the dopamine transporter is low during clinical treatment. Psychopharmacology (Berl) 2002, 163, 102–105. [Google Scholar]
- Baraban, J.M.; Aghajanian, G.K. Suppression of serotonergic neuronal firing by alpha-adrenoceptor antagonists: evidence against GABA mediation. Eur. J. Pharmacol. 1980, 66, 287–294. [Google Scholar]
- Baraban, J.M.; Wang, R.Y.; Aghajanian, G. Reserpine suppression of dorsal raphe neuronal firing: mediation by adrenergic system. Eur. J. Pharmacol. 1978, 52, 27–36. [Google Scholar]
- Svensson, T.H.; Bunney, B.S.; Aghajanian, G.K. Inhibition of both noradrenergic and serotonergic neurons in brain by the alpha-adrenergic agonist clonidine. Brain Res. 1975, 92, 291–306. [Google Scholar]
- Mazei, M.S.; Pluto, C.P.; Kirkbride, B.; Pehek, E.A. Effects of catecholamine uptake blockers in the caudate-putamen and subregions of the medial prefrontal cortex of the rat. Brain Res. 2002, 936, 58–67. [Google Scholar]
- Chernoloz, O.; El Mansari, M.; Blier, P. Sustained administration of pramipexole modifies the spontaneous firing of dopamine, norepinephrine, and serotonin neurons in the rat brain. Neuropsychopharmacology 2009, 34, 651–661. [Google Scholar]
- Aman, T.K.; Shen, R.Y.; Haj-Dahmane, S. D2-like dopamine receptors depolarize dorsal raphe serotonin neurons through the activation of nonselective cationic conductance. J. Pharmacol. Exp. Ther. 2007, 320, 376–385. [Google Scholar]
- Haj-Dahmane, S. D2-like dopamine receptor activation excites rat dorsal raphe 5-HT neurons in vitro. Eur. J. Neurosci. 2001, 14, 125–134. [Google Scholar]
- Caldarone, B.J.; Paterson, N.E.; Zhou, J.; Brunner, D.; Kozikowski, A.P.; Westphal, K.G.; Korte-Bouws, G.A.; Prins, J.; Korte, S.M.; Olivier, B.; Ghavami, A. The novel triple reuptake inhibitor, JZAD-IV-22, exhibits an antidepressant pharmacological profile without locomotor stimulant or sensitization properties. J. Pharmacol. Exp. Ther. 2010. [Google Scholar] [CrossRef]
- Carter, D.S.; Cai, H.Y.; Lee, E.K.; Iyer, P.S.; Lucas, M.C.; Roetz, R.; Schoenfeld, R.C.; Weikert, R.J. 2-Substituted N-aryl piperazines as novel triple reuptake inhibitors for the treatment of depression. Bioorg. Med. Chem. Lett. 2010, 20, 3941–3945. [Google Scholar]
- Lee, K.H.; Park, C.E.; Min, K.H.; Shin, Y.J.; Chung, C.M.; Kim, H.H.; Yoon, H.J.; Won, K.; Ryu, E.J.; Nam, H.S.; Cho, J.W.; Lee, H.Y. Synthesis and pharmacological evaluation of 3-aryl-3-azolylpropan-1-amines as selective triple serotonin/norepinephrine/dopamine reuptake inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 5567–5571. [Google Scholar]
- Lucas, M.C.; Weikert, R.J.; Carter, D.S.; Cai, H.Y.; Greenhouse, R.; Iyer, P.S.; Lin, C.J.; Lee, E.K.; Madera, A.M.; Moore, A.; Ozboya, K.; Schoenfeld, R.C.; Steiner, S.; Zhai, Y.; Lynch, S.M. Design, synthesis, and biological evaluation of new monoamine reuptake inhibitors with potential therapeutic utility in depression and pain. Bioorg. Med. Chem. Lett. 2010, 20, 5559–5566. [Google Scholar]
- Micheli, F.; Cavanni, P.; Andreotti, D.; Arban, R.; Benedetti, R.; Bertani, B.; Bettati, M.; Bettelini, L.; Bonanomi, G.; Braggio, S.; et al. 6-(3,4-dichlorophenyl)-1-[(methyloxy)methyl]-3-azabicyclo[4.1.0]heptane: a new potent and selective triple reuptake inhibitor. J. Med. Chem. 2010, 53, 4989–5001. [Google Scholar]
- Micheli, F.; Cavanni, P.; Arban, R.; Benedetti, R.; Bertani, B.; Bettati, M.; Bettelini, L.; Bonanomi, G.; Braggio, S.; Checchia, A.; et al. 1-(Aryl)-6-[alkoxyalkyl]-3-azabicyclo[3.1.0] hexanes and 6-(aryl)-6-[alkoxyalkyl]-3-azabicyclo[3.1.0]hexanes: a new series of potent and selective triple reuptake inhibitors. J. Med. Chem. 2010, 53, 2534–2551. [Google Scholar]
- Schoedel, K.A.; Meier, D.; Chakraborty, B.; Manniche, P.M.; Sellers, E.M. Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin. Pharmacol. Ther. 2010, 88, 69–78. [Google Scholar]
- Prins, J.; Denys, D.A.; Westphal, K.G.; Korte-Bouws, G.A.; Quinton, M.S.; Schreiber, R.; Groenink, L.; Olivier, B.; Korte, S.M. The putative antidepressant DOV 216,303, a triple reuptake inhibitor, increases monoamine release in the prefrontal cortex of olfactory bulbectomized rats. Eur. J. Pharmacol. 2010, 633, 55–61. [Google Scholar]
- Kuikka, J.T.; Tiihonen, J.; Bergstrom, K.A.; Karhu, J.; Hartikainen, P.; Viinamaki, H.; Lansimies, E.; Lehtonen, J.; Hakola, P. Imaging of serotonin and dopamine transporters in the living human brain. Eur. J. Nucl. Med. 1995, 22, 346–350. [Google Scholar]
- Devoto, P.; Flore, G.; Pira, L.; Longu, G.; Gessa, G.L. Mirtazapine-induced corelease of dopamine and noradrenaline from noradrenergic neurons in the medial prefrontal and occipital cortex. Eur. J. Pharmacol. 2004, 487, 105–111. [Google Scholar]
- Devoto, P.; Flore, G.; Pira, L.; Longu, G.; Gessa, G.L. Alpha2-adrenoceptor mediated co-release of dopamine and noradrenaline from noradrenergic neurons in the cerebral cortex. J. Neurochem. 2004, 88, 1003–1009. [Google Scholar]
- Shoblock, J.R.; Maisonneuve, I.M.; Glick, S.D. Differential interactions of desipramine with amphetamine and methamphetamine: evidence that amphetamine releases dopamine from noradrenergic neurons in the medial prefrontal cortex. Neurochem. Res. 2004, 29, 1437–1442. [Google Scholar]
- Valentini, V.; Frau, R.; Di Chiara, G. Noradrenaline transporter blockers raise extracellular dopamine in medial prefrontal but not parietal and occipital cortex: differences with mianserin and clozapine. J. Neurochem. 2004, 88, 917–927. [Google Scholar]
- Gresch, P.J.; Sved, A.F.; Zigmond, M.J.; Finlay, J.M. Local influence of endogenous norepinephrine on extracellular dopamine in rat medial prefrontal cortex. J. Neurochem. 1995, 65, 111–116. [Google Scholar]
