Proteolytically Derived Endogenous Angioinhibitors Originating from the Extracellular Matrix

Abstract
: Angiogenesis, a neovascularization process induced from the existing parent blood vessels, is a prerequisite for many physiological and pathological conditions. Under physiological conditions it is regulated by a balance between endogenous angioinhibitors and angioactivators, and an imbalance between them would lead to pathological conditions such as cancer, age-related macular degeneration (AMD), diabetic retinopathy, cardiovascular diseases, etc. Several proteolytically generated endogenous molecules have been identified which exhibit angioinhibition and/or antitumor activities. These angioinhibitors interact with endothelial and tumor cells by binding to distinct integrins and initiate many of their intracellular signaling mechanisms regulating the cell survival and or apoptotic pathways. The present review will focus on the extracellular matrix derived angioinhibitors, and their mechanisms of actions that point to the clinical significance and therapeutic implications.1. Introduction
The term angiogenesis refers to the growth of new blood vessels from the pre-existing ones [1]. Angiogenesis is required for normal growth and body development that involves several physiological processes such as wound healing, tissue and organ regeneration, embryonic development, etc. This physiological angiogenesis is regulated with a tight control on endothelial cells and growth factors, however under pathological conditions such as cancer, AMD, diabetic retinopathy, cardiovascular diseases, etc., an abnormal growth of new blood vessels occurs, which essentially contributes to the severity of the disease. In early 70s Professor Judah Folkman hypothesized that tumors require additional oxygen and nutrients for their rapid growth and thus induce growth of new blood vessels towards the growing tumors (tumor angiogenesis). Since under normal physiological conditions angiogenesis is tightly controlled, it was obvious that there exists endogenous angioinhibitors that play vital role in this tight control of physiological angiogenesis. Thus with the discovery of the thrombospondins (the first endogenous angiogenesis inhibitor to be identified) a new branch of basic research has emerged and at present about 27 endogenous antiangiogenic molecules have been identified, of which, many of them are in preclinical trials. Antiangiogenic therapy is now gaining its significance as the fourth treatment modality for cancer besides surgery, chemotherapy and radiotherapy. Several inhibitors and monoclonal antibodies were developed to prevent tumor angiogenesis and are currently in different phase trials proving their efficacy in cancer treatment. However, tumors may counterbalance the inhibitory effects of angioinhibitors by secreting increased amounts of angiogenic factors [2-4]. Thus to overcome such inhibition endogenous angioinhibitors appear more promising as they would not invoke any defense mechanism. However in cancer cells when thrombospondin-1, endostatin, and tumstatin are over expressed, tumor cells were found to escape angiogenesis inhibition by up-regulation of various proangiogenic factors [5]. Yet with the increasing list of endogenous antiangiogenic molecules each with a unique mechanism of action and with varied potential of inhibiting de novo angiogenesis in different pathological conditions, the chances of preventing tumor angiogenesis are high, from this perspective the present review discusses on the endogenous angioinhibitors that are derived from the extracellular matrix as a result of the proteolytic activity of several endoproteases and details the possible integrin receptors and their mechanism of actions.
With the exception of PEX domain and vasohibins, the below described angioinhibitors could be classified as either extracellular matrix derived (such as arresten, canstatin: tumstatin: endostatin: endorepellin) or plasma derived molecules (that include: angiostatin: prothrombin kringle domain-2: thrombospondins).
