Opioid Actions in Primary-Afferent Fibers—Involvement in Analgesia and Anesthesia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Opioids inhibit glutamatergic excitatory transmission from the periphery by activating G-protein coupled opioid receptors in the central terminals of primary-afferent neurons in the spinal substantia gelatinosa, resulting in antinociception. Opioid receptor activation in the peripheral terminals of primary-afferent neurons inhibits the production of action potentials in response to nociceptive stimuli given to the periphery, leading to antinociception. Opioids also exhibit a local anesthetic effect without opioid receptor activation in peripheral nerve fibers. This review article will focus on analgesia and anesthesia produced by the actions of opioids on primary-afferent fibers.1. Introduction
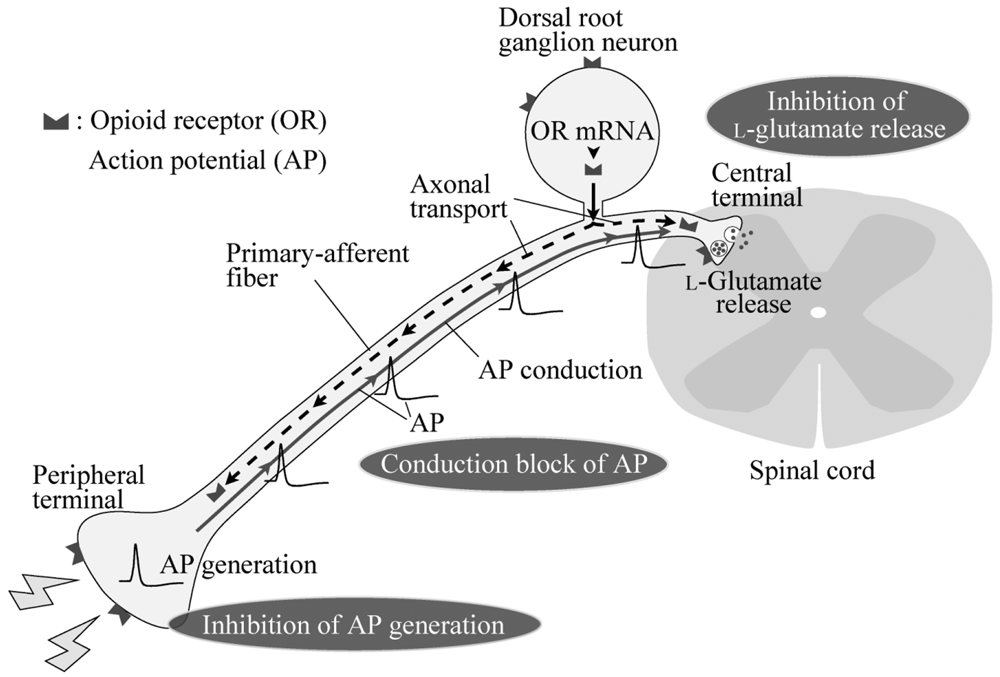
Nociceptive stimuli given to the skin produce an action potential (AP) in the peripheral terminals of primary-afferent neurons. Such an AP conducts through fine myelinated Aδ and unmyelinated C primary-afferent glutamatergic fibers contained in the dorsal root to the superficial laminae of the dorsal horn, especially the substantia gelatinosa (SG; lamina II of Rexed; [1,2]; Figure 1). This nociceptive information flows to the thalamus through a connection with projection neurons in lamina I and deeper laminae of the spinal dorsal horn [3], and then to the primary sensory area of the cerebral cortex, eliciting nociceptive sensation. A neuronal circuitry in the SG is thought to play an important role in the modulation of nociceptive transmission by various endogenous substances including opioids [4]. SG neurons preferentially receive in a mono- or polysynaptic manner Aδ- and C-primary afferent fibers, which carry fast and slow nociceptive information, respectively [5]. Inhibitory neurotransmitters, glycine and GABA, as well as l-glutamate, are involved in synaptic transmission in SG neurons in a polysynaptic manner from primary-afferent fibers.
Intrathecal administration of opioids produces a powerful analgesia in rats [6] and humans [7,8]. This analgesic effect is thought to be mediated by three principal subtypes of G-protein coupled opioid receptors, μ-, δ- and κ-type, the activation of which inhibits voltage-gated Ca2+ channels, activates inwardly-rectifying K+ channels or inhibits adenylate cyclase through the activation of G proteins [9]. These opioid receptors have been found in the superficial dorsal horn, especially the SG, in rats [10-13] and in humans [14]. Radioligand-binding experiments in the rat spinal cord have demonstrated that the most prevalent type of opioid receptors in laminae I-II is μ-type (63% or more) with considerably fewer of δ- (23% or less) and κ-type (15% or less; [10,15,16]). A partial (by 40–70%) reduction in the number of opioid-binding sites has been observed in the dorsal horn after the disruption of primary afferents by mechanical (dorsal rhizotomy; [10,11,17]) or chemical (the pretreatment with a selective fine-afferent neurotoxin, capsaicin; [18]) methods, indicating the localization of opioid receptors to both nerve terminals and postsynaptic neurons [19].
It has been reported that μ- or δ-opioid receptor agonists presynaptically inhibit glutamatergic synaptic transmissions in CNS neurons including superficial spinal dorsal horn [20-22], spinal trigeminal nucleus [23] and midbrain periaqueductal gray (PAG) neurons [24]. Alternatively, opioids may open one or more K+ channels through the activation of each of μ-, δ- and κ-opioid receptors and thus hyperpolarize membranes, resulting in an inhibition of excitatory transmission in CNS neurons including rat spinal cord [20,25] and spinal trigeminal nucleus neurons [23,26,27].
Although administration of opioids into the nerve sheath has been reported to lead to pain relief [28], many of pain treatments by use of opioids are based on systemic administration of conventional centrally-penetrating opioids, resulting in their actions in the PNS and CNS, both of which contribute to analgesia [29]. It is possible that centrally-injected opioids act on not only the CNS, but also the PNS, because King et al. [30] have demonstrated that opioids are transported from the brain to periphery by P-glycoprotein. In support of an important role of opioids in the PNS, subcutaneously-applied N-methylmorphine, which was shown not to penetrate the blood brain barrier, exhibited antinociception in an acetic acid-writhing model in mice [31]. Shannon and Lutz [32] have demonstrated that subcutaneous administration of an opioid loperamide, which hardly penetrates into the brain, produces antinociception in the formalin test in rats. Such an action of opioids in the PNS appears to be mediated by opioid receptors in the peripheral terminals of primary-afferent neurons [31-37].
Conduction of APs in peripheral nerve fibers is generally blocked by opioids, although Yuge et al. [38] have reported that there is not any significant change in the amplitude of compound action potential (CAP) in the superficial radial nerve following perineural application of an opioid morphine in decerebrate cats. For instance, opioids such as fentanyl and sufentanil reduce the peak amplitudes of CAPs recorded from peripheral nerve fibers [39] and inhibit peripheral nerve AP conduction [40]. Such a CAP inhibition is also produced by a non-narcotic opioid tramadol, (1RS; 2RS)-2-[(dimethyl-amino)methyl]-1-(3-methoxyphenyl)-cyclohexanol hydrochloride [41]. Jurna and Grossmann [42] have reported that an inhibitory effect of morphine on CAPs in mammalian peripheral nerve fibers is antagonized by a nonspecific opioid-receptor antagonist naloxone, indicating an involvement of opioid receptors. Consistent with this idea, binding and immunohistochemical studies have demonstrated the presence of opioid receptors in mammalian peripheral nerve fibers [43-45]. Hunter and Frank [46] also have reported a naloxone-sensitive inhibition of CAPs produced by opioids in frog sciatic nerve fibers. On the contrary, there are reports showing that opioids reduce CAP peak amplitudes [39] and inhibit nerve conduction [40] in a manner insensitive to naloxone.
This review article will mention our data about the actions of opioids on the central terminals of primary-afferent neurons and on peripheral nerve fibers and also other investigators' results about their actions on the peripheral terminals of primary-afferent neurons.
2. Opioid Actions on the Central Terminals of Primary-Afferent Neurons
It is possible that an opioid-induced inhibition of pain transmission is due to a negative modulation of glutamatergic transmission in the central terminals of primary-afferent neurons in the SG. In support of this idea, opioids administrated into the SG in anesthetized cats inhibited an excitation of deeper dorsal horn neurons caused by noxious peripheral stimuli without a change in their responses to innocuous stimuli such as touch [47]. In order to know a role of opioids in the nociceptive transmission, we examined their effects on l-glutamate-mediated excitatory postsynaptic currents (EPSCs) by applying the whole-cell patch-clamp technique to SG neurons in spinal cord slices dissected from the adult rat.