- Aluisio, L.; Lord, B.; Barbier, A.J.; Fraser, I.C.; Wilson, S.J.; Boggs, J.; Dvorak, L.K.; Letavic, M.A.; Maryanoff, B.E.; Carruthers, N.I.; Bonaventure, P.; Lovenberg, T.W. In-vitro and in-vivo characterization of JNJ-7925476, a novel triple monoamine uptake inhibitor. Eur. J. Pharmacol. 2008, 587, 141–146. [Google Scholar]
- Giros, B.; Wang, Y.M.; Suter, S.; McLeskey, S.B.; Pifl, C.; Caron, M.G. Delineation of discrete domains for substrate, cocaine, and tricyclic antidepressant interactions using chimeric dopamine-norepinephrine transporters. J. Biol. Chem. 1994, 269, 15985–15988. [Google Scholar]
- Ren, K.; Dubner, R. Descending modulation in persistent pain: an update. Pain 2002, 100, 1–6. [Google Scholar]
- Zhuo, M.; Gebhart, G.F. Spinal serotonin receptors mediate descending facilitation of a nociceptive reflex from the nuclei reticularis gigantocellularis and gigantocellularis pars alpha in the rat. Brain Res. 1991, 550, 35–48. [Google Scholar]
- Ardid, D.; Marty, H.; Fialip, J.; Privat, A.M.; Eschalier, A.; Lavarenne, J. Comparative effects of different uptake inhibitor antidepressants in two pain tests in mice. Fundam. Clin. Pharmacol. 1992, 6, 75–82. [Google Scholar]
- Mathews, T.A.; Fedele, D.E.; Coppelli, F.M.; Avila, A.M.; Murphy, D.L.; Andrews, A.M. Gene dose-dependent alterations in extraneuronal serotonin but not dopamine in mice with reduced serotonin transporter expression. J. Neurosci. Methods 2004, 140, 169–181. [Google Scholar]
- Fox, M.A.; Jensen, C.L.; Murphy, D.L. Tramadol and another atypical opioid meperidine have exaggerated serotonin syndrome behavioural effects, but decreased analgesic effects, in genetically deficient serotonin transporter (SERT) mice. Int. J. Neuropsychopharmacol. 2009, 12, 1055–1065. [Google Scholar]
- Otsuka, N.; Kiuchi, Y.; Yokogawa, F.; Masuda, Y.; Oguchi, K.; Hosoyamada, A. Antinociceptive efficacy of antidepressants: assessment of five antidepressants and four monoamine receptors in rats. J. Anesth. 2001, 15, 154–158. [Google Scholar]
- Korzeniewska-Rybicka, I.; Plaznik, A. Supraspinally mediated analgesic effect of antidepressant drugs. Pol. J. Pharmacol. 2000, 52, 93–99. [Google Scholar]
- Korzeniewska-Rybicka, I.; Plaznik, A. Analgesic effect of antidepressant drugs. Pharmacol. Biochem. Behav. 1998, 59, 331–338. [Google Scholar]
- Schreiber, S.; Pick, C.G. From selective to highly selective SSRIs: a comparison of the antinociceptive properties of fluoxetine, fluvoxamine, citalopram and escitalopram. Eur. Neuropsychopharmacol. 2006, 16, 464–468. [Google Scholar]
- Begovic, A.; Zulic, I.; Becic, F. Testing of analgesic effect of fluoxetine. Bosn. J. Basic. Med. Sci. 2004, 4, 79–81. [Google Scholar]
- Singh, V.P.; Jain, N.K.; Kulkarni, S.K. On the antinociceptive effect of fluoxetine, a selective serotonin reuptake inhibitor. Brain Res. 2001, 915, 218–226. [Google Scholar]
- Yokogawa, F.; Kiuchi, Y.; Ishikawa, Y.; Otsuka, N.; Masuda, Y.; Oguchi, K.; Hosoyamada, A. An investigation of monoamine receptors involved in antinociceptive effects of antidepressants. Anesth. Analg. 2002, 95, 163–168. [Google Scholar]
- Mahmood, D.; Akhtar, M.; Vohora, D.; Khanam, R. Comparison of antinociceptive and antidiabetic effects of sertraline and amitriptyline on streptozotocin-induced diabetic rats. Hum. Exp. Toxicol. 2010, 29, 881–886. [Google Scholar]
- Duman, E.N.; Kesim, M.; Kadioglu, M.; Yaris, E.; Kalyoncu, N.I.; Erciyes, N. Possible involvement of opioidergic and serotonergic mechanisms in antinociceptive effect of paroxetine in acute pain. J. Pharmacol. Sci. 2004, 94, 161–165. [Google Scholar]
- Duman, E.N.; Kesim, M.; Kadioglu, M.; Ulku, C.; Kalyoncu, N.I.; Yaris, E. Effect of gender on antinociceptive effect of paroxetine in hot plate test in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 292–296. [Google Scholar]
- Pakulska, W. Influence of sertraline on the antinociceptive effect of morphine, metamizol and indomethacin in mice. Acta Pol Pharm 2004, 61, 157–163. [Google Scholar]
- Nayebi, A.R.; Hassanpour, M.; Rezazadeh, H. Effect of chronic and acute administration of fluoxetine and its additive effect with morphine on the behavioural response in the formalin test in rats. J. Pharm. Pharmacol. 2001, 53, 219–225. [Google Scholar]
- Sounvoravong, S.; Nakashima, M.N.; Wada, M.; Nakashima, K. Modification of antiallodynic and antinociceptive effects of morphine by peripheral and central action of fluoxetine in a neuropathic mice model. Acta. Biol. Hung. 2007, 58, 369–379. [Google Scholar]
- Kesim, M.; Duman, E.N.; Kadioglu, M.; Yaris, E.; Kalyoncu, N.I.; Erciyes, N. The different roles of 5-HT(2) and 5-HT(3) receptors on antinociceptive effect of paroxetine in chemical stimuli in mice. J. Pharmacol. Sci. 2005, 97, 61–66. [Google Scholar]
- Gutierrez, M.; Ortega-Alvaro, A.; Gibert-Rahola, J.; Mico, J.A. Interactions of acute morphine with chronic imipramine and fluvoxamine treatment on the antinociceptive effect in arthritic rats. Neurosci. Lett. 2003, 352, 37–40. [Google Scholar]
- Ise, Y.; Katayama, S.; Hirano, M.; Aoki, T.; Narita, M.; Suzuki, T. Effects of fluvoxamine on morphine-induced inhibition of gastrointestinal transit, antinociception and hyperlocomotion in mice. Neurosci. Lett. 2001, 299, 29–32. [Google Scholar]
- Akunne, H.C.; Soliman, K.F. Serotonin modulation of pain responsiveness in the aged rat. Pharmacol. Biochem. Behav. 1994, 48, 411–416. [Google Scholar]
- Hynes, M.D.; Lochner, M.A.; Bemis, K.G.; Hymson, D.L. Fluoxetine, a selective inhibitor of serotonin uptake, potentiates morphine analgesia without altering its discriminative stimulus properties or affinity for opioid receptors. Life Sci. 1985, 36, 2317–2323. [Google Scholar]
- Langman, N.J.; Smith, C.G.; Whitehead, K.J. Selective serotonin re-uptake inhibition attenuates evoked glutamate release in the dorsal horn of the anaesthetised rat in vivo. Pharmacol. Res. 2006, 53, 149–155. [Google Scholar]