2. Extracellular Matrix Derived Angioinhibitors
2.1. Arresten (α1(IV)NC1)
Arresten was isolated as a 26-kDa endogenous molecule from the C-terminal noncollagenous (NC1) domain of the α1 chain of type IV collagen [6]. It was shown to inhibit bFGF-induced, endothelial cell proliferation and tube formation besides reducing tumor metastases and human xenograft tumors in arresten treated nude mice. They also showed decreased endothelial cell binding to arresten coated plates when cells were treated with α1 and β1 integrin antibodies, suggesting that α1β1 is a possible integrin receptor for arresten. Bacculovirus-expressed recombinant arresten was found to inhibit tube formation, proliferation and migration of HUVECs in an α1β1 integrin dependent manner, also pretreatment of arresten to HUVECs inhibited FAK/c-Raf/MEK/ERK1/2/p38 MAPK pathway [7]. The authors also showed that arresten, when treated to endothelial cells cultured on type IV collagen inhibited hypoxia induced expression of HIFα and VEGF by inhibiting ERK1/2 and p38 MAPK activation. Their study also reports that SCC-PSA1 tumors had decreased number of CD31 positive vasculature when treated with recombinant arresten, indicating that arresten inhibits tumor angiogenesis in vivo. Arresten was also shown to induce apoptosis by decreasing the expression of Bcl-2 and Bcl-xL in endothelial and tumor cells in mice [8]. More interestingly the active site of arresten was identified through deletion mutagenesis to be localized within the C-terminal subunit. Endothelial cell proliferation induced by bFGF was found to be significantly inhibited by arresten in a dose and time dependent manner [9]. Increased secretion and activation of MMP-2 by bFGF was also shown to be inhibited when endothelial cells were treated with arresten in a dose-dependent manner, however the levels of MMP-2 mRNA were not affected, indicating that arresten interacts with MMP-2 and inhibits its activation. The mechanism shown here was that arresten covalently binds to proMMP-2 and inhibits its auto activation. Arresten was also reported to promote apoptosis by caspase-3/PARP activation and by negatively regulating FAK, p38-MAPK phosphorylation, Bcl-2, and Bcl-xL expression in mouse retinal endothelial cells (MREC) [10]. Interestingly they also found that arresten inhibited VEGF-induced MREC migration and proliferation, but not MRPEC proliferation.
2.2. Canstatin (α2(IV)NC1)
Canstatin was isolated as a 24-kDa fragment from the C-terminal noncollagenous domain of the α2 chain of type IV collagen, which significantly inhibited endothelial cell migration and tube formation without affecting nonendothelial cells, besides inducing apoptosis and suppressing in vivo human xenograft prostate adenocarcinoma tumors in nude mice [11]. The apoptotic activity of canstatin was shown to be mediated by binding to αVβ3 and αVβ5 integrins which initiate cell death via activation of procaspase 8 and 9 which in turn lead to activation of caspase-3 [11-13]. Treatment with canstatin increased expression of Fas ligand and decreased FLIP protein binding to FADD and caspase-8, inducing death receptor mediated apoptosis [11,13,14]. Canstatin localizes on the MDA-MB-231 tumor cells and increases mitochondrial caspase-9 activity, thereby inducing apoptosis [12]. Through immunoprecipitation studies using antibodies against αVβ3 and αVβ5 it was shown that canstatin binds to both these integrins on the endothelial surface, and has a higher antiangiogenic potential than angiostatin [12]. When endothelial cells were treated with canstatin, phosphorylation of FAK, Akt, and downstream targets such as mTOR, 4E-BP1, and p70s6k were found to be inhibited, indicating the caspase-9 mediated apoptotic activity of canstatin [13]. The amino acids 1–89 of canstatin was shown to be more potent that canstatin itself and this region was found to specifically inhibit endothelial cell proliferation and induced apoptosis, besides suppressing growth of B16 murine melanoma tumors [15]. The same group also showed that the C-terminal 157–227 amino acid region of canstatin inhibits endothelial cell proliferation and apoptosis, but the apoptosis-inducing activity was much lower than the 1–89 amino acid region of canstatin with similar tumor suppression activity [16]. In another interesting study which is a first report of its kind, the 131I radiotherapy was combined with angiogenesis inhibition, using both sodium iodide symporter (NIS) and canstatin that was delivered by adenovirus. This dual therapy was found to strongly impede the growth of xenograft and spontaneous tumors in mice [17]. The recombinant canstatin not only was shown to inhibit tube formation in HUVECs and lymphatic endothelial cells, but also reduced the growth of oral squamous cell carcinoma tumors in mice models [18]. Using the novel oncolytic conditionally-replicating adenovirus (CRAd) in which the E1B-55kDa gene for selective replication in tumor cells was replaced with canstatin, the synergistic effects of oncolytic therapy and anti-angiogenesis therapy for pancreatic cancer was also reported [19]. By combining tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene therapy and canstatin, inhibition of human breast tumors in nude mice was observed [20]. Recently, the same group has identified that recombinant canstatin inhibits angiopoietin-1-induced angiogenesis and lymphangiogenesis [21]. In their study they also identified that expression of angiopoietin-1 in CT-26 cells under hypoxic conditions is inhibited by canstatin and affects both angiogenic and lymphangiogenic signaling induced by angiopoietin-1, which is presumed to be mediated through integrin-dependent FAK signaling induced by angiopoietin-1/Tie-2 and/or VEGFR-3. They also showed the antiangiogenic effects of canstatin in inhibiting alkali burn-induced corneal neovascularization in mice [21].