2.1. Actions of Opioid-Receptor Agonists
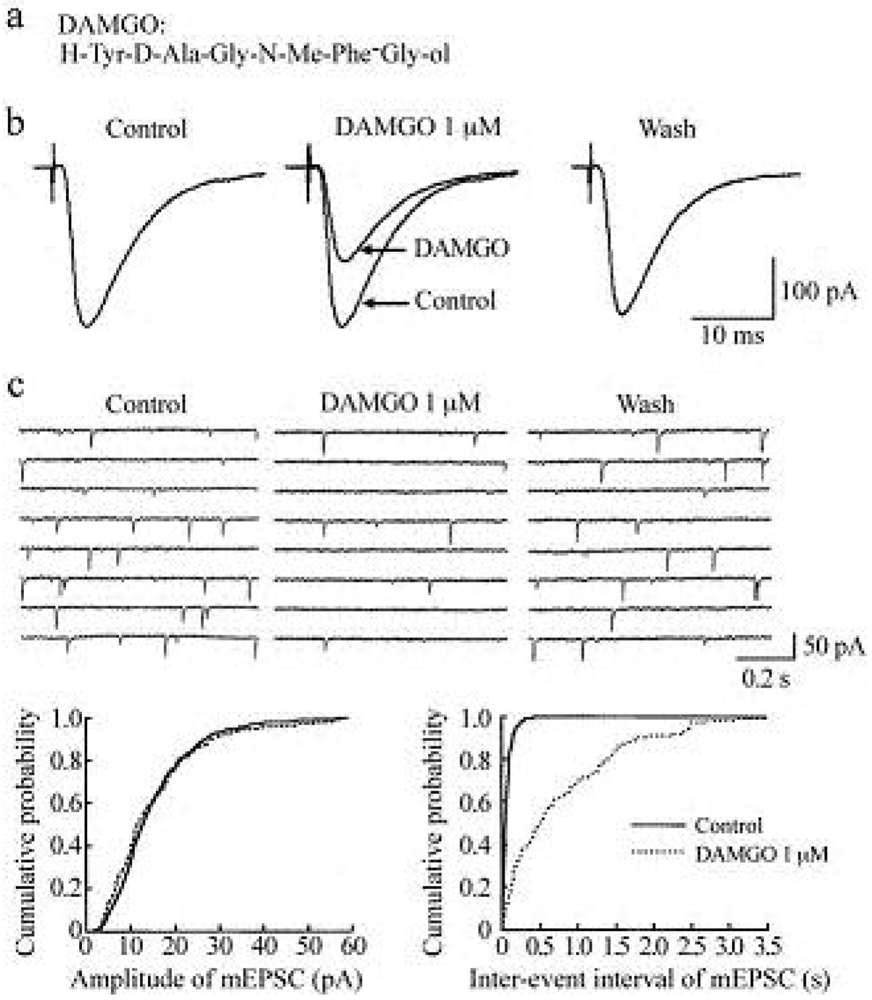
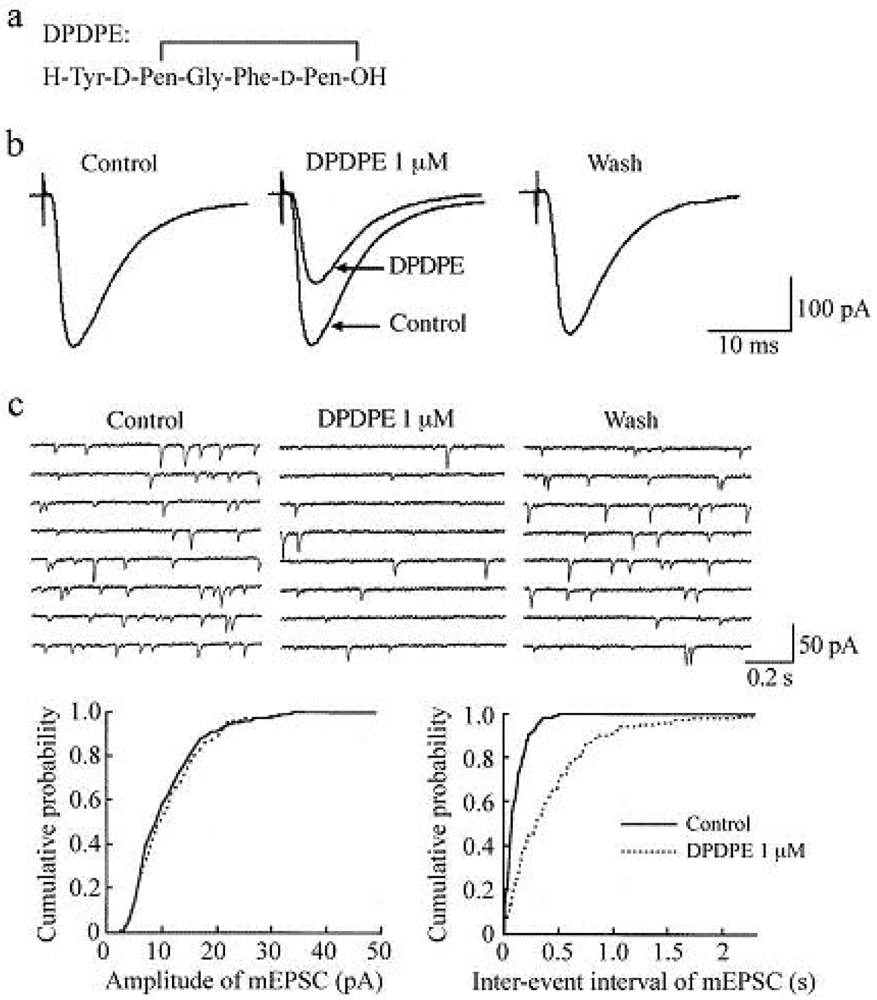
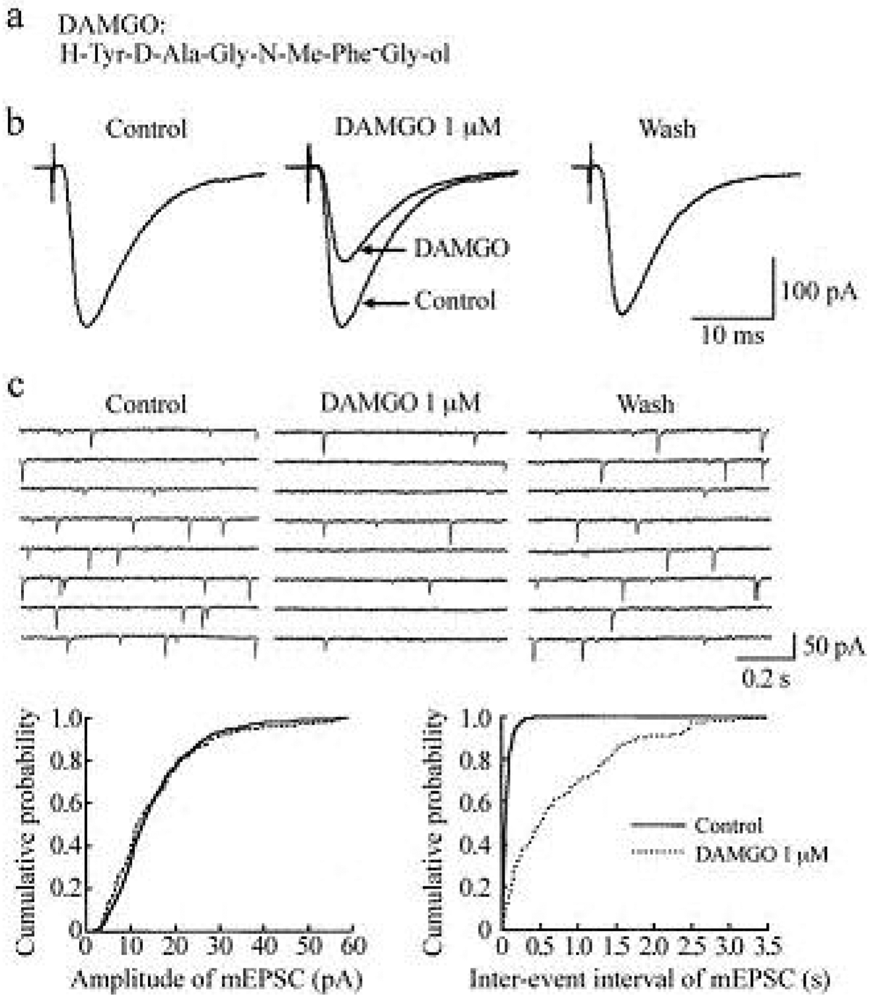
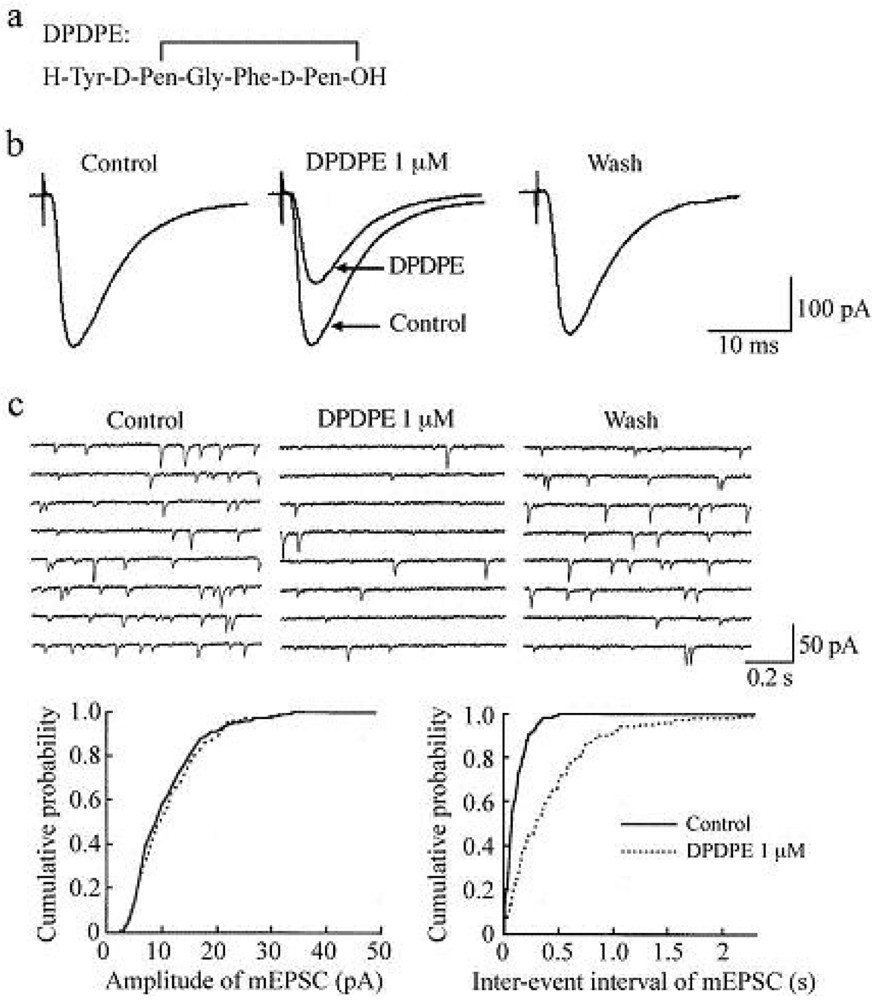
In the presence of a GABAA- and glycine-receptor antagonist, which are bicuculline (20 μM) and strychnine (2 μM), respectively, stimulation of a dorsal root with a low stimulus strength evoked a monosynaptic Aδ-fiber EPSC in SG neurons, as shown in Figure 2 and Figure 3. This Aδ-fiber evoked EPSC was completely blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μM), indicating an activation of non-N-methyl-d-aspartate (non-NMDA) receptors [48]. In 95% of neurons examined, superfusing a μ-opioid receptor agonist DAMGO (1 μM; Figure 2a) reversibly reduced the peak amplitude of the Aδ-fiber evoked EPSC, as shown in Figure 2b. The magnitude of this depression was 27%. In 71% of neurons tested, a δ-opioid receptor agonist DPDPE (1 μM; Figure 3a) also decreased the peak amplitude of Aδ-fiber evoked EPSC (Figure 3b) with an extent of 17%. When examined in neurons exhibiting inhibition of more than 5%, the DAMGO and DPDPE actions were not seen in the presence of a μ-opioid receptor antagonist CTAP (1 μM) and a δ-opioid receptor antagonist naltrindole (1 μM), respectively. Such a reduction in Aδ-fiber evoked EPSC amplitude was presynaptic in origin, because neither DAMGO (1 μM) nor DPDPE (1 μM) affected the peak amplitude of a response of SG neurons to a non-NMDA receptor agonist AMPA (10 μM). In contrast, a κ-opioid receptor agonist U-69593 (1 μM) had little effect on the Aδ-fiber evoked EPSC [49], although Randić et al. [50] have reported that κ agonists cause both potentiation and inhibition of excitatory transmission in SG neurons. These results indicate the presence of μ- and δ-type opioid receptors involved in inhibiting the release of l-glutamate from primary-afferent central terminals. A cellular mechanism for the presynaptic inhibition of evoked EPSC would be an inhibition of voltage-gated Ca2+ channels by opioids in nerve terminals, because opioids suppress Ca2+-channel currents in rat dorsal root ganglion (DRG) neurons [27,51]. DAMGO-sensitive adult rat DRG neurons seem not to express T-type Ca2+ (Cav 3.2) channels [52].
Although the central terminals of primary-afferent neurons express not only opioid receptors but also various types of neurotransmitter receptors, recent studies have revealed an interaction among the receptors in a synergistic (greater-than-additive) manner in inhibiting nociceptive transmission. For instance, Riedl et al. [53] have suggested that δ-opioid receptors may interact with α2A adrenoceptors, the activation of which inhibits monosynaptic primary-afferent Aδ-fiber and C-fiber glutamatergic transmission in SG neurons [54].