- Ikeda, T.; Ishida, Y.; Naono, R.; Takeda, R.; Abe, H.; Nakamura, T.; Nishimori, T. Effects of intrathecal administration of newer antidepressants on mechanical allodynia in rat models of neuropathic pain. Neurosci. Res. 2009, 63, 42–46. [Google Scholar]
- Taiwo, Y.O.; Fabian, A.; Pazoles, C.J.; Fields, H.L. Potentiation of morphine antinociception by monoamine reuptake inhibitors in the rat spinal cord. Pain 1985, 21, 329–337. [Google Scholar]
- Irwin, S.; Houde, R.W.; Bennett, D.R.; Hendershot, L.C.; Seevers, M.H. The effects of morphine methadone and meperidine on some reflex responses of spinal animals to nociceptive stimulation. J. Pharmacol. Exp. Ther. 1951, 101, 132–143. [Google Scholar]
- Caggiula, A.R.; Epstein, L.H.; Perkins, K.A.; Saylor, S. Different methods of assessing nicotine-induced antinociception may engage different neural mechanisms. Psychopharmacology (Berl) 1995, 122, 301–306. [Google Scholar]
- Rubinstein, M.; Mogil, J.S.; Japon, M.; Chan, E.C.; Allen, R.G.; Low, M.J. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3995–4000. [Google Scholar]
- Pakulska, W.; Czarnecka, E. Effect of citalopram and buspirone on the antinociceptive action of analgesic drugs. Acta. Pol. Pharm. 2001, 58, 299–305. [Google Scholar]
- Bomholt, S.F.; Mikkelsen, J.D.; Blackburn-Munro, G. Antinociceptive effects of the antidepressants amitriptyline, duloxetine, mirtazapine and citalopram in animal models of acute, persistent and neuropathic pain. Neuropharmacology 2005, 48, 252–263. [Google Scholar]
- Kall, E.; Lindstrom, E.; Martinez, V. The serotonin reuptake inhibitor citalopram does not affect colonic sensitivity or compliance in rats. Eur. J. Pharmacol. 2007, 570, 203–211. [Google Scholar]
- Jochum, T.; Boettger, M.K.; Wigger, A.; Beiderbeck, D.; Neumann, I.D.; Landgraf, R.; Sauer, H.; Bar, K.J. Decreased sensitivity to thermal pain in rats bred for high anxiety-related behaviour is attenuated by citalopram or diazepam treatment. Behav. Brain Res. 2007, 183, 18–24. [Google Scholar]
- Korzeniewska-Rybicka, I.; Plaznik, A. Role of serotonergic and noradrenergic systems in a model of visceral pain. Pol. J. Pharmacol. 2001, 53, 475–480. [Google Scholar]
- Jesse, C.R.; Wilhelm, E.A.; Nogueira, C.W. Depression-like behavior and mechanical allodynia are reduced by bis selenide treatment in mice with chronic constriction injury: a comparison with fluoxetine, amitriptyline, and bupropion. Psychopharmacology (Berl) 2010, 22, 513–522. [Google Scholar]
- Rephaeli, A.; Gil-Ad, I.; Aharoni, A.; Tarasenko, I.; Tarasenko, N.; Geffen, Y.; Halbfinger, E.; Nisemblat, Y.; Weizman, A.; Nudelman, A. Gamma-aminobutyric acid amides of nortriptyline and fluoxetine display improved pain suppressing activity. J. Med. Chem. 2009, 52, 3010–3017. [Google Scholar]
- Zhao, Z.Q.; Chiechio, S.; Sun, Y.G.; Zhang, K.H.; Zhao, C.S.; Scott, M.; Johnson, R.L.; Deneris, E.S.; Renner, K.J.; Gereau, R.W.t.; Chen, Z.F. Mice lacking central serotonergic neurons show enhanced inflammatory pain and an impaired analgesic response to antidepressant drugs. J. Neurosci. 2007, 27, 6045–6053. [Google Scholar]
- Anjaneyulu, M.; Chopra, K. Fluoxetine attenuates thermal hyperalgesia through 5-HT1/2 receptors in streptozotocin-induced diabetic mice. Eur. J. Pharmacol. 2004, 497, 285–292. [Google Scholar]
- Kesim, M.; Duman, E.N.; Kadioglu, M.; Ulku, C.; Muci, E.; Kalyoncu, N.I.; Yaris, E. Antinociceptive effects of fluoxetine and paroxetine with their related actions on glycemia in mice. Neuro. Endocrinol. Lett. 2006, 27, 281–287. [Google Scholar]
- Leventhal, L.; Smith, V.; Hornby, G.; Andree, T.H.; Brandt, M.R.; Rogers, K.E. Differential and synergistic effects of selective norepinephrine and serotonin reuptake inhibitors in rodent models of pain. J. Pharmacol. Exp. Ther. 2007, 320, 1178–1185. [Google Scholar]
- Pichon, X.; Wattiez, A.S.; Becamel, C.; Ehrlich, I.; Bockaert, J.; Eschalier, A.; Marin, P.; Courteix, C. Disrupting 5-HT(2A) receptor/PDZ protein interactions reduces hyperalgesia and enhances SSRI efficacy in neuropathic pain. Mol. Ther. 2010, 18, 1462–1470. [Google Scholar]
- Garcia, X.; del Valle, J.; Escribano, E.; Domenech, J.; Queralt, J. Analgesic and antiallodynic effects of antidepressants after infiltration into the rat. Pharmacology 2010, 86, 216–223. [Google Scholar]
- LaBuda, C.J.; Little, P.J. Pharmacological evaluation of the selective spinal nerve ligation model of neuropathic pain in the rat. J. Neurosci. Methods 2005, 144, 175–181. [Google Scholar]
- Abdel-Salam, O.M.; Baiuomy, A.R.; Arbid, M.S. Studies on the anti-inflammatory effect of fluoxetine in the rat. Pharmacol. Res. 2004, 49, 119–131. [Google Scholar]
- Abdel-Salam, O.M.; Nofal, S.M.; El-Shenawy, S.M. Evaluation of the anti-inflammatory and anti-nociceptive effects of different antidepressants in the rat. Pharmacol. Res. 2003, 48, 157–165. [Google Scholar]
- Sawynok, J.; Esser, M.J.; Reid, A.R. Peripheral antinociceptive actions of desipramine and fluoxetine in an inflammatory and neuropathic pain test in the rat. Pain 1999, 82, 149–158. [Google Scholar]
- Dirksen, R.; Van Luijtelaar, E.L.; Van Rijn, C.M. Selective serotonin reuptake inhibitors may enhance responses to noxious stimulation. Pharmacol. Biochem. Behav. 1998, 60, 719–725. [Google Scholar]
- Jett, M.F.; McGuirk, J.; Waligora, D.; Hunter, J.C. The effects of mexiletine, desipramine and fluoxetine in rat models involving central sensitization. Pain 1997, 69, 161–169. [Google Scholar]
- Pedersen, L.H.; Nielsen, A.N.; Blackburn-Munro, G. Anti-nociception is selectively enhanced by parallel inhibition of multiple subtypes of monoamine transporters in rat models of persistent and neuropathic pain. Psychopharmacology (Berl) 2005, 182, 551–561. [Google Scholar]