2.3. Tumstatin (α3(IV)NC1)
Tumstatin was isolated as a 28-kDa noncollagenous NC1 domain that was proteolytically cleaved from the C-terminal region of α3 chain of type IV collagen [22]. The region between 185–203 amino acids of tumstatin was found to inhibit activation of human polymorphonuclear monocytes [23]. Also the region between 54–132-amino acids corresponding to Tum-5 peptide was shown to inhibit tube formation and induce cell cycle arrest at G1 phase in endothelial cells, besides inhibiting human prostate cancer growth and angiogenesis in nude mice [24]. Tumstatin was reported to inhibit bFGF-induced proliferation of HUVECs, and melanoma cells, besides inducing apoptosis in endothelial cells and inhibiting neovascularization in matrigel plugs and in vivo tumor growth in different murine cancer types [22,24-27]. The antiangiogenic properties of tumstatin have been reported through several different pathways. Tumstatin binds to αVβ3 integrins through an RGD-independent mechanism and inhibits CAP-dependent protein translation by FAK/PI3K/Akt pathway down regulating mTOR, 4E-BP1, and eIF-4E [26]. This specific activity of Tumstatin was found in the region between 69–98 amino acids. The same integrins were also reported to be involved in regulating the antiangiogenic functions through PTEN/Akt pathway [28]. Deletion of tumstatin and thrombospondin-1 in mice lacking the p53 tumor suppressor gene showed increased incidence and reduced latency of angiogenic lymphomas [29]. Also intratumoral expression of Tum1 showed significant repression of the growth of Huh-7 (hepatocellular carcinoma) tumors in nude mice with decreased CD34 positive vessels indicating the antiangiogenic potential of Tum1 that could be used in gene therapy [30]. A fusion protein comprising the 88 amino acid sequence from tumstatin 45–132 with TNFα showed inhibition of angiogenesis and tumor-cell viability in vitro, also intratumoral injection of this Tumstatin45-132-TNFα protein showed decreased blood-vessel density in xenograft F6 tumors in mice [31]. In oral squamous cell carcinoma animal model, the effects of tumstatin in inhibiting tumor growth was shown in vivo, although the tumors did not show total remission, the authors found decreased tumor microvessel density indicating that tumstatin delays the tumor growth and metastasis of oral squamous cell carcinomas [32]. The antiangiogenic properties of tumstatin were also shown to be mediated by its binding to integrins αVβ3 and α3β1, and regulation of the PI3-K/4E-BP1 pathway [33,34]. The expression of the proinflamatory molecule COX-2 was reported to be inhibited in integrin β3-null MLECs, and not in α3-null MLECs when treated with tumstatin, indicating that integrin α3β1 is a functional receptor for, umstatin in inhibiting hypoxic COX2 expression which is also a proangiogenic factor [35]. A ,umstatin peptide was shown to bind to αVβ3 integrins on proliferating endothelial cells in tumor endothelium, and in combination with anti-VEGF antibody (bevacizumab) it was shown to suppress renal cell carcinoma tumors [36]. The YSNSG cyclopeptide derived from umstatin showed inhibition of endothelial cell migration in vitro without affecting cell proliferation, this inhibition of cellular migration was reported to be mediated by a decrease in active MT1-MMP, u-PA and u-PAR expression [37]. Expression of Tumstatin45-132-TNFα showed inhibition of cellular proliferation and induction of apoptosis in prostate cancer cells in xenograft tumors [38,39].
2.4. Hexastatin (α6(IV)NC1)
Another noncollagenous domain derived from the sixth chain of type IV Collagen was identified as antiangiogenic, is about 228 amino acids in length and was found to inhibit angiogenesis and tumor growth affecting endothelial cell adhesion and migration which was mediated by αV and β1 integrins [33]. Hexastatin was also shown to inhibit proliferation of HUVECs and neovascularization in matrigel plugs besides inhibiting the growth of LLC and pancreatic tumors in mice [40]. However a detailed mechanistic study in identifying the signaling mechanisms involved in its antiangiogenic functions is yet to be identified.