Miniature EPSCs (mEPSCs) were isolated by adding a voltage-gated Na+-channel blocker tetrodotoxin (TTX; 0.5 μM) together with bicuculline (20 μM) and strychnine (2 μM) to superfusing Krebs solution. These were abolished by the addition of CNQX (20 μM), indicating an involvement of non-NMDA receptors [48,55]. The mEPSCs are produced by l-glutamate released onto SG neurons from the central terminals of primary-afferent neurons and glutamatergic interneuron terminals. In all neurons examined, superfusion of DAMGO (1 μM) resulted in a rapid and reversible reduction in the frequency of mEPSC without a change in the amplitude, as shown in the upper part of Figure 2c. The extent of this reduction was 61%. The lower part of Figure 2c demonstrates the action of DAMGO on cumulative distributions of the amplitude and inter-event interval of mEPSC.
While DAMGO increased a proportion of mEPSCs having a longer inter-event interval, it had no consistent effect on the cumulative distribution of mEPSC amplitude. In 75% of neurons tested, DPDPE (1 μM) also exhibited a similar action, as seen in Figure 3c; the frequency of mEPSC was reduced with an extent of 23% without a change in the amplitude. These effects on sEPSC frequency and amplitude indicate a decrease in the spontaneous release of l-glutamate from nerve terminals without a change in the sensitivity of non-NMDA receptors to l-glutamate. The DAMGO and DPDPE actions were not seen in the presence of CTAP and naltrindole (each 1 μM), respectively. On the contrary, U-69593 (1 μM) had little effect on the frequency of mEPSC [49]. These results indicate that μ- and δ-type opioid receptors also exist in glutamatergic interneuron terminals.
When examined at the same concentration of 1 μM, the sequence of efficacies of opioids in reducing either evoked EPSC amplitude or mEPSC frequency was μ- > δ- ≫ κ-type. This rank order was the same as that for the proportion of neurons in which each of the opioids exhibits evoked EPSC amplitude and mEPSC frequency reductions [49]. These results are in good agreement with that of analgesic effect caused by intrathecal injection of opioids in the rat, the potency order of which is μ- > δ- ≫ κ-type [56-58]. These appear to be due to a difference in density among the three types of opioid receptors expressed in the superficial dorsal horn (see above).
Recently, Zhou et al. [59] have reported that DAMGO (1 μM) produced not only a decrease in monosynaptic EPSC amplitude and mEPSC frequency but also a long-lasting increase in the amplitude and in the frequency in about a half of adult rat SG neurons examined. This heterosynaptic long-term potentiation has been attributed to opioid-induced hyperalgesia and tolerance.
2.2. Actions of Endomorphins
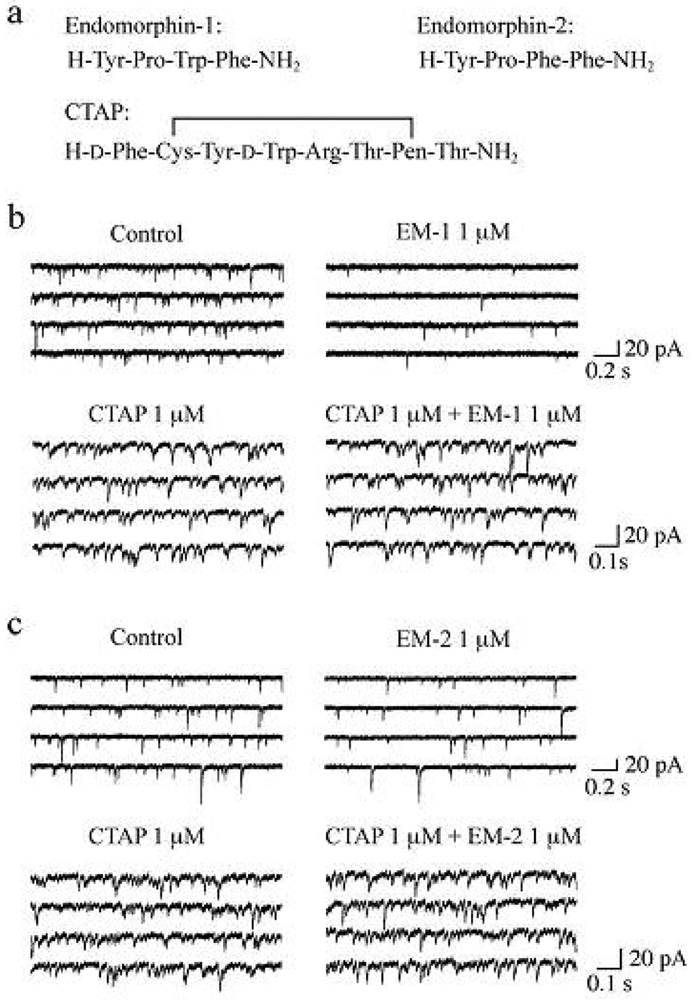
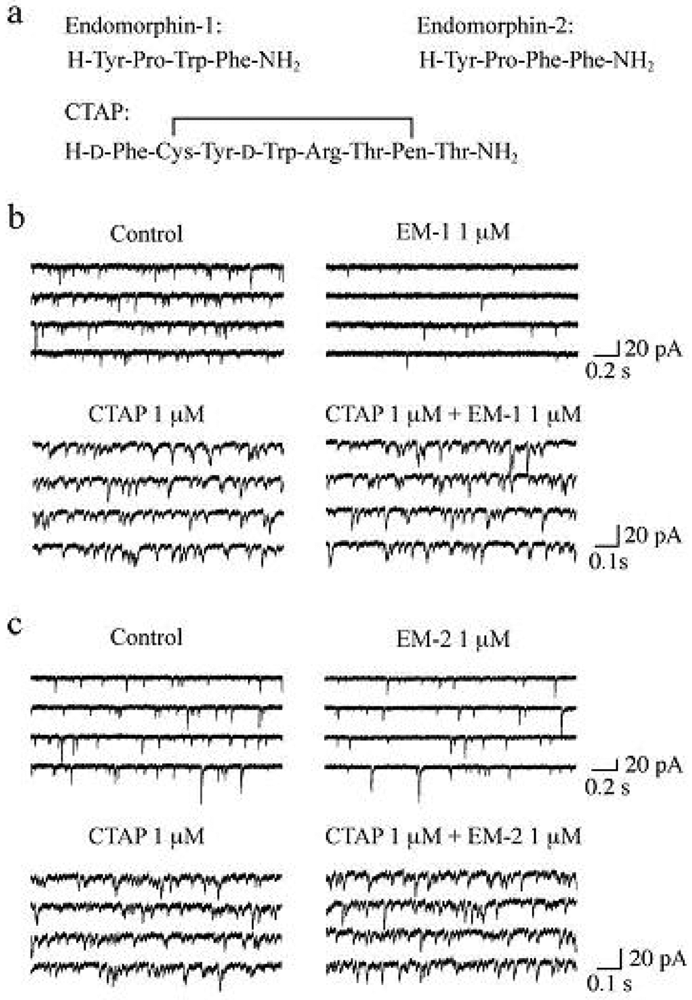
It has been demonstrated that the rat SG contains endogenous opioid peptides such as enkephalins [60,61], endomorphin-1 and endomorphin-2 (EM-1 and EM-2, respectively; see Figure 4a for their amino-acid sequences). EM-1 and EM-2, which were isolated from mammalian brain in 1997, possess high affinity and selectivity for the μ-opioid receptor as compared to the δ- and κ-opioid receptors [62-64]. There is much evidence showing that EM-1 and EM-2 play a pivotal role in inhibiting nociceptive transmission at the spinal cord level. Intrathecal administration of EM-1 and EM-2 produced antinociception in the tail-flick, paw-withdrawal, tail-pressure and flexor-reflex tests in adult rodents [62,65-72]. EM-1 and EM-2 like immunoreactive fibers have been shown to exist in the superficial laminae of the rat spinal cord [73-76] and in rat primary-afferent fibers [77,78]. EM-2 like immunoreactive fibers and terminals in the rat spinal dorsal horn have been demonstrated to originate from ipsilateral primary afferents and bilateral descending fibers from the nucleus tractus solitarii [79]. Furthermore, EM-2 like substances are released from the rat spinal dorsal horn in response to electrical stimulation applied to the dorsal root entry zone [80]. Axon terminals containing EM-2 like immunoreactivity make synapses with neurons immunostained for μ-opioid receptors in the rat spinal dorsal horn [81]. EM-1 and EM-2 are different by only one amino acid residue and thus exhibit similar antinociceptive potency at the spinal cord level in mice [65,69] and rats [67,72]. On the other hand, antinociceptive effects produced by them are distinct in the development of acute tolerance [65], in the extent [66,68] and in the duration [72] from each other.
Both EM-1 and EM-2 reduce primary-afferent C-fiber mediated responses while EM-1 but not EM-2 inhibits Aβ-fiber ones in rat dorsal horn neurons [82]. Such a distinction may not be unexpected, because there is a difference between EM-1 and EM-2 in the affinity to μ-opioid receptors, determined from binding experiments [62]. Gong et al. [83] have reported that the activation of K+ channels through cloned μ-opioid receptors differs in extent between EM-1 and EM-2 actions. Human μ-opioid receptors fused to Gi1α or Gi2α in transfected HEK 293 cells exhibited binding affinities which were different by 3-8-fold between EM-1 and EM-2 [84]. Behavioral studies have suggested that each of EM-1 and EM-2 may activate different μ-opioid receptor subtypes such as μ1 and μ2 [85], which are pharmacologically distinct, in the spinal dorsal horn [69,70], although there is no evidence for the presence of the μ-opioid receptor subtypes. A similar idea has been also applied to a difference between EM-1 and EM-2 in motivational effects and conditioned place preference responses, which are produced by their intracerebroventricular administrations [86,87]. There is a difference between EM-1 and EM-2 immunoreactivities in the distribution in the spinal dorsal horn in such that EM-2 exists at higher density than EM-1, suggesting a different role of EM-1 and EM-2 in spinal antinociception [74].
Spontaneous EPSCs (sEPSCs) were unaffected in frequency and amplitude by TTX (0.5 μM), indicating that the production of the sEPSCs was independent of the spontaneous activities of neurons presynaptic to SG neurons and thus sEPSCs were equivalent to mEPSCs. In the following, we examined the effects of EMs on sEPSCs, which were observed in Krebs solution without TTX. Superfusing EM-1 or EM-2 (each 1 μM) for 2 min resulted in a reduction in the occurrence of sEPSC, as shown in Figure 4b,c. A maximal reduction in sEPSC frequency was seen around 2 min after washout of EM-1 or EM-2; this magnitude was about 45%. On the other hand, sEPSC amplitude was unaffected by EM-1 or EM-2. Consistent with no change in sEPSC amplitude, EM-1 and EM-2 did not affect the peak amplitude of the response of SG neurons to AMPA (5 μM). Such a reduction in sEPSC frequency by EM-1 or EM-2 was not seen in the presence of CTAP (1 μM; see Figure 4a for its amino-acid sequence; Figure 4b,c). The sEPSC frequency reductions by EM-1 and EM-2 were not different in extent from each other. Similar reduction in sEPSC frequency by EM-1 in adult rat SG neurons has been reported by Yajiri and Huang [88].