- Hayashi, T.; Miyata, M.; Nagata, T.; Izawa, Y.; Kawakami, Y. Intracerebroventricular fluvoxamine administration inhibited pain behavior but increased Fos expression in affective pain pathways. Pharmacol. Biochem. Behav. 2009, 91, 441–446. [Google Scholar]
- Nozaki, C.; Kamei, J. Possible involvement of opioidergic systems in the antinociceptive effect of the selective serotonin reuptake inhibitors in sciatic nerve-injured mice. Eur. J. Pharmacol. 2006, 552, 99–104. [Google Scholar]
- Honda, M.; Uchida, K.; Tanabe, M.; Ono, H. Fluvoxamine, a selective serotonin reuptake inhibitor, exerts its antiallodynic effects on neuropathic pain in mice via 5-HT2A/2C receptors. Neuropharmacology 2006, 51, 866–872. [Google Scholar]
- Schreiber, S.; Backer, M.M.; Yanai, J.; Pick, C.G. The antinociceptive effect of fluvoxamine. Eur. Neuropsychopharmacol. 1996, 6, 281–284. [Google Scholar]
- Gray, A.M.; Spencer, P.S.; Sewell, R.D. The involvement of the opioidergic system in the antinociceptive mechanism of action of antidepressant compounds. Br. J. Pharmacol. 1998, 124, 669–674. [Google Scholar]
- Obata, H.; Saito, S.; Koizuka, S.; Nishikawa, K.; Goto, F. The monoamine-mediated antiallodynic effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a rat model of neuropathic pain. Anesth. Analg. 2005, 100, 1406–1410. [Google Scholar]
- Yalcin, I.; Tessier, L.H.; Petit-Demouliere, N.; Doridot, S.; Hein, L.; Freund-Mercier, M.J.; Barrot, M. Beta2-adrenoceptors are essential for desipramine, venlafaxine or reboxetine action in neuropathic pain. Neurobiol. Dis. 2009, 33, 386–394. [Google Scholar]
- Rojas-Corrales, M.O.; Casas, J.; Moreno-Brea, M.R.; Gibert-Rahola, J.; Mico, J.A. Antinociceptive effects of tricyclic antidepressants and their noradrenergic metabolites. Eur. Neuropsychopharmacol. 2003, 13, 355–363. [Google Scholar]
- Jones, K.L.; Finn, D.P.; Governo, R.J.; Prior, M.J.; Morris, P.G.; Kendall, D.A.; Marsden, C.A.; Chapman, V. Identification of discrete sites of action of chronic treatment with desipramine in a model of neuropathic pain. Neuropharmacology 2009, 56, 405–413. [Google Scholar]
- Sawynok, J.; Reid, A. Antinociception by tricyclic antidepressants in the rat formalin test: differential effects on different behaviours following systemic and spinal administration. Pain 2001, 93, 51–59. [Google Scholar]
- Kawamata, T.; Omote, K.; Kawamata, M.; Namiki, A. Analgesic effect of intrathecal desipramine on carrageenan-induced thermal hyperalgesia in the rat. Br. J. Anaesth. 1999, 83, 449–452. [Google Scholar]
- Reimann, W.; Schlutz, H.; Selve, N. The antinociceptive effects of morphine, desipramine, and serotonin and their combinations after intrathecal injection in the rat. Anesth. Analg. 1999, 88, 141–145. [Google Scholar]
- Sawynok, J.; Reid, A. Desipramine potentiates spinal antinociception by 5-hydroxytryptamine, morphine and adenosine. Pain 1992, 50, 113–118. [Google Scholar]
- Ardid, D.; Guilbaud, G. Antinociceptive effects of acute and ‘chronic’ injections of tricyclic antidepressant drugs in a new model of mononeuropathy in rats. Pain 1992, 49, 279–287. [Google Scholar]
- Lund, A.; Tjolsen, A.; Hole, K. The apparent antinociceptive effect of desipramine and zimelidine in the tail flick test in rats is mainly caused by changes in tail skin temperature. Pain 1989, 38, 65–69. [Google Scholar]
- Lund, A.; Mjellem-Joly, N.; Hole, K. Chronic administration of desipramine and zimelidine changes the behavioural response in the formalin test in rats. Neuropharmacology 1991, 30, 481–487. [Google Scholar]
- Danysz, W.; Minor, B.G.; Post, C.; Archer, T. Chronic treatment with antidepressant drugs and the analgesia induced by 5-methoxy-N,N-dimethyltryptamine: attenuation by desipramine. Acta. Pharmacol. Toxicol. (Copenh) 1986, 59, 103–112. [Google Scholar]
- Schreiber, S.; Frishtick, R.; Volis, I.; Rubovitch, V.; Pick, C.G.; Weizman, R. The antinociceptive properties of reboxetine in acute pain. Eur. Neuropsychopharmacol. 2009, 19, 735–739. [Google Scholar]
- Obata, H.; Conklin, D.; Eisenach, J.C. Spinal noradrenaline transporter inhibition by reboxetine and Xen2174 reduces tactile hypersensitivity after surgery in rats. Pain 2005, 113, 271–276. [Google Scholar]
- Kuhad, A.; Bishnoi, M.; Chopra, K. Anti-nociceptive effect of duloxetine in mouse model of diabetic neuropathic pain. Indian J. Exp. Biol. 2009, 47, 193–197. [Google Scholar]
- Kiso, T.; Watabiki, T.; Tsukamoto, M.; Okabe, M.; Kagami, M.; Nishimura, K.; Aoki, T.; Matsuoka, N. Pharmacological characterization and gene expression profiling of an L5/L6 spinal nerve ligation model for neuropathic pain in mice. Neuroscience 2008, 153, 492–500. [Google Scholar]
- Wantuch, C.; Piesla, M.; Leventhal, L. Pharmacological validation of a model of cystitis pain in the mouse. Neurosci. Lett. 2007, 421, 250–252. [Google Scholar]
- Jones, C.K.; Peters, S.C.; Shannon, H.E. Synergistic interactions between the dual serotonergic, noradrenergic reuptake inhibitor duloxetine and the non-steroidal anti-inflammatory drug ibuprofen in inflammatory pain in rodents. Eur. J. Pain 2007, 11, 208–215. [Google Scholar]
- Jones, C.K.; Peters, S.C.; Shannon, H.E. Efficacy of duloxetine, a potent and balanced serotonergic and noradrenergic reuptake inhibitor, in inflammatory and acute pain models in rodents. J. Pharmacol. Exp. Ther. 2005, 312, 726–732. [Google Scholar]
- Bardin, L.; Gregoire, S.; Aliaga, M.; Malfetes, N.; Vitton, O.; Ladure, P.; Newman-Tancredi, A.; Depoortere, R. Comparison of milnacipran, duloxetine and pregabalin in the formalin pain test and in a model of stress-induced ultrasonic vocalizations in rats. Neurosci. Res. 2010, 66, 135–140. [Google Scholar]