2.5. Endostatin
Endostatin was discovered as a 20 kDa protein derived from the C-terminal fragment of type XVIII collagen which inhibited endothelial cell proliferation, angiogenesis and tumor growth [41]. The authors also identified the antitumor potential of endostatin in inhibiting growth of several tumor types such as Lewis lung carcinomas, T241 fibrosarcomas, B16F10 melanomas and hemangioendothelioma. Recently, tumor growth in many cancer types was found to be significantly reduced upon treatment with endostatin [42]. Lack of endostatin in humans results in Knobloch syndrome, an autosomal recessive disorder that results in blindness at birth due to failure of retinal development [43]. A similar condition was also reported in mice deficient in endostatin that fail to develop a vascularized retina [44]. The circulating levels of endostatin in healthy individuals was reported to be between 10 to 50 ng/mL which is equivalent to 0.5–2.5 nM however, elevated levels of endostatin were reported in several cancer types that include osteosarcoma, NSCLC, hepatocellular carcinoma, ovarian cancer, bladder cancer, head and neck squamous cell carcinoma, renal cell carcinoma, soft tissue sarcoma, acute myeloid leukemia and colorectal cancer. Such higher levels of endostatin in cancer patients was implicated as a prognostic factor indicating that endostatin may be used a therapeutic target for the treatment of these cancer types [45-56]. Murine hemangioendothelioma tumor cells (EOMA) from which endostatin was originally isolated secrete MMPs and procathepsin that gets activated to cathepsin in acidic medium and generates endostatin from type XVIII collagen which then exerts its antiangiogenic activity [57]. One of the many mechanisms by which endostatin regulates its anitangiogenic activity was thorough inhibition of matrix metalloproteinases2 (MMP-2)-mediated endothelial cell invasion, where endostatin binds to the catalytic domain of proMMP2 forming a stable complex [58-60]. Endostatin inhibits endothelial cell survival and migration by inhibiting VEGF121, VEGF165 and their cognate receptor KDR/FLK-1 [61,62]. Endostatin was also shown to inhibit PDGF mediated recruitment of perivascular cells affecting maturation of new blood vessels [63]. The many other mechanisms which enhance the antiangiogenic properties of endostatin include, blocking VEGFR2 signaling, suppressing Wnt signaling, catenin destabilization, or altering catenin/VE cadherin interactions in interendothelial cell junctions. A comprehensive signaling mechanism of endostatin involving Id/AP-1, HIF-1α, ephrin and TNF-α, NF-κB, STAT and Ets in cell proliferation, migration, survival, tube formation and apoptosis, besides coagulation cascades and adhesion molecule pathways was also reported [64]. Nucleolin on the cell surface to which endostatin binds with high affinity, was identified to be a vital receptor for endostatin to mediate its antiangiogenic and antitumor functions [65]. Nucleolin is a ubiquitous multifunctional protein critically involved in cell proliferation, chromatin organization, packaging of pre-RNA, rDNA transcription, and ribosome assembly [66-68]. Nucleolin is mobilized from nucleus to endothelial cell surface and is expressed only in new angiogenic blood vessels which is modulated by VEGF and ECM proteins [69]. Other mechanism by which endostatin functions were reported by inducing endothelial cell apoptosis, down regulating Bcl-2, and up regulating caspase-3 expression in vitro [70,71]. Endostatin binds to the endothelial cell surface integrins α5 and αV and prevents integrin dependent cell migration [72,73]. More precisely the integrin α5β1 was identified as a potential receptor for endostatin by which it regulates its outside in signaling [34]. Also the tumor suppressor functions of endostatin in knockout mice were reported where tumors grew 2- to 3-fold faster [74].
2.6. Endorepellin
Endorepellin was identified from the C-terminal functional region of Perlecan with the potential to inhibit angiogenesis, and was found to inhibit endothelial cell migration, tube morphogenesis and adhesion in vitro, besides inhibiting angiogenesis in matrigel plug and CAM assay in vivo. Endorepellin also affects cell adhesion in several cancer cells such as those derived from colon, neuroectoderm and mesenchyme [75]. The authors also showed that endorepellin is comprised of three LG modules and LG2 binds to endostatin, but this binding does not affect the antiangiogenic activity of either endostatin or endorepellin. Enzymes belonging to the BMP-1/tolloid-like proteinase family were reported to cleave endorepellin and liberate LG3 module, which contributes the antiangiogenic functions of endorepellin [76]. Endorepellin when administered intraperitoneally was reported to target the tumor vasculature in squamous cell carcinoma xenografts and in syngeneic LLC tumors, inhibiting tumor growth. A possible mechanism of caspase-3 activation during apoptosis initiating the release of cathepsin-L was identified to be vital for endorepellin proteolysis and LG3 production [77]. Endorepellin belongs to the RGD-independent and cation-independent class of molecules that bind to α2β1 integrin receptors.