The magnitudes (45%) of sEPSC frequency reductions produced by EM-1 and EM-2 (each 1 μM) were similar to a maximal reductive one (40%) of monosynaptic Aδ-fiber evoked EPSC amplitude [88] and also to those (about 40%) by EMs (1 μM) of short-latency evoked EPSCs in young rat SG neurons by stimulating the dorsal root entry zone [89]. Wu et al. [89] have reported that there is no difference between EM-1 and EM-2 in efficacy for reducing evoked EPSC amplitudes in young rat SG neurons, as seen for EMs-induced outward current (hyperpolarization) in postsynaptic membranes of adult rat SG neurons [90]. In conclusion, the difference in behaviorally-examined antinociceptive effects between EM-1 and EM-2 could not be attributed to a distinction in their pre- and postsynaptic effects on excitatory transmission in SG neurons, and may be explained by a difference in their enzymatic degradation [90]. This idea may be consistent with the observations that EM-1 required a longer pretreatment time than EM-2 before tolerance was observed [65] and that the duration of spinal antinociceptive effects was significantly longer for EM-1 than EM-2 [72]. A cellular mechanism for the presynaptic inhibition of evoked EPSC would be an inhibition of voltage-gated Ca2+ channels by opioids in nerve terminals, because EM-1 and EM-2 are reported to reduce Ca2+-channel currents in NGMO-251 cells expressing μ-opioid receptors [91].
2.3. Actions of Tramadol Metabolite M1
Tramadol is a clinically-used, orally-active drug, which is considered to act as an analgesic in the CNS [92]. The activation of μ-opioid receptors by tramadol has been revealed from the experimental results of a μ-opioid receptor binding of tramadol [93] and its [35S]GTP-γ-S binding stimulation [94]. Although tramadol is metabolized to various compounds including mono-O-demethyl tramadol (M1) via N- and O-demethylation in humans and animals [95], M1 is thought to be a therapeutically active drug as central analgesics [92]. M1 has the highest affinity for the cloned μ-opioid receptors among the metabolites of tramadol [94].
Under the condition where M1-induced postsynaptic current mediated by μ-opioid receptors [96] was inhibited, M1 (1 mM) superfused for 2 min reduced the frequency (by about 30%) but not amplitude of sEPSCs recorded at −70 mV; a response of SG neurons to bath-applied AMPA (10 μM) was unaffected by M1 (1 mM). This sEPSC frequency reduction persisted for at least 20 min after washout of M1. This inhibitory action of M1 was not seen in the presence of CTAP (1 μM). M1 (1 mM) also reduced the peak amplitudes of EPSCs which were monosynaptically evoked at −70 mV in SG neurons by stimulating primary-afferent Aδ-fiber and/or C-fiber in a spinal cord slice with an attached dorsal root. Each of the Aδ-fiber and C-fiber EPSC amplitude was reduced by M1 with a similar extent of 40–50%. This was so in a single neuron where both of the monosynaptic Aδ-fiber and C-fiber EPSCs were seen. It was concluded that M1 inhibits the quantal release of l-glutamate from the central terminals of primary-afferent neurons through the activation of μ-opioid receptors in the SG; this action is not distinct in extent between primary-afferent Aδ-fiber and C-fiber glutamatergic transmission [97].
3. Opioid Actions on the Peripheral Terminals of Primary-Afferent Neurons
There is much evidence supporting the idea that APs produced in the peripheral terminals of primary-afferent neurons in response to nociceptive stimuli given to the periphery are inhibited by an action of opioids. A shift in a voltage dependency of currents (Ihs) through hyperpolarization-activated channels involved in neuronal excitability was produced by an adenylate cyclase activator forskolin, cyclic AMP analogue and one of membrane phospholipid metabolites, prostaglandin E2 (PGE2; which is involved in a sensitization of nociception in the peripheral terminal) in cultured guinea-pig nodose ganglion neurons. Such a change in Ih was reversed by opioids in a manner sensitive to naloxone [98]. DAMGO inhibited a modulation by PGE2 of TTX-resistant Na+ channels, which play a pivotal role in sensitization of nociceptors, in rat DRG neurons; this DAMGO action was inhibited by naloxone [99]. The TTX-resistant Na+ channels have been shown to be involved in the spontaneous and ectopic production of APs in chronic pain rat models [100] and also in visceral pain and referred hyperalgesia [36,37,101].
4. Opioid Actions on Peripheral Nerve Fibers
AP, which plays an important role in transmitting neuronal information in nerve fibers, is generally mediated by voltage-gated Na+ and K+ channels located in neuronal membranes. AP produced at a point of nerve fiber membrane electrotonically conducts to a nearby membrane where a depolarization is produced. This depolarization opens voltage-gated Na+ channels, resulting in Na+ influx, i.e., inward current caused by the gradient of the electrochemical potential of Na+, and then in the production of AP. APs thus produced disappear owing to the inactivation of voltage-gated Na+ channels following their opening and also the activation of a delayed-rectifier type of voltage-gated K+ channels. When the opening and closing of Na+ and K+ channels occur in the longitudinal direction of a nerve fiber, a current flows on the extracellular surface of the fiber [102]. When many of nerve fibers contained in the nerve trunk are simultaneously stimulated by using an electrode put on the trunk, currents flowing on the surface of the fibers can be recorded as CAP by using a recording electrode put near the stimulating electrode. We examined the effect of opioids on fast-conducting frog sciatic nerve CAPs (conduction velocity: 30–40 m/s; this corresponds to that of A fibers) which are easy to be measured by using the air-gap method and well characterized in property.
4.1. Actions of Opioids on CAPs
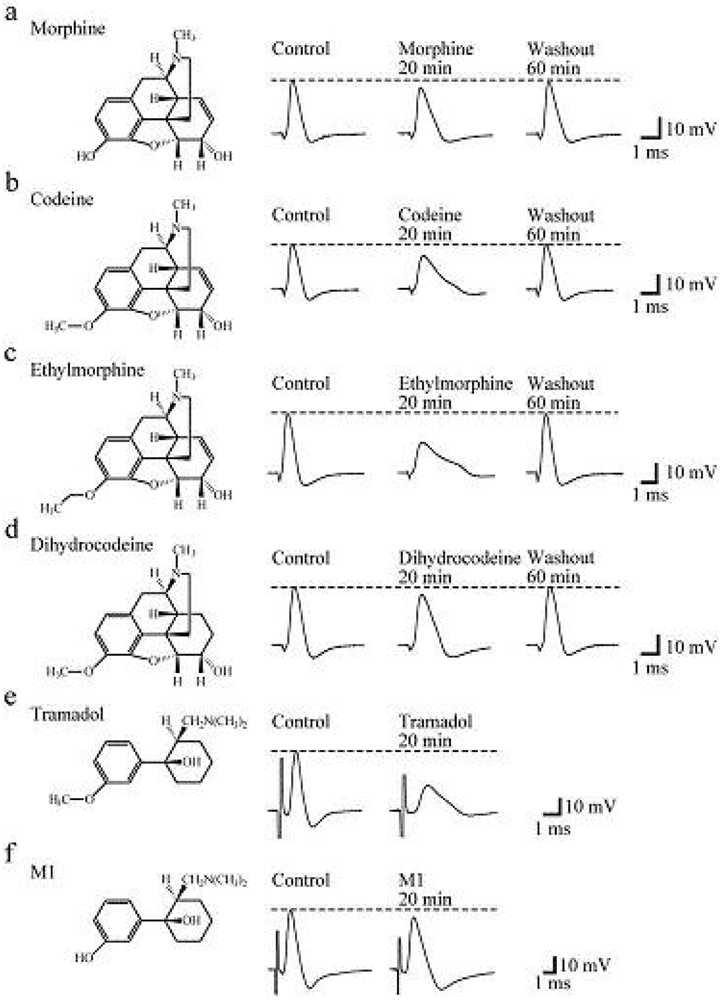
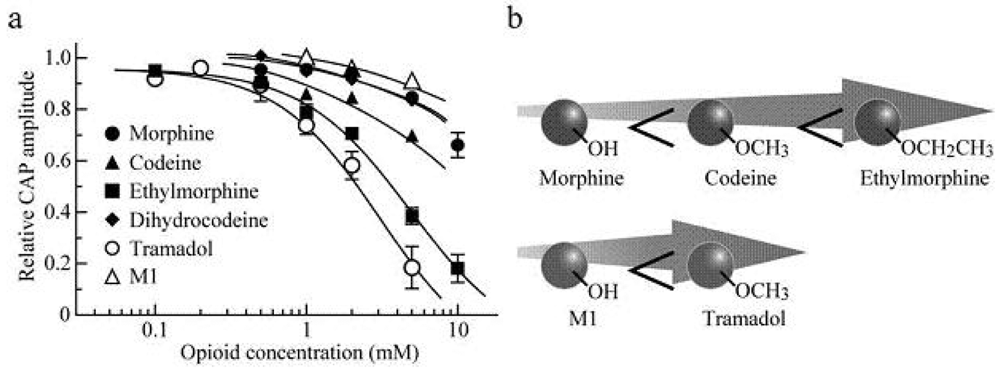
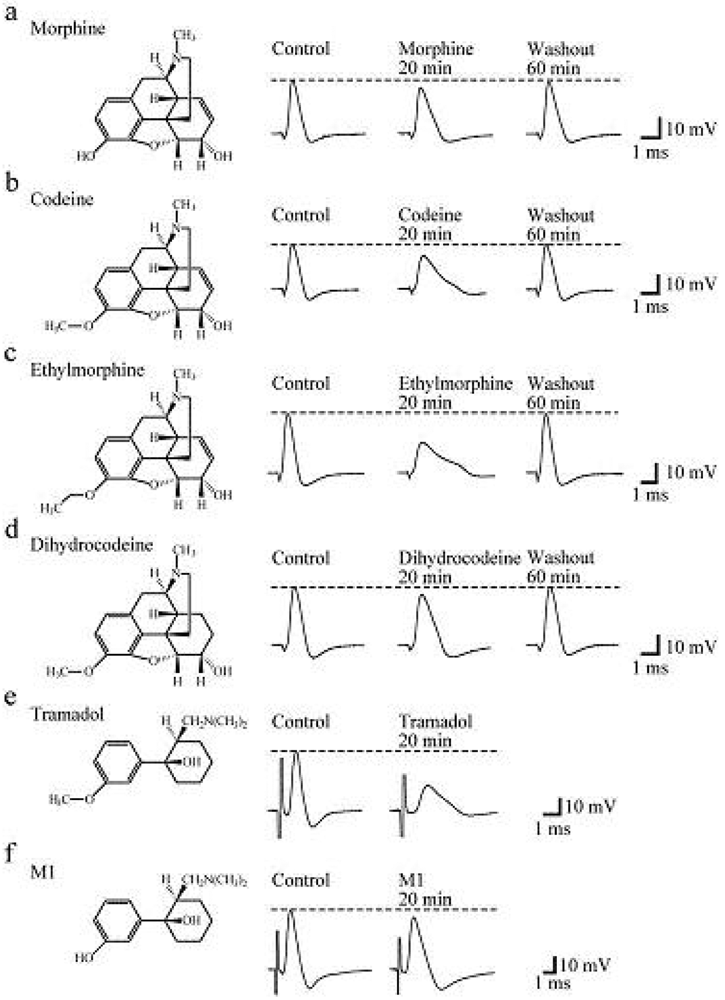
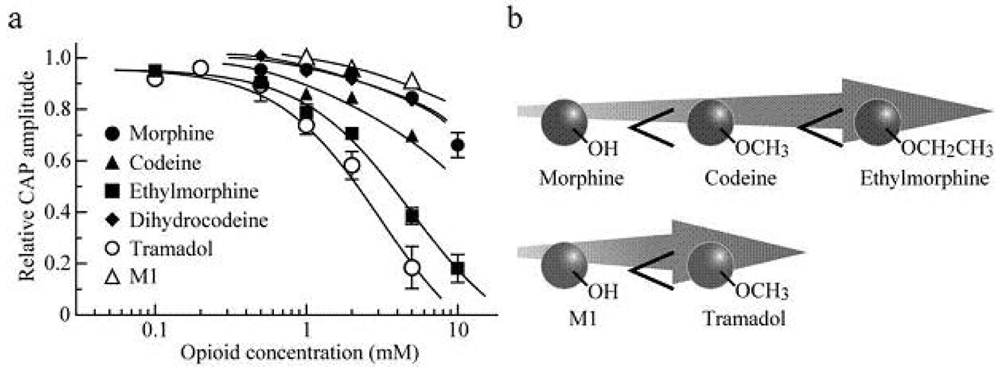
Soaking the sciatic nerve into morphine (5 mM)-containing Ringer solution reversibly reduced the peak amplitude of the CAP (Figure 5a). The morphine-induced reduction in CAP peak amplitude attained a steady effect within 20 min after the soaking. CAP amplitude reduction at a steady state increased in extent with an increase in morphine concentration. The concentration-response curve for the morphine-induced CAP amplitude reduction obtained from many nerve trunks is given in Figure 6a [103]. CAPs in the frog sciatic nerve were less sensitive to morphine than those in the guinea-pig and rabbit vagus nerves (formed by C fibers) in such that the peak amplitudes in the vagus nerves are reduced by 20–32% at 0.5 mM [42].