- Chandran, P.; Pai, M.; Blomme, E.A.; Hsieh, G.C.; Decker, M.W.; Honore, P. Pharmacological modulation of movement-evoked pain in a rat model of osteoarthritis. Eur. J. Pharmacol. 2009, 613, 39–45. [Google Scholar]
- Munro, G. Pharmacological assessment of the rat formalin test utilizing the clinically used analgesic drugs gabapentin, lamotrigine, morphine, duloxetine, tramadol and ibuprofen: influence of low and high formalin concentrations. Eur. J. Pharmacol. 2009, 605, 95–102. [Google Scholar]
- Iyengar, S.; Webster, A.A.; Hemrick-Luecke, S.K.; Xu, J.Y.; Simmons, R.M. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J. Pharmacol. Exp. Ther. 2004, 311, 576–584. [Google Scholar]
- Thibault, K.; Calvino, B.; Pezet, S. Characterisation of sensory abnormalities observed in an animal model of multiple sclerosis: A behavioural and pharmacological study. Eur. J. Pain 2010. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Zou, S.; Wei, F.; Dubner, R.; Ren, K. Long lasting pain hypersensitivity following ligation of the tendon of the masseter muscle in rats: a model of myogenic orofacial pain. Mol. Pain 2010, 6, 40. [Google Scholar]
- Suzuki, T.; Ueta, K.; Tamagaki, S.; Mashimo, T. Antiallodynic and antihyperalgesic effect of milnacipran in mice with spinal nerve ligation. Anesth. Analg. 2008, 106, 1309–1315. [Google Scholar]
- Aoki, M.; Tsuji, M.; Takeda, H.; Harada, Y.; Nohara, J.; Matsumiya, T.; Chiba, H. Antidepressants enhance the antinociceptive effects of carbamazepine in the acetic acid-induced writhing test in mice. Eur. J. Pharmacol. 2006, 550, 78–83. [Google Scholar]
- Obata, H.; Kimura, M.; Nakajima, K.; Tobe, M.; Nishikawa, K.; Saito, S. Monoamine-dependent, opioid-independent antihypersensitivity effects of intrathecally administered milnacipran, a serotonin noradrenaline reuptake inhibitor, in a postoperative pain model in rats. J. Pharmacol. Exp. Ther. 2010, 334, 1059–1065. [Google Scholar]
- Takeda, R.; Watanabe, Y.; Ikeda, T.; Abe, H.; Ebihara, K.; Matsuo, H.; Nonaka, H.; Hashiguchi, H.; Nishimori, T.; Ishida, Y. Analgesic effect of milnacipran is associated with c-Fos expression in the anterior cingulate cortex in the rat neuropathic pain model. Neurosci. Res. 2009, 64, 380–384. [Google Scholar]
- Kim, S.H.; Song, J.; Mun, H.; Park, K.U. Effect of the combined use of tramadol and milnacipran on pain threshold in an animal model of fibromyalgia. Korean J. Int. Med. 2009, 24, 139–142. [Google Scholar]
- Onal, A.; Parlar, A.; Ulker, S. Milnacipran attenuates hyperalgesia and potentiates antihyperalgesic effect of tramadol in rats with mononeuropathic pain. Pharmacol. Biochem. Behav. 2007, 88, 171–178. [Google Scholar]
- Shin, S.W.; Eisenach, J.C.; Rao, S.G.; Tong, C. The monoamine reuptake inhibitor milnacipran does not affect nociception to acute visceral distension in rats. Anesth. Analg. 2004, 98, 1365–1369. [Google Scholar]
- Berrocoso, E.; Mico, J.A. Role of serotonin 5-HT1A receptors in the antidepressant-like effect and the antinociceptive effect of venlafaxine in mice. Int. J. Neuropsychopharmacol. 2009, 12, 61–71. [Google Scholar]
- Yaba, G.; Sezer, Z.; Tekol, Y. Interaction between venlafaxine and caffeine on antinociception in mice. Pharmazie 2006, 61, 60–62. [Google Scholar]
- Schreiber, S.; Bleich, A.; Pick, C.G. Venlafaxine and mirtazapine: different mechanisms of antidepressant action, common opioid-mediated antinociceptive effects--a possible opioid involvement in severe depression? J. Mol. Neurosci. 2002, 18, 143–149. [Google Scholar]
- Schreiber, S.; Backer, M.M.; Pick, C.G. The antinociceptive effect of venlafaxine in mice is mediated through opioid and adrenergic mechanisms. Neurosci. Lett. 1999, 273, 85–88. [Google Scholar]
- Aricioglu, F.; Buldanlioglu, U.; Salanturoglu, G.; Ozyalcin, N.S. Evaluation of antinociceptive and anti-inflammatory effects of venlafaxine in the rat. Agri 2005, 17, 41–46. [Google Scholar]
- Marchand, F.; Alloui, A.; Pelissier, T.; Hernandez, A.; Authier, N.; Alvarez, P.; Eschalier, A.; Ardid, D. Evidence for an antihyperalgesic effect of venlafaxine in vincristine-induced neuropathy in rat. Brain Res. 2003, 980, 117–120. [Google Scholar]
- Ling, B.; Authier, N.; Balayssac, D.; Eschalier, A.; Coudore, F. Behavioral and pharmacological description of oxaliplatin-induced painful neuropathy in rat. Pain 2007, 128, 225–234. [Google Scholar]
- Uyar, M.; Onal, A.; Dogru, A.; Soykan, N. The antinociceptive effect of tramadol-venlafaxine combination on the paw withdrawal threshold in a rat model of neuropathic pain. Methods Find. Exp. Clin. Pharmacol. 2003, 25, 361–365. [Google Scholar]
- Gultekin, H.; Ahmedov, V. Role of the opioidergic system and nitric oxide in the analgesic effect of venlafaxine. Yakugaku Zasshi 2006, 126, 117–121. [Google Scholar]
- Rode, F.; Brolos, T.; Blackburn-Munro, G.; Bjerrum, O.J. Venlafaxine compromises the antinociceptive actions of gabapentin in rat models of neuropathic and persistent pain. Psychopharmacology (Berl) 2006, 187, 364–375. [Google Scholar]
- Bonnefont, J.; Chapuy, E.; Clottes, E.; Alloui, A.; Eschalier, A. Spinal 5-HT1A receptors differentially influence nociceptive processing according to the nature of the noxious stimulus in rats: effect of WAY-100635 on the antinociceptive activities of paracetamol, venlafaxine and 5-HT. Pain 2005, 114, 482–490. [Google Scholar]
- Hernandez, A.; Constandil, L.; Laurido, C.; Pelissier, T.; Marchand, F.; Ardid, D.; Alloui, A.; Eschalier, A.; Soto-Moyano, R. Venlafaxine-induced depression of wind-up activity in mononeuropathic rats is potentiated by inhibition of brain 5-HT1A receptor expression in vivo. Int. J. Neurosci. 2004, 114, 229–242. [Google Scholar]
- Lang, E.; Hord, A.H.; Denson, D. Venlafaxine hydrochloride (Effexor) relieves thermal hyperalgesia in rats with an experimental mononeuropathy. Pain 1996, 68, 151–155. [Google Scholar]