Codeine at a concentration of 5 mM also reduced CAP peak amplitude in a reversible manner (Figure 5b). Like morphine, codeine (5 mM) exhibited a steady effect of CAP amplitude reduction within 20 min after the soaking. The extent of the CAP peak amplitude reduction produced by codeine was enhanced with an increase in its concentration (Figure 6a; [103]). When compared at a concentration of 5 mM, codeine-induced reduction (about 30%) in CAP amplitude in the frog sciatic nerve was much smaller than that (about 70%) in the rat phrenic nerve (formed by A fibers; [104]), while there was not so a large difference in morphine action (about 10%).
Ethylmorphine at a concentration of 5 mM reversibly reduced CAP peak amplitude (Figure 5c). Figure 6a demonstrates the effects of ethylmorphine in a wide range of 0.1–10 mM on CAPs. The extent of the CAP peak amplitude reduction produced by ethylmorphine was enhanced with an increase in its concentration. Analysis based on the Hill equation showed that half-maximal inhibitory concentration (IC50) value for ethylmorphine is 4.6 mM with the Hill coefficient (nH) of 1.2 [103].
To more know a relationship between CAP inhibition by opioids and their chemical structures, we examined the effect of dihydrocodeine, where codeine was hydrogenated, on CAPs. Like other opioids, dihydrocodeine at a concentration of 5 mM reversibly reduced CAP peak amplitude (Figure 5d). The extent of this reduction was smaller than those of codeine and ethylmorphine while being almost comparable to that of morphine. Figure 6a demonstrates the effects of dihydrocodeine in a wide range of 0.5–5 mM on CAPs. The extent of the CAP peak amplitude reduction produced by dihydrocodeine was enhanced with an increase in its concentration [103].
In order to know whether the opioid-induced reduction of CAP peak amplitude is mediated by opioid receptors, we examined the effects of naloxone on the CAP amplitude reductions produced by opioids. The CAP peak reductions produced by morphine (10 mM), codeine (5 mM), ethylmorphine (2 mM) and dihydrocodeine (5 mM) in the presence of naloxone (0.01 mM) were not significantly different in extent from those in the absence of this opioid-receptor antagonist [103]. Thus, the opioid actions in frog sciatic nerve fibers were resistant to naloxone, although naloxone-sensitive CAP inhibition has been reported in mammalian and amphibian peripheral nerve fibers [42,46]. Our result is consistent with previous reports [39,40,104] showing naloxone-insensitive CAP amplitude reduction and nerve conduction inhibition produced by opioids.
When the inhibitory action of ethylmorphine was compared with those of local anesthetics, the IC50 value for this opioid was larger by about six-fold than those of lidocaine and cocaine (0.74 mM and 0.80 mM, respectively; [103,105]).
4.2. Actions of Tramadol and M1 on CAPs
Soaking the sciatic nerve into tramadol (1 mM)-containing solution resulted in reducing the peak amplitude of the CAP (Figure 5e). The tramadol-induced reduction in CAP peak amplitude attained a steady effect within 20 min after the soaking. At least 1 h after soaking the sciatic nerve into tramadol-free solution, the CAP amplitude did not recover to control level. The CAP peak amplitude reduction produced by tramadol was enhanced in extent with an increase in its concentration. Figure 6a demonstrates the effect of tramadol in a wide concentration range of 0.2 to 5 mM on CAPs. Analysis based on the Hill equation showed that IC50 value for tramadol is 2.3 mM with the nH of 1.7 [105]. A similar CAP amplitude reduction produced by tramadol has been obtained by applying the sucrose-gap method to frog [41] and rat sciatic nerves [106,107]. The tramadol action in the rat sciatic nerve has been suggested to be larger for fast-conducting than slow-conducting CAPs [108]. IC50 value (2.3 mM) for tramadol in our study was smaller by about three-fold than that (6.6 mM) obtained previously for the frog sciatic nerve [41]. Since tramadol exhibits a high affinity for opioid receptors [109], we next investigated whether the tramadol effect is mediated by opioid receptors. Pretreatment for 20 min of sciatic nerves with naloxone (0.01 mM) did not affect the tramadol-induced inhibition of CAP [105]. Consistent with this observation, Tsai et al. [110] have reported that a reduction in spinal somatosensory evoked potentials following the application of tramadol on rat sciatic nerves in vivo is resistant to naloxone. Moreover, a μ-opioid receptor agonist DAMGO at 0.001 mM, a concentration maximally activating μ-opioid receptors in rat SG neurons [90], did not affect CAPs.
In order to know further whether the tramadol-induced reduction in CAP peak amplitude is related to μ-opioid receptor activation, we examined the effect on sciatic nerve CAPs of M1, which is similar in chemical structure to tramadol (see Figures 5e,f) while having a higher affinity for the receptors than tramadol does [95]. CAPs were not affected by soaking the sciatic nerve into Ringer solution containing M1 at a concentration of 1 or 2 mM for 20 min [105]. M1 at a higher concentration such as 5 mM reduced CAP peak amplitude by about 9% (Figure 5f and Figure 6a).
No involvement of G-protein coupled opioid receptors in CAP inhibition by various opioids is consistent with the fact that our preparation is the dissected sciatic nerve devoid from the neuronal cell body and the neuronal terminals.
The CAP peak amplitude reduction produced by tramadol in frog sciatic nerves may provide a basis for a local anesthetic effect following its intradermal injection in patients [111-113]. Consistent with the fact that the IC50 value for tramadol in reducing CAP peak amplitude is larger by 3.1-fold than that of lidocaine [105], a sensory block at the intradermal injection site of 5% tramadol was similar to that of 1% lidocaine [111].
Since alcohols, anticonvulsants, barbiturates and narcotics block AP conduction in peripheral nerve fibers [114], the effects of opioids on CAPs in our study may be due to nonspecific interactions with membrane bilayers and also ion channels such as voltage-gated Na+ and K+ channels [115]. In support of the latter idea, Wagner et al. [116] have reported that an opioid meperidine, which is used for AP conduction blockade and thus analgesia, reduces voltage-gated Na+-channel current amplitudes. Tsai et al. [117] have demonstrated that tramadol attenuates the current amplitude of delayed-rectifier K+ channels (Kv3.1a type) expressed in NG 108-15 cells. Tramadol also inhibits heterologously expressed neuronal voltage-gated Na+ channels (Nav1.2 type; [118]). Hu and Rubly [119] have reported in single myelinated nerve fibers isolated from the frog sciatic nerve that morphine depresses steady-state K+ currents and peak Na+ currents, resulting in the prolongation of APs. Frazier et al. [120] have demonstrated that intracellularly-applied morphine reduces voltage-gated Na+- and K+-channel current amplitudes in squid giant axons.
Figure 6b demonstrates a schematic illustration of a difference in the extent of conduction inhibition among morphine, codeine and ethylmorohine and also between M1 and tramadol. Although morphine, codeine and ethylmorphine are distinct in chemical structure in terms of only a group attached to a benzene ring in such that they have -OH, -OCH3 and -OCH2CH3, respectively, this reduction enhanced in extent with an increase in the number of -CH2. Alternatively, tramadol having -OCH3 in a benzene ring reduced CAP peak amplitudes more effectively than M1 which is different from tramadol only in terms of the presence of -OH in the ring. This is so, although the chemical structures of morphine, codeine and ethylmorphine are quite different from those of tramadol and M1 (Figure 5). Since the increase in the number of -CH2 is expected to enhance lipophilicity of opioids, it is suggested that lipophilic opioid-channel interactions may play an important role in nerve conduction block, as shown for local anesthetics [121,122]. In support of this idea, the potency in inhibiting CAPs in the rat sciatic nerve was in the order of cocaine < cocaethylene (where the methyl ester group of cocaine was replaced with an ethyl ester group) < isopropylcocaine (where its methyl ester group was replaced with an isopropyl ester group; [123]). It would be of interest to note that the sequence of the affinity of opioids for μ-opioid receptors is morphine > codeine > ethylmorphine [124], the order of which is reversed to one for CAP inhibition. If the opioid-induced inhibition of CAPs is mediated by μ-opioid receptors, CAP inhibition sequence will be expected to be morphine > codeine > ethylmorphine. However, this is not the case, a result being consistent with our idea that the opioids-induced CAP inhibition in the frog sciatic nerve is not mediated by opioid receptors. Such a difference in chemical structure could serve as information to know molecular mechanisms for the inhibition of AP conduction by opioids.
The inhibition of AP conduction by opioids in large diameter (A-type) fibers might contribute to local anesthesia following peripheral perineural injections of opioids which are expected to result in a direct action of opioids at high doses on peripheral nerves, such as intradermal injection. Although sensory information is transmitted by not only fast- but also slow-conducting fibers in sciatic nerves, the present study does not examine the effects of opioids on slow-conducting APs in small caliber and unmyelinated axons which are involved in peripheral analgesia [125]. In order to more firmly establish a clinical significance of CAP amplitude reduction produced by opioids, their effects on slow-conducting CAPs such as TTX-resistant ones [126] remain to be examined. Moreover, it remains to be examined whether the structure-function relationship of opioids is applied to slow-conducting APs. Since codeine is metabolized to morphine via O-demethylation in animals and humans [29,127,128], peripherally-applied codeine might have a similar effect to that of morphine.
It remains to be examined how APs in primary-afferent fibers are affected by opioids in order to clearly know their actions on sensory transmission, because the sciatic nerve contains not only afferent (sensory) but also efferent (motor) fibers.
5. Conclusions
Opioids inhibit nociceptive transmission in primary-afferent neurons with opioid-receptor activation. Opioid receptors located in the central and peripheral terminals of primary-afferent neurons are involved in a decrease in the release of l-glutamate onto spinal SG neurons and an inhibition of the production of APs in response to nociceptive stimuli given to the periphery, respectively (Figure 1). These opioid-receptor activation results in antinocicetion, i.e., analgesia. Opioids at concentrations much higher than required to activate opioid receptors inhibit AP conduction in primary-afferent fibers (see Figure 1), possibly by inhibiting voltage-gated Na+ channels, and thus exhibit an anesthetic action. Comparison in the extent of conduction inhibition among opioids revealed that a chemical structure of opioids plays an important role in inhibiting nerve conduction. Although this inhibitory effect of ethylmorphine is smaller than that of lidocaine, the structure-activity relationship revealed in this study may serve to develop an opioid which has a local anesthetic effect more effective than lidocaine.
Acknowledgements
This work was supported in part by Grants-in-aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (KAKENHI: 21500370 for E.K.; 14780619 for T.F.; 22.7148 for K.M., JSPS Research Fellow).
References
- Rexed, B. The cytoarchitectonic organization of the spinal cord in the cat. J. Comp. Neurol. 1952, 96, 415–495. [Google Scholar]
- Kumazawa, T.; Perl, E.R. Excitation of marginal and substantia gelatinosa neurons in the primate spinal cord: indications of their place in dorsal horn functional organization. J. Comp. Neurol. 1978, 177, 417–434. [Google Scholar]
- Willis, W.D., Jr.; Coggeshall, R.E. Sensory Mechanisms of the Spinal Cord, 2nd ed.; Plenum: New York, YSA, 1991. [Google Scholar]
- Fürst, S. Transmitters involved in antinociception in the spinal cord. Brain Res. Bull. 1999, 48, 129–141. [Google Scholar]
- Yoshimura, M.; Jessell, T.M. Primary afferent-evoked synaptic responses and slow potential generation in rat substantia gelatinosa neurons in vitro. J. Neurophysiol. 1989, 62, 96–108. [Google Scholar]
- Yaksh, T.L.; Rudy, T.A. Analgesia mediated by a direct spinal action of narcotics. Science 1976, 192, 1357–1358. [Google Scholar]
- Onofrio, B.M.; Yaksh, T.L. Long-term pain relief produced by intrathecal morphine infusion in 53 patients. J. Neurosurg. 1990, 72, 200–209. [Google Scholar]
- Cousins, M.J.; Mather, L.E. Intrathecal and epidural administration of opioids. Anesthesiology 1984, 61, 276–310. [Google Scholar]
- Minami, M.; Satoh, M. Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci. Res. 1995, 23, 121–145. [Google Scholar]
- Besse, D.; Lombard, M.C.; Zajac, J.M.; Roques, B.P.; Besson, J.M. Pre- and postsynaptic distribution of μ, δ and κ opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res. 1990, 521, 15–22. [Google Scholar]
- Gouardères, C.; Beaudet, A.; Zajac, J.-M.; Cros, J.; Quirion, R. High resolution radioautographic localization of [125I] FK-33-824-labelled mu opioid receptors in the spinal cord of normal and deafferented rats. Neuroscience 1991, 43, 197–209. [Google Scholar]
- Mansour, A.; Fox, C.A.; Burke, S.; Meng, F.; Thompson, R.C.; Akil, H.; Watson, S.J. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J. Comp. Neurol. 1994, 350, 412–438. [Google Scholar]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar]
- Faull, R.L.M.; Villiger, J.W. Opiate receptors in the human spinal cord: a detailed anatomical study comparing the autoradiographic localization of [3H] diprenorphine binding sites with the laminar pattern of substance P, myelin and nissl staining. Neuroscience 1987, 20, 395–407. [Google Scholar]
- Stevens, C.W.; Lacey, C.B.; Miller, K.E.; Elde, R.P.; Seybold, V.S. Biochemical characterization and regional quantification of μ, δ and κ opioid binding sites in rat spinal cord. Brain Res. 1991, 550, 77–85. [Google Scholar]
- Rahman, W.; Dashwood, M.R.; Fitzgerald, M.; Aynsley-Green, A.; Dickenson, A.H. Postnatal development of multiple opioid receptors in the spinal cord and development of spinal morphine analgesia. Develop. Brain Res. 1998, 108, 239–254. [Google Scholar]
- Ninkovic, M.; Hunt, S.P.; Kelly, J.S. Effect of dorsal rhizotomy on the autoradiographic distribution of opiate and neurotensin receptors and neurotensin-like immunoreactivity within the rat spinal cord. Brain Res. 1981, 230, 111–119. [Google Scholar]
- Gamse, R.; Holzer, P.; Lembeck, F. Indirect evidence for presynaptic location of opiate receptors on chemosensitive primary sensory neurons. Naunyn-Schmiedeberg's Arch. Pharmacol. 1979, 308, 281–285. [Google Scholar]
- Coggeshall, R.E.; Carlton, S.M. Receptor localization in the mammalian dorsal horn and primary afferent neurons. Brain Res. Rev. 1997, 24, 28–66. [Google Scholar]
- Jeftinija, S. Enkephalins modulate excitatory synaptic transmission in the superficial dorsal horn by acting at μ-opioid receptor sites. Brain Res. 1988, 460, 260–268. [Google Scholar]
- Hori, Y.; Endo, K.; Takahashi, T. Presynaptic inhibitory action of enkephalin on excitatory transmission in superficial dorsal horn of rat spinal cord. J. Physiol. 1992, 450, 673–685. [Google Scholar]
- Glaum, S.R.; Miller, R.J.; Hammond, D.L. Inhibitory actions of δ1-, δ2-, and μ-opioid receptor agonists on excitatory transmission in lamina II neurons of adult rat spinal cord. J. Neurosci. 1994, 14, 4965–4971. [Google Scholar]
- Grudt, T.J.; Williams, J.T. μ-Opioid agonists inhibit spinal trigeminal substantia gelatinosa neurons in guinea pig and rat. J. Neurosci. 1994, 14, 1646–1654. [Google Scholar]
- Vaughan, C.W.; Christie, M.J. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J. Physiol. 1997, 498, 463–472. [Google Scholar]
- Yoshimura, M.; North, R.A. Substantia gelatinosa neurones hyperpolarized in vitro by enkephalin. Nature 1983, 305, 529–530. [Google Scholar]
- Grudt, T.J.; Williams, J.T. κ-Opioid receptors also increase potassium conductance. Proc. Natl. Acad. Sci. USA 1993, 90, 11429–11432. [Google Scholar]
- North, R.A. Opioid actions on membrane ion channels. In Handbook of Experimental Pharmacology; Herz, A., Ed.; Springer: Berlin, Germany, 1993; Volume 104, pp. 773–797. [Google Scholar]
- Viel, E.J.; Eledjam, J.J.; De La Coussaye, J.E.; D'Athis, F. Brachial plexus block with opioids for postoperative pain relief: comparison between buprenorphine and morphine. Reg. Anesth. 1989, 14, 274–278. [Google Scholar]
- Gutstein, H.B.; Akil, H. Opioid analgesics. In Goodman & Gilman's The Pharmacological Basis of Therapeutics, 11th ed.; Brunton, L.L., Lazo, J.S., Parker, K.L., Eds.; McGraw-Hill, Medical Publishing Division: New York, USA, 2006; pp. 547–590. [Google Scholar]
- King, M.; Su, W.; Chang, A.; Zuckerman, A.; Pasternak, G.W. Transport of opioids from the brain to the periphery by P-glycoprotein: peripheral actions of central drugs. Nat. Neurosci. 2001, 4, 268–274. [Google Scholar]
- Smith, T.W.; Buchan, P.; Parsons, D.N.; Wilkinson, S. Peripheral antinociceptive effects of N-methyl morphine. Life Sci. 1982, 31, 1205–1208. [Google Scholar]
- Shannon, H.E.; Lutz, E.A. Comparison of the peripheral and central effects of the opioid agonists loperamide and morphine in the formalin test in rats. Neuropharmacology 2002, 42, 253–261. [Google Scholar]
- Stein, C.; Comisel, K.; Haimerl, E.; Yassouridis, A.; Lehrberger, K.; Herz, A.; Peter, K. Analgesic effect of intraarticular morphine after arthroscopic knee surgery. New Eng. J. Med. 1991, 325, 1123–1126. [Google Scholar]
- Wenk, H.N.; Brederson, J.-D.; Honda, C.N. Morphine directly inhibits nociceptors in inflamed skin. J. Neurophysiol. 2006, 95, 2083–2097. [Google Scholar]
- Labuz, D.; Mousa, S.A.; Schäfer, M.; Stein, C.; Machelska, H. Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res. 2007, 1160, 30–38. [Google Scholar]
- Stein, C.; Schäfer, M.; Machelska, H. Attacking pain at its source: new perspectives on opioids. Nat. Med. 2003, 9, 1003–1008. [Google Scholar]
- Stein, C.; Lang, L.J. Peripheral mechanisms of opioid analgesia. Curr. Opin. Pharmacol. 2009, 9, 3–8. [Google Scholar]
- Yuge, O.; Matsumoto, M.; Kitahata, L.M.; Collins, J.G.; Senami, M. Direct opioid application to peripheral nerves does not alter compound action potentials. Anesth. Analg. 1985, 64, 667–671. [Google Scholar]
- Gissen, A.J.; Gugino, L.D.; Datta, S.; Miller, J.; Covino, B.G. Effects of fentanyl and sufentanil on peripheral mammalian nerves. Anesth. Analg. 1987, 66, 1272–1276. [Google Scholar]
- Jaffe, R.A.; Rowe, M.A. A comparison of the local anesthetic effects of meperidine, fentanyl, and sufentanil on dorsal root axons. Anesth. Analg. 1996, 83, 776–781. [Google Scholar]
- Mert, T.; Gunes, Y.; Guven, M.; Gunay, I.; Ozcengiz, D. Comparison of nerve conduction blocks by an opioid and a local anesthetic. Eur. J. Pharmacol. 2002, 439, 77–81. [Google Scholar]
- Jurna, I.; Grossmann, W. The effect of morphine on mammalian nerve fibres. Eur. J. Pharmacol. 1977, 44, 339–348. [Google Scholar]
- Fields, H.L.; Emson, P.C.; Leigh, B.K.; Gilbert, R.F.T.; Iversen, L.L. Multiple opiate receptor sites on primary afferent fibres. Nature 1980, 284, 351–353. [Google Scholar]
- Coggeshall, R.E.; Zhou, S.; Carlton, S.M. Opioid receptors on peripheral sensory axons. Brain Res. 1997, 764, 126–132. [Google Scholar]
- Wenk, H.N.; Honda, C.N. Immunohistochemical localization of delta opioid receptors in peripheral tissues. J. Comp. Neurol. 1999, 408, 567–579. [Google Scholar]
- Hunter, E.G.; Frank, G.B. An opiate receptor on frog sciatic nerve axons. Can. J. Physiol. Pharmacol. 1979, 57, 1171–1174. [Google Scholar]
- Duggan, A.W.; Hall, J.G.; Headley, P.M. Suppression of transmission of nociceptive impulses by morphine: selective effects of morphine administered in the region of the substantia gelatinosa. Br. J. Pharmacol. 1977, 61, 65–76. [Google Scholar]
- Kumamoto, E.; Fujita, T. Role of adenosine in regulating nociceptive transmission in the spinal dorsal horn. In Recent Research Developments in Physiology; Pandalai, S.G., Ed.; Research Signpost: Kerala, India, 2005; Volume 3, pp. 39–57. [Google Scholar]
- Kohno, T.; Kumamoto, E.; Higashi, H.; Shimoji, K.; Yoshimura, M. Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J. Physiol. 1999, 518, 803–813. [Google Scholar]