- Gilbert, A.K.; Franklin, K.B. Characterization of the analgesic properties of nomifensine in rats. Pharmacol. Biochem. Behav. 2001, 68, 783–787. [Google Scholar]
- Rigal, F.; Eschalier, A.; Devoize, J.L.; Pechadre, J.C. Activities of five antidepressants in a behavioral pain test in rats. Life Sci. 1983, 32, 2965–2971. [Google Scholar]
- Gonzalez, J.P.; Sewell, R.D.; Spencer, P.S. Dopaminergic involvement in the hyperalgesic effect of nomifensine. Life Sci. 1981, 28, 951–956. [Google Scholar]
- Gonzalez, J.P.; Sewell, R.D.; Spencer, P.S. Antinociceptive activity of opiates in the presence of the antidepressant agent nomifensine. Neuropharmacology 1980, 19, 613–618. [Google Scholar]
- Basile, A.S.; Janowsky, A.; Golembiowska, K.; Kowalska, M.; Tam, E.; Benveniste, M.; Popik, P.; Nikiforuk, A.; Krawczyk, M.; Nowak, G.; Krieter, P.A.; Lippa, A.S.; Skolnick, P.; Koustova, E. Characterization of the antinociceptive actions of bicifadine in models of acute, persistent, and chronic pain. J. Pharmacol. Exp. Ther. 2007, 321, 1208–1225. [Google Scholar]
- Mork, A.; Kreilgaard, M.; Sanchez, C. The R-enantiomer of citalopram counteracts escitalopram-induced increase in extracellular 5-HT in the frontal cortex of freely moving rats. Neuropharmacology 2003, 45, 167–173. [Google Scholar]
- Sanchez, C.; Gruca, P.; Bien, E.; Papp, M. R-citalopram counteracts the effect of escitalopram in a rat conditioned fear stress model of anxiety. Pharmacol. Biochem. Behav. 2003, 75, 903–907. [Google Scholar]
- McCarson, K.E.; Duric, V.; Reisman, S.A.; Winter, M.; Enna, S.J. GABA(B) receptor function and subunit expression in the rat spinal cord as indicators of stress and the antinociceptive response to antidepressants. Brain Res. 2006, 1068, 109–117. [Google Scholar]
- Gray, E. Linking chronic pain and depression. Nurs. Stand. 2001, 15, 33–36. [Google Scholar]
- Bohn, L.M.; Xu, F.; Gainetdinov, R.R.; Caron, M.G. Potentiated opioid analgesia in norepinephrine transporter knock-out mice. J. Neurosci. 2000, 20, 9040–9045. [Google Scholar]
- Jasmin, L.; Tien, D.; Janni, G.; Ohara, P.T. Is noradrenaline a significant factor in the analgesic effect of antidepressants? Pain 2003, 106, 3–8. [Google Scholar]
- Kehl, L.J.; Wilcox, G.L. Anti-nociceptive effect of tricyclic anti-depressants following intrathecal administration. Anesth. Prog. 1984, 31, 82–84. [Google Scholar]
- Kellstein, D.E.; Malseed, R.T.; Goldstein, F.J. Contrasting effects of acute vs. chronic tricyclic antidepressant treatment on central morphine analgesia. Pain 1984, 20, 323–334. [Google Scholar]
- Goldstein, F.J.; Mojaverian, P.; Ossipov, M.H.; Swanson, B.N. Elevation in analgetic effect and plasma levels of morphine by desipramine in rats. Pain 1982, 14, 279–282. [Google Scholar]
- Nielsen, C.K.; Lewis, R.J.; Alewood, D.; Drinkwater, R.; Palant, E.; Patterson, M.; Yaksh, T.L.; McCumber, D.; Smith, M.T. Anti-allodynic efficacy of the chi-conopeptide, Xen2174, in rats with neuropathic pain. Pain 2005, 118, 112–124. [Google Scholar]
- Whiteside, G.T.; Dwyer, J.M.; Harrison, J.E.; Beyer, C.E.; Cummons, T.; Manzino, L.; Mark, L.; Johnston, G.H.; Strassle, B.W.; Adedoyin, A.; et al. WAY-318068: a novel, potent and selective noradrenaline re-uptake inhibitor with activity in rodent models of pain and depression. Br. J. Pharmacol. 2010, 160, 1105–1118. [Google Scholar]
- Marchand, F.; Alloui, A.; Chapuy, E.; Jourdan, D.; Pelissier, T.; Ardid, D.; Hernandez, A.; Eschalier, A. Evidence for a monoamine mediated, opioid-independent, antihyperalgesic effect of venlafaxine, a non-tricyclic antidepressant, in a neurogenic pain model in rats. Pain 2003, 103, 229–235. [Google Scholar]
- Marchand, F.; Alloui, A.; Chapuy, E.; Hernandez, A.; Pelissier, T.; Ardid, D.; Eschalier, A. The antihyperalgesic effect of venlafaxine in diabetic rats does not involve the opioid system. Neurosci. Lett. 2003, 342, 105–108. [Google Scholar]
- Fishbain, D.A.; Cutler, R.; Rosomoff, H.L.; Rosomoff, R.S. Evidence-based data from animal and human experimental studies on pain relief with antidepressants: a structured review. Pain Med. 2000, 1, 310–316. [Google Scholar]
- Gatch, M.B.; Negus, S.S.; Mello, N.K. Antinociceptive effects of monoamine reuptake inhibitors administered alone or in combination with mu opioid agonists in rhesus monkeys. Psychopharmacology (Berl) 1998, 135, 99–106. [Google Scholar]
- Rogers, L.W.; Giordano, J. Effects of systemically administered monoamine reuptake blocking agents on patterns of buspirone-induced analgesia in rats. Life Sci. 1990, 47, 961–969. [Google Scholar]
- Burkey, A.R.; Carstens, E.; Jasmin, L. Dopamine reuptake inhibition in the rostral agranular insular cortex produces antinociception. J. Neurosci. 1999, 19, 4169–4179. [Google Scholar]
- Wong, E.H.; Sonders, M.S.; Amara, S.G.; Tinholt, P.M.; Piercey, M.F.; Hoffmann, W.P.; Hyslop, D.K.; Franklin, S.; Porsolt, R.D.; Bonsignori, A.; Carfagna, N.; McArthur, R.A. Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol. Psychiatry 2000, 47, 818–829. [Google Scholar]
- Miller, D.K.; Wong, E.H.; Chesnut, M.D.; Dwoskin, L.P. Reboxetine: functional inhibition of monoamine transporters and nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2002, 302, 687–695. [Google Scholar]
- Hajos, M.; Fleishaker, J.C.; Filipiak-Reisner, J.K.; Brown, M.T.; Wong, E.H. The selective norepinephrine reuptake inhibitor antidepressant reboxetine: pharmacological and clinical profile. CNS Drug Rev. 2004, 10, 23–44. [Google Scholar]
- Wood, P.B. Role of central dopamine in pain and analgesia. Expert. Rev. Neurother. 2008, 8, 781–797. [Google Scholar]
- Pelissier, T.; Laurido, C.; Hernandez, A.; Constandil, L.; Eschalier, A. Biphasic effect of apomorphine on rat nociception and effect of dopamine D2 receptor antagonists. Eur. J. Pharmacol. 2006, 546, 40–47. [Google Scholar]
- Taylor, B.K.; Joshi, C.; Uppal, H. Stimulation of dopamine D2 receptors in the nucleus accumbens inhibits inflammatory pain. Brain Res. 2003, 987, 135–143. [Google Scholar]
- Aimone, L.D.; Jones, S.L.; Gebhart, G.F. Stimulation-produced descending inhibition from the periaqueductal gray and nucleus raphe magnus in the rat: mediation by spinal monoamines but not opioids. Pain 1987, 31, 123–136. [Google Scholar]
- Pastoriza, L.N.; Morrow, T.J.; Casey, K.L. Medial frontal cortex lesions selectively attenuate the hot plate response: possible nocifensive apraxia in the rat. Pain 1996, 64, 11–17. [Google Scholar]
- Jensen, T.S.; Schroder, H.D.; Smith, D.F. The role of spinal pathways in dopamine mediated alteration in the tail-flick reflex in rats. Neuropharmacology 1984, 23, 149–153. [Google Scholar]
- Barasi, S.; Duggal, K.N. The effect of local and systemic application of dopaminergic agents on tail flick latency in the rat. Eur. J. Pharmacol. 1985, 117, 287–294. [Google Scholar]
- Gao, X.; Zhang, Y.; Wu, G. Effects of dopaminergic agents on carrageenan hyperalgesia after intrathecal administration to rats. Eur. J. Pharmacol. 2001, 418, 73–77. [Google Scholar]
- Tamae, A.; Nakatsuka, T.; Koga, K.; Kato, G.; Furue, H.; Katafuchi, T.; Yoshimura, M. Direct inhibition of substantia gelatinosa neurones in the rat spinal cord by activation of dopamine D2-like receptors. J. Physiol. 2005, 568, 243–253. [Google Scholar]
- Munro, G. Dopamine D(1) and D(2) receptor agonism enhances antinociception mediated by the serotonin and noradrenaline reuptake inhibitor duloxetine in the rat formalin test. Eur. J. Pharmacol. 2007, 575, 66–74. [Google Scholar]
- Rosland, J.H.; Hole, K. The effect of nefopam and its enantiomers on the uptake of 5-hydroxytryptamine, noradrenaline and dopamine in crude rat brain synaptosomal preparations. J. Pharm. Pharmacol. 1990, 42, 437–438. [Google Scholar]
- Esposito, E.; Romandini, S.; Merlo-Pich, E.; Mennini, T.; Samanin, R. Evidence of the involvement of dopamine in the analgesic effect of nefopam. Eur. J. Pharmacol. 1986, 128, 157–164. [Google Scholar]
- Girard, P.; Coppe, M.C.; Verniers, D.; Pansart, Y.; Gillardin, J.M. Role of catecholamines and serotonin receptor subtypes in nefopam-induced antinociception. Pharmacol. Res. 2006, 54, 195–202. [Google Scholar]
- Munro, G.; Baek, C.A.; Erichsen, H.K.; Nielsen, A.N.; Nielsen, E.O.; Scheel-Kruger, J.; Weikop, P.; Peters, D. The novel compound (+/−)-1-[10-((E)-3-Phenyl-allyl)-3,10-diaza-bicyclo[4.3.1]dec-3-yl]-propan -1-one (NS7051) attenuates nociceptive transmission in animal models of experimental pain; a pharmacological comparison with the combined mu-opioid receptor agonist and monoamine reuptake inhibitor tramadol. Neuropharmacology 2008, 54, 331–343. [Google Scholar]
- Efficacy and safety of bicifadine in the treatment of chronic low back pain. (January)
- long-term safety study of bicifadine for the treatment of chroic low back pain. http://clinicaltrials.gov/ct2/show/NCT00281645, accessed on 26 January 2011.
- Double Blind RCT of Bicifadine SR in outpatients with chronic neuropathic pain associated with diabetes. http://clinicaltrials.gov/ct2/show/NCT00553592, accessed on 26 January 2011.
- A 52-week study of bicifadine in patients with chronic neuropathic pain associated with diabetic periphral neurpathy. (Jun, 29)
- Hill, R. NK1 (substance P) receptor antagonists--why are they not analgesic in humans? Trends Pharmacol. Sci. 2000, 21, 244–246. [Google Scholar]
- Mao, J. Translational pain research: achievements and challenges. J. Pain 2009, 10, 1001–1011. [Google Scholar]
- Mao, J. Translational pain research: bridging the gap between basic and clinical research. Pain 2002, 97, 183–187. [Google Scholar]
- Villanueva, L. Is there a gap between preclinical and clinical studies of analgesia? Trends Pharmacol. Sci. 2000, 21, 461–462, author reply 465. [Google Scholar]
- Langford, D.J.; Bailey, A.L.; Chanda, M.L.; Clarke, S.E.; Drummond, T.E.; Echols, S.; Glick, S.; Ingrao, J.; Klassen-Ross, T.; Lacroix-Fralish, M.L.; et al. Coding of facial expressions of pain in the laboratory mouse. Nat. Methods 2010, 7, 447–449. [Google Scholar]
- Chesler, E.J.; Wilson, S.G.; Lariviere, W.R.; Rodriguez-Zas, S.L.; Mogil, J.S. Identification and ranking of genetic and laboratory environment factors influencing a behavioral trait, thermal nociception, via computational analysis of a large data archive. Neurosci. Biobehav. Rev. 2002, 26, 907–923. [Google Scholar]
- Mogil, J.S.; Davis, K.D.; Derbyshire, S.W. The necessity of animal models in pain research. Pain 2010, 151, 12–17. [Google Scholar]
- Rice, A.S.; Cimino-Brown, D.; Eisenach, J.C.; Kontinen, V.K.; Lacroix-Fralish, M.L.; Machin, I.; Mogil, J.S.; Stohr, T. Animal models and the prediction of efficacy in clinical trials of analgesic drugs: a critical appraisal and call for uniform reporting standards. Pain 2008, 139, 243–247. [Google Scholar]
- Lepine, J.P.; Briley, M. The epidemiology of pain in depression. Hum. Psychopharmacol. 2004, 19 Suppl. 1, S3–S7. [Google Scholar]
- Ansari, A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv. Rev. Psychiatry 2000, 7, 257–277. [Google Scholar]
- Max, M.B.; Lynch, S.A.; Muir, J.; Shoaf, S.E.; Smoller, B.; Dubner, R. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. N. Engl. J. Med. 1992, 326, 1250–1256. [Google Scholar]
- Staiger, T.O.; Gaster, B.; Sullivan, M.D.; Deyo, R.A. Systematic review of antidepressants in the treatment of chronic low back pain. Spine (Phila Pa 1976) 2003, 28, 2540–2545. [Google Scholar]
- Max, M.B. Treatment of post-herpetic neuralgia: antidepressants. Ann. Neurol. 1994, 35, S50–53. [Google Scholar]