- Randić, M.; Cheng, G.; Kojic, L. κ-Opioid receptor agonists modulate excitatory transmission in substantia gelatinosa neurons of the rat spinal cord. J. Neurosci. 1995, 15, 6809–6826. [Google Scholar]
- Schroeder, J.E.; Fischbach, P.S.; Zheng, D.; McCleskey, E.W. Activation of μ opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron 1991, 6, 13–20. [Google Scholar]
- Wu, Z.-Z.; Cai, Y.-Q.; Pan, H.-L. A functional link between T-type calcium channels and μ-opioid receptor expression in adult primary sensory neurons. J. Neurochem. 2009, 109, 867–878. [Google Scholar]
- Riedl, M.S.; Schnell, S.A.; Overland, A.C.; Chabot-Doré, A.-J.; Taylor, A.M.; Ribeiro-da-Silva, A.; Elde, R.P.; Wilcox, G.L.; Stone, L.S. Coexpression of α2A-adrenergic and δ-opioid receptors in substance P-containing terminals in rat dorsal horn. J. Comp. Neurol. 2009, 513, 385–398. [Google Scholar]
- Kawasaki, Y.; Kumamoto, E.; Furue, H.; Yoshimura, M. α2 Adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology 2003, 98, 682–689. [Google Scholar]
- Yoshimura, M.; Nishi, S. Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neuroscience 1993, 53, 519–526. [Google Scholar]
- Dickenson, A.H.; Sullivan, A.F.; Knox, R.; Zajac, J.M.; Roques, B.P. Opioid receptor subtypes in the rat spinal cord: electrophysiological studies with μ- and δ-opioid receptor agonists in the control of nociception. Brain Res. 1987, 413, 36–44. [Google Scholar]
- Leighton, G.E.; Rodriguez, R.E.; Hill, R.G.; Hughes, J. κ-Opioid agonists produce antinociception after i.v. and i.c.v. but not intrathecal administration in the rat. Br. J. Pharmacol. 1988, 93, 553–560. [Google Scholar]
- Danzebrink, R.M.; Green, S.A.; Gebhart, G.F. Spinal mu and delta, but not kappa, opioid-receptor agonists attenuate responses to noxious colorectal distension in the rat. Pain 1995, 63, 39–47. [Google Scholar]
- Zhou, H.-Y.; Chen, S.-R.; Chen, H.; Pan, H.-L. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J. Neurosci. 2010, 30, 4460–4466. [Google Scholar]
- Hunt, S.P.; Kelly, J.S.; Emson, P.C. The electron microscopic localization of methionine-enkephalin within the superficial layers (I and II) of the spinal cord. Neuroscience 1980, 5, 1871–1890. [Google Scholar]
- Merchenthaler, I.; Maderdrut, J.L.; Altschuler, R.A.; Petrusz, P. Immunocytochemical localization of proenkephalin-derived peptides in the central nervous system of the rat. Neuroscience 1986, 17, 325–348. [Google Scholar]
- Zadina, J.E.; Hackler, L.; Ge, L.-J.; Kastin, A.J. A potent and selective endogenous agonist for the μ-opiate receptor. Nature 1997, 386, 499–502. [Google Scholar]
- Horvath, G. Endomorphin-1 and endomorphin-2: pharmacology of the selective endogenous μ-opioid receptor agonists. Pharmacol. Ther. 2000, 88, 437–463. [Google Scholar]
- Gelman, P.L.; Herrera, N.E.G.; Ortega, M.E.M.; Romero, L.P.; Santillán, C.T.; Juárez, A.S.; Palma, B.A. Endomorphin peptides: pharmacological and functional implications of these opioid peptides in the brain of mammals. Part one. Salud Mental 2010, 33, 179–196. [Google Scholar]
- Stone, L.S.; Fairbanks, C.A.; Laughlin, T.M.; Nguyen, H.O.; Bushy, T.M.; Wessendorf, M.W.; Wilcox, G.L. Spinal analgesic actions of the new endogenous opioid peptides endomorphin-1 and -2. NeuroReport 1997, 8, 3131–3135. [Google Scholar]
- Goldberg, I.E.; Rossi, G.C.; Letchworth, S.R.; Mathis, J.P.; Ryan-Moro, J.; Leventhal, L.; Su, W.; Emmel, D.; Bolan, E.A.; Pasternak, G.W. Pharmacological characterization of endomorphin-1 and endomorphin-2 in mouse brain. J. Pharmacol. Exp. Ther. 1998, 286, 1007–1013. [Google Scholar]
- Horvath, G.; Szikszay, M.; Tömböly, C.; Benedek, G. Antinociceptive effects of intrathecal endomorphin-1 and -2 in rats. Life Sci. 1999, 65, 2635–2641. [Google Scholar]
- Ohsawa, M.; Mizoguchi, H.; Narita, M.; Nagase, H.; Kampine, J.P.; Tseng, L.F. Differential antinociception induced by spinally administered endomorphin-1 and endomorphin-2 in the mouse. J. Pharmacol. Exp. Ther. 2001, 298, 592–597. [Google Scholar]
- Sakurada, S.; Zadina, J.E.; Kastin, A.J.; Katsuyama, S.; Fujimura, T.; Murayama, K.; Yuki, M.; Ueda, H.; Sakurada, T. Differential involvement of μ-opioid receptor subtypes in endomorphin-1-and -2-induced antinociception. Eur. J. Pharmacol. 1999, 372, 25–30. [Google Scholar]
- Sakurada, S.; Hayashi, T.; Yuhki, M.; Fujimura, T.; Murayama, K.; Yonezawa, A.; Sakurada, C.; Takeshita, M.; Zadina, J.E.; Kastin, A.J.; Sakurada, T. Differential antagonism of endomorphin-1 and endomorphin-2 spinal antinociception by naloxonazine and 3-methoxynaltrexone. Brain Res. 2000, 881, 1–8. [Google Scholar]
- Sakurada, S.; Hayashi, T.; Yuhki, M.; Orito, T.; Zadina, J.E.; Kastin, A.J.; Fujimura, T.; Murayama, K.; Sakurada, C.; Sakurada, T.; Narita, M.; Suzuki, T.; Tan-no, K.; Tseng, L.F. Differential antinociceptive effects induced by intrathecally administered endomorphin-1 and endomorphin-2 in the mouse. Eur. J. Pharmacol. 2001, 427, 203–210. [Google Scholar]
- Grass, S.; Xu, I.S.; Wiesenfeld-Hallin, Z.; Xu, X.-J. Comparison of the effect of intrathecal endomorphin-1 and endomorphin-2 on spinal cord excitability in rats. Neurosci. Lett. 2002, 324, 197–200. [Google Scholar]
- Martin-Schild, S.; Zadina, J.E.; Gerall, A.A.; Vigh, S.; Kastin, A.J. Localization of endomorphin-2-like immunoreactivity in the rat medulla and spinal cord. Peptides 1997, 18, 1641–1649. [Google Scholar]
- Martin-Schild, S.; Gerall, A.A.; Kastin, A.J.; Zadina, J.E. Differential distribution of endomorphin 1- and endomorphin 2-like immunoreactivities in the CNS of the rodent. J. Comp. Neurol. 1999, 405, 450–471. [Google Scholar]
- Wu, S.-Y.; Dun, S.L.; Wright, M.T.; Chang, J.-K.; Dun, N.J. Endomorphin-like immunoreactivity in the rat dorsal horn and inhibition of substantia gelatinosa neurons in vitro. Neuroscience 1999, 89, 317–321. [Google Scholar]
- Wang, Q.-P.; Zadina, J.E.; Guan, J.-L.; Kastin, A.J.; Funahashi, H.; Shioda, S. Endomorphin-2 immunoreactivity in the cervical dorsal horn of the rat spinal cord at the electron microscopic level. Neuroscience 2002, 113, 593–605. [Google Scholar]
- Pierce, T.L.; Grahek, M.D.; Wessendorf, M.W. Immunoreactivity for endomorphin-2 occurs in primary afferents in rats and monkey. NeuroReport 1998, 9, 385–389. [Google Scholar]
- Martin-Schild, S.; Gerall, A.A.; Kastin, A.J.; Zadina, J.E. Endomorphin-2 is an endogenous opioid in primary sensory afferent fibers. Peptides 1998, 19, 1783–1789. [Google Scholar]
- Hui, R.; Wang, W.; Chen, T.; Lü, B.C.; Li, H.; Zhang, T.; Wu, S.-X.; Li, Y.-Q. Origins of endomorphin-2 immunopositive fibers and terminals in the spinal dorsal horn of the rat. Neuroscience 2010, 169, 422–430. [Google Scholar]
- Williams, C.A.; Wu, S.Y.; Dun, S.L.; Kwok, E.H.; Dun, N.J. Release of endomorphin-2 like substances from the rat spinal cord. Neurosci. Lett. 1999, 273, 25–28. [Google Scholar]
- Wang, Q.-P.; Zadina, J.E.; Guan, J.-L.; Shioda, S. Morphological evidence of endomorphin as an agonist for the mu-opioid receptor in the rat spinal cord. Neurosci. Lett. 2003, 341, 107–110. [Google Scholar]
- Chapman, V.; Diaz, A.; Dickenson, A.H. Distinct inhibitory effects of spinal endomorphin-1 and endomorphin-2 on evoked dorsal horn neuronal responses in the rat. Br. J. Pharmacol. 1997, 122, 1537–1539. [Google Scholar]
- Gong, J.; Strong, J.A.; Zhang, S.; Yue, X.; DeHaven, R.N.; Daubert, J.D.; Cassel, J.A.; Yu, G.; Mansson, E.; Yu, L. Endomorphins fully activate a cloned human mu opioid receptor. FEBS Lett. 1998, 439, 152–156. [Google Scholar]
- Massotte, D.; Brillet, K.; Kieffer, B.L.; Milligan, G. Agonists activate Gi1α or Gi2α fused to the human mu opioid receptor differently. J. Neurochem. 2002, 81, 1372–1382. [Google Scholar]
- Pasternak, G.W.; Wood, P.J. Multiple mu opiate receptors. Life Sci. 1986, 38, 1889–1898. [Google Scholar]
- Narita, M.; Ozaki, S.; Ioka, M.; Mizoguchi, H.; Nagase, H.; Tseng, L.F.; Suzuki, T. Different motivational effects induced by the endogenous μ-opioid receptor ligands endomorphin-1 and -2 in the mouse. Neuroscience 2001, 105, 213–218. [Google Scholar]
- Wu, H.-E.; MacDougall, R.S.; Clithero, A.D.; Leitermann, R.J.; Terashvili, M.; Tseng, L.F. Opposite conditioned place preference responses to endomorphin-1 and endomorphin-2 in the mouse. Neurosci. Lett. 2004, 365, 157–161. [Google Scholar]
- Yajiri, Y.; Huang, L.-Y.M. Actions of endomorphins on synaptic transmission of Aδ-fibers in spinal cord dorsal horn neurons. J. Biomed. Sci. 2000, 7, 226–231. [Google Scholar]
- Wu, S.-Y.; Ohtubo, Y.; Brailoiu, G.C.; Dun, N.J. Effects of endomorphin on substantia gelatinosa neurons in rat spinal cord slices. Br. J. Pharmacol. 2003, 140, 1088–1096. [Google Scholar]
- Fujita, T.; Kumamoto, E. Inhibition by endomorphin-1 and endomorphin-2 of excitatory transmission in adult rat substantia gelatinosa neurons. Neuroscience 2006, 139, 1095–1105. [Google Scholar]
- Higashida, H.; Hoshi, N.; Knijnik, R.; Zadina, J.E.; Kastin, A.J. Endomorphins inhibit high-threshold Ca2+ channel currents in rodent NG108-15 cells overexpressing μ-opioid receptors. J. Physiol. 1998, 507, 71–75. [Google Scholar]
- Klotz, U. Tramadol - the impact of its pharmacokinetic and pharmacodynamic properties on the clinical management of pain. Arzneimittelforschung 2003, 53, 681–687. [Google Scholar]
- Frink, M.C.; Hennies, H.-H.; Englberger, W.; Haurand, M.; Wilffert, B. Influence of tramadol on neurotransmitter systems of the rat brain. Arzneimittelforschung 1996, 46, 1029–1036. [Google Scholar]
- Gillen, C.; Haurand, M.; Kobelt, D.J.; Wnendt, S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human μ-opioid receptor. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000, 362, 116–121. [Google Scholar]
- Lintz, W.; Erlaçin, S.; Frankus, E.; Uragg, H. Metabolismus von Tramadol bei Mensch und Tier. Arzneimittelforschung 1981, 31, 1932–1943. [Google Scholar]
- Koga, A.; Fujita, T.; Totoki, T.; Kumamoto, E. Tramadol produces outward currents by activating μ-opioid receptors in adult rat substantia gelatinosa neurones. Br. J. Pharmacol. 2005, 145, 602–607. [Google Scholar]
- Koga, A.; Fujita, T.; Liu, T.; Mizuta, K.; Nakatsuka, T.; Hasuo, H.; Kumamoto, E. Tramadol presynaptically inhibits glutamatergic excitatory synaptic transmission in rat spinal dorsal horn neurons by activating μ-opioid receptors. Society for Neuroscience 37th Annual Meeting, Japan, 2007, November 3-7.
- Ingram, S.L.; Williams, J.T. Opioid inhibition of Ih via adenylyl cyclase. Neuron 1994, 13, 179–186. [Google Scholar]
- Gold, M.S.; Levine, J.D. DAMGO inhibits prostaglandin E2-induced potentiation of a TTX-resistant Na+ current in rat sensory neurons in vitro. Neurosci. Lett. 1996, 212, 83–86. [Google Scholar]
- Porreca, F.; Lai, J.; Bian, D.; Wegert, S.; Ossipov, M.H.; Eglen, R.M.; Kassotakis, L.; Novakovic, S.; Rabert, D.K.; Sangameswaran, L.; Hunter, J.C. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc. Natl. Acad. Sci. USA 1999, 96, 7640–7644. [Google Scholar]
- Laird, J.M.A.; Souslova, V.; Wood, J.N.; Cervero, F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J. Neurosci. 2002, 22, 8352–8356. [Google Scholar]
- Nicholls, J.G.; Martin, A.R.; Wallace, B.G.; Fuchs, P.A. From Neuron to Brain, 4th ed.; Sinauer Associates, Inc.: Sunderland, Massachusetts, USA, 2001. [Google Scholar]
- Mizuta, K.; Fujita, T.; Nakatsuka, T.; Kumamoto, E. Inhibitory effects of opioids on compound action potentials in frog sciatic nerves and their chemical structures. Life Sci. 2008, 83, 198–207. [Google Scholar]
- Brodin, P.; Skoglund, L.A. Dose-response inhibition of rat compound nerve action potential by dextropropoxyphene and codeine compared to morphine and cocaine in vitro. Gen. Pharmacol. 1990, 21, 551–553. [Google Scholar]
- Katsuki, R.; Fujita, T.; Koga, A.; Liu, T.; Nakatsuka, T.; Nakashima, M.; Kumamoto, E. Tramadol, but not its major metabolite (mono-O-demethyl tramadol) depresses compound action potentials in frog sciatic nerves. Br. J. Pharmacol. 2006, 149, 319–327. [Google Scholar]
- Mert, T.; Gunes, Y.; Guven, M.; Gunay, I.; Gocmen, C. Differential effects of lidocaine and tramadol on modified nerve impulse by 4-aminopyridine in rats. Pharmacology 2003, 69, 68–73. [Google Scholar]
- Guven, M.; Mert, T.; Günay, I. Effects of tramadol on nerve action potentials in rat: comparisons with benzocaine and lidocaine. Int. J. Neurosci. 2005, 115, 339–349. [Google Scholar]
- Dalkilic, N.; Tuncer, S.; Bariskaner, H.; Kiziltan, E. The effect of tramadol on the rat sciatic nerve conduction: a numerical analysis and conduction velocity distribution study. Yakugaku Zasshi 2009, 129, 485–493. [Google Scholar]
- Hennies, H.-H.; Friderichs, E.; Schneider, J. Receptor binding, analgesic and antitussive potency of tramadol and other selected opioids. Arzneimittelforschung 1988, 38, 877–880. [Google Scholar]
- Tsai, Y.-C.; Chang, P.-J.; Jou, I.-M. Direct tramadol application on sciatic nerve inhibits spinal somatosensory evoked potentials in rats. Anesth. Analg. 2001, 92, 1547–1551. [Google Scholar]
- Pang, W.-W.; Mok, M.S.; Chang, D.-P.; Huang, M.-H. Local anesthetic effect of tramadol, metoclopramide, and lidocaine following intradermal injection. Reg. Anesth. Pain Med. 1998, 23, 580–583. [Google Scholar]
- Altunkaya, H.; Ozer, Y.; Kargi, E.; Babuccu, O. Comparison of local anaesthetic effects of tramadol with prilocaine for minor surgical procedures. Br. J. Anaesth. 2003, 90, 320–322. [Google Scholar]
- Altunkaya, H.; Ozer, Y.; Kargi, E.; Ozkocak, I.; Hosnuter, M.; Demirel, C.B.; Babuccu, O. The postoperative analgesic effect of tramadol when used as subcutaneous local anesthetic. Anesth. Analg. 2004, 99, 1461–1464. [Google Scholar]
- Staiman, A.; Seeman, P. The impulse-blocking concentrations of anesthetics, alcohols, anticonvulsants, barbiturates, and narcotics on phrenic and sciatic nerves. Can. J. Physiol. Pharmacol. 1974, 52, 535–550. [Google Scholar]
- Scholz, A. Mechanisms of (local) anaesthetics on voltage-gated sodium and other ion channels. Br. J. Anaesth. 2002, 89, 52–61. [Google Scholar]
- Meperidine and lidocaine block of recombinant voltage-dependent Na+ channels: evidence that meperidine is a local anesthetic. Anesthesiology 1999, 91, 1481–1490.
- Tsai, T.-Y.; Tsai, Y.-C.; Wu, S.-N.; Liu, Y.-C. Tramadol-induced blockade of delayed rectifier potassium current in NG108-15 neuronal cells. Eur. J. Pain 2006, 10, 597–601. [Google Scholar]
- Haeseler, G.; Foadi, N.; Ahrens, J.; Dengler, R.; Hecker, H.; Leuwer, M. Tramadol, fentanyl and sufentanil but not morphine block voltage-operated sodium channels. Pain 2006, 126, 234–244. [Google Scholar]
- Hu, S.; Rubly, N. Effects of morphine on ionic currents in frog node of Ranvier. Eur. J. Pharmacol. 1983, 95, 185–192. [Google Scholar]
- Frazier, D.T.; Murayama, K.; Abbott, N.J.; Narahashi, T. Effects of morphine on internally perfused squid giant axons. Proc. Soc. Exp. Biol. Med. 1972, 139, 434–438. [Google Scholar]
- Bräu, M.E.; Nau, C.; Hempelmann, G.; Vogel, W. Local anesthetics potently block a potential insensitive potassium channel in myelinated nerve. J. Gen. Physiol. 1995, 105, 485–505. [Google Scholar]
- Bräu, M.E.; Vogel, W.; Hempelmann, G. Fundamental properties of local anesthetics: half-maximal blocking concentrations for tonic block of Na+ and K+ channels in peripheral nerve. Anesth. Analg. 1998, 87, 885–889. [Google Scholar]
- Tokuno, H.A.; Bradberry, C.W.; Everill, B.; Agulian, S.K.; Wilkes, S.; Baldwin, R.M.; Tamagnan, G.D.; Kocsis, J.D. Local anesthetic effects of cocaethylene and isopropylcocaine on rat peripheral nerves. Brain Res. 2004, 996, 159–167. [Google Scholar]
- Chen, Z.R.; Irvine, R.J.; Somogyi, A.A.; Bochner, F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991, 48, 2165–2171. [Google Scholar]
- Mays, K.S.; Lipman, J.J.; Schnapp, M. Local analgesia without anesthesia using peripheral perineural morphine injections. Anesth. Analg. 1987, 66, 417–420. [Google Scholar]
- Kobayashi, J.; Ohta, M.; Terada, Y. C fiber generates a slow Na+ spike in the frog sciatic nerve. Neurosci. Lett. 1993, 162, 93–96. [Google Scholar]
- Mikus, G.; Somogyi, A.A.; Bochner, F.; Eichelbaum, M. Codeine O-demethylation: rat strain differences and the effects of inhibitors. Biochem. Pharmacol. 1991, 41, 757–762. [Google Scholar]
- Cleary, J.; Mikus, G.; Somogyi, A.; Bochner, F. The influence of pharmacogenetics on opioid analgesia: studies with codeine and oxycodone in the Sprague-Dawley/Dark Agouti rat model. J. Pharmacol. Exp. Ther. 1994, 271, 1528–1534. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kumamoto, E.; Mizuta, K.; Fujita, T. Opioid Actions in Primary-Afferent Fibers—Involvement in Analgesia and Anesthesia. Pharmaceuticals 2011, 4, 343-365. https://doi.org/10.3390/ph4020343
Kumamoto E, Mizuta K, Fujita T. Opioid Actions in Primary-Afferent Fibers—Involvement in Analgesia and Anesthesia. Pharmaceuticals. 2011; 4(2):343-365. https://doi.org/10.3390/ph4020343
Chicago/Turabian StyleKumamoto, Eiichi, Kotaro Mizuta, and Tsugumi Fujita. 2011. "Opioid Actions in Primary-Afferent Fibers—Involvement in Analgesia and Anesthesia" Pharmaceuticals 4, no. 2: 343-365. https://doi.org/10.3390/ph4020343
APA StyleKumamoto, E., Mizuta, K., & Fujita, T. (2011). Opioid Actions in Primary-Afferent Fibers—Involvement in Analgesia and Anesthesia. Pharmaceuticals, 4(2), 343-365. https://doi.org/10.3390/ph4020343