- Anderberg, U.M.; Marteinsdottir, I.; von Knorring, L. Citalopram in patients with fibromyalgia--a randomized, double-blind, placebo-controlled study. Eur. J. Pain 2000, 4, 27–35. [Google Scholar]
- Fishbain, D. Evidence-based data on pain relief with antidepressants. Ann. Med. 2000, 32, 305–316. [Google Scholar]
- Entsuah, R.; Gao, B. Global benefit-risk evaluation of antidepressant action: Comparison of pooled data for venlafaxine, SSRIs, and placebo. CNS Spectr. 2002, 7, 882–888. [Google Scholar]
- Sindrup, S.H.; Jensen, T.S. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain 1999, 83, 389–400. [Google Scholar]
- Dworkin, R.H.; O'Connor, A.B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; Portenoy, R.K.; Rice, A.S.; Stacey, B.R.; Treede, R.D.; Turk, D.C.; Wallace, M.S. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar]
- Lunn, M. P.; Hughes, R. A.; Wiffen, P. J. Duloxetine for treating painful neuropathy or chronic pain. Cochrane Database Syst. Rev. 2009, CD007115. [Google Scholar]
- Davidson, J.R.; France, R.D. Bupropion in chronic low back pain. J. Clin. Psychiatry 1994, 55, 362. [Google Scholar]
- Semenchuk, M.R.; Davis, B. Efficacy of sustained-release bupropion in neuropathic pain: an open-label study. Clin. J. Pain 2000, 16, 6–11. [Google Scholar]
- Semenchuk, M.R.; Sherman, S.; Davis, B. Double-blind, randomized trial of bupropion SR for the treatment of neuropathic pain. Neurology 2001, 57, 1583–1588. [Google Scholar]
- Papakostas, G.I. The efficacy, tolerability, and safety of contemporary antidepressants. J. Clin. Psychiatry 2010, 71 Suppl. E1, e03. [Google Scholar]
- Schatzberg, A.F. Safety and tolerability of antidepressants: weighing the impact on treatment decisions. J. Clin. Psychiatry 2007, 68 Suppl. 8, 26–34. [Google Scholar]
- Gitlin, M.J.; Suri, R.; Altshuler, L.; Zuckerbrow-Miller, J.; Fairbanks, L. Bupropion-sustained release as a treatment for SSRI-induced sexual side effects. J. Sex Marital. Ther. 2002, 28, 131–138. [Google Scholar]
- Zisook, S.; Rush, A.J.; Haight, B.R.; Clines, D.C.; Rockett, C.B. Use of bupropion in combination with serotonin reuptake inhibitors. Biol. Psychiatry 2006, 59, 203–210. [Google Scholar]
- Safarinejad, M.R. The effects of the adjunctive bupropion on male sexual dysfunction induced by a selective serotonin reuptake inhibitor: a double-blind placebo-controlled and randomized study. BJU Int. 2010, 106, 840–847. [Google Scholar]
- Fukui, H.; Murai, T. Severe weight gain induced by combination treatment with risperidone and paroxetine. Clin. Neuropharmacol. 2002, 25, 269–271. [Google Scholar]
- Dording, C.M.; Mischoulon, D.; Petersen, T.J.; Kornbluh, R.; Gordon, J.; Nierenberg, A.A.; Rosenbaum, J.E.; Fava, M. The pharmacologic management of SSRI-induced side effects: a survey of psychiatrists. Ann. Clin. Psychiatry 2002, 14, 143–147. [Google Scholar]
- Volkow, N.D. What do we know about drug addiction? Am. J. Psychiatry. 2005, 162, 1401–1402. [Google Scholar]
- Rothman, R.B. High affinity dopamine reuptake inhibitors as potential cocaine antagonists: a strategy for drug development. Life Sci. 1990, 46, PL17–PL21. [Google Scholar]
- Beer, B.; Stark, J.; Krieter, P.; Czobor, P.; Beer, G.; Lippa, A.; Skolnick, P. DOV 216,303, a “triple” reuptake inhibitor: safety, tolerability, and pharmacokinetic profile. J. Clin. Pharmacol. 2004, 44, 1360–1367. [Google Scholar]
- Rascol, O.; Poewe, W.; Lees, A.; Aristin, M.; Salin, L.; Juhel, N.; Waldhauser, L.; Schindler, T. Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced Parkinson disease and motor fluctuations: the ADVANS Study. Arch. Neurol. 2008, 65, 577–583. [Google Scholar]
- Hauser, R.A.; Salin, L.; Juhel, N.; Konyago, V.L. Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson's disease. Mov. Disord. 2007, 22, 359–365. [Google Scholar]
Abbreviations
| ACC | anterior cingulate cortex |
| 5-HT | 5-hydroxytryptamine or serotonin |
| 5-HTT | serotonin transporter |
| CPu | caudate putamen |
| CRF | corticotropin-releasing factor |
| DA | dopamine |
| DAT | dopamine transporter |
| DRN | dorsal raphe nucleus |
| FC | frontal cortex |
| IC | insular cortex |
| LC | locus coeruleus |
| mPFC | medial prefrontal cortex |
| NAcc | nucleus accumbens |
| NE | norepinephrine |
| NET | norepinephrine transporter |
| PAG | periaqueductal grey |
| PFN | parafascicular nucleus |
| RVM | rostral ventromedial medulla |
| SN | substantia nigra |
| SNL | sciatic nerve ligature |
| SNRI | serotonin and norepinephrine reuptake inhibitor |
| SP | substance P |
| SSRI | serotonin selective reuptake inhibitor |
| STZ | streptozotocin |
| TRI | triple reuptake inhibitor |
| VTA | ventral tegmental area |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hache, G.; Coudore, F.; Gardier, A.M.; Guiard, B.P. Monoaminergic Antidepressants in the Relief of Pain: Potential Therapeutic Utility of Triple Reuptake Inhibitors (TRIs). Pharmaceuticals 2011, 4, 285-342. https://doi.org/10.3390/ph4020285
Hache G, Coudore F, Gardier AM, Guiard BP. Monoaminergic Antidepressants in the Relief of Pain: Potential Therapeutic Utility of Triple Reuptake Inhibitors (TRIs). Pharmaceuticals. 2011; 4(2):285-342. https://doi.org/10.3390/ph4020285
Chicago/Turabian StyleHache, Guillaume, François Coudore, Alain M. Gardier, and Bruno P. Guiard. 2011. "Monoaminergic Antidepressants in the Relief of Pain: Potential Therapeutic Utility of Triple Reuptake Inhibitors (TRIs)" Pharmaceuticals 4, no. 2: 285-342. https://doi.org/10.3390/ph4020285
APA StyleHache, G., Coudore, F., Gardier, A. M., & Guiard, B. P. (2011). Monoaminergic Antidepressants in the Relief of Pain: Potential Therapeutic Utility of Triple Reuptake Inhibitors (TRIs). Pharmaceuticals, 4(2), 285-342. https://doi.org/10.3390/ph4020285