Knockdown of integrin α2 subunit using siRNA significantly affected migration of endothelial and fibrosarcoma cells across collagen. Using a LLC xenograft model, it was also confirmed that the antitumorangiogenic activity of endorepellin is mediated through α2β1 integrins [78]. The angiostatic activity of endorepellin was reported to be greatly confined within the LG3 module which binds to α2-I integrin domain, a major binding site for collagen I [79]. It was concluded that interactions of endorepellin with α2β1 integrins causes increase in cAMP and activation of PKA and FAK, but not Erk1/Erk2, and leads to transient phosphorylation of p38 MAPK and Hsp27. Blockade of α2β1 integrins in fibroblasts was shown to inhibit the antiapoptotic response initiated by recombinant LG3 [80]. Endorepellin supports α2β1 integrin-mediated and Src-kinase-dependent platelet adhesion, but does not contribute to activation or platelet aggregation [81]. Endorepellin was shown to cause a rapid activation of the tyrosine phosphatase Src homology-2 protein phosphatase-1 (SHP-1), and provokes global dephosphorylation of several RTKs that are dependent on the presence of the integrin α2β1 [82]. TIMP-2 was reported to induce a substantial increase in cAMP levels with the activation of cAMP-dependent PKA in microvascular endothelial cells, fibroblasts, as well as in several transformed cells. The ability of endorepellin to activate SHP-1 with TIMP-2 was shown to induce a signaling cascade of events involving key angiogenic regulators such as RTKs, VEGFR2 and FGFR1 [83-86]. Recently, similar to TIMP-2, which binds to the α3β1 integrin and induces SHP-1, and in turn dephosphorylates several receptor tyrosine kinases, including VEGFR2 and FGFR1, endorepellin was also shown to activate the phosphatase SHP-1 but its antiangiogenic signaling was found to be mediated through α2β1 integrins unlike TIMP-2 which is mediated by α3β1 [87]. Also, endorepellin treated tumors were shown to be hypoxic with decreased metabolism, and decreased cell proliferation without inducing apoptosis or inhibiting wound healing [88]. The LG3 module of endorepellin was reported as a serological biomarker for breast cancer since its plasma levels were found low in breast cancer patients [89]. Signaling mechanisms of the extracellular matrix derived angioinhibitors are shown in Figure 1.
3. Plasma Derived Endogenous Angioinhibitors
3.1. Angiostatin
Angiostatin was identified as a 38 kDa fragment from the elastase digest of plasminogen which was isolated from the urine of LLC tumor bearing mice, with an half-life of 2.5 days [90]. Although with little ambiguity, angiostatin refers to the proteolytic fragment of plasminogen comprising five kringle domains, and depending on the protease used its molecular weight ranges between 38–55 kDa. Among the five kringle domains, kringle domain-4 was ineffective, unlike the other four domains that showed strong antiangiogenic functions in endothelial cells. A number of antiangiogenic properties of angiostatin have been identified, in endothelial cells angiostatin was shown to inhibit cellular proliferation, migration and tube formation on matrigel matrix. However, angiostatin was reported to have no effect on a variety of normal, neoplastic and nonendothelial cell lines such as 3T3 fibroblasts, bovine aorta smooth muscle cells, bovine retinal pigment epithelial cells, human fetal fibroblasts, and LLC carcinoma cells [91,92]. Angiostatin was shown to induce endothelial cell apoptosis in vitro by RGD independent activation of FAK besides inhibiting VEGF and bFGF mediated cellular migration and tube formation [93]. However the signal transduction mechanism of angiostatin upon treatment to microvascular endothelial cells, resulted in decreased activation of ERK 1 & 2 MAP kinases that were activated by VEGF and bFGF [94]. Intracranial administration of angiostatin was also shown to result in suppression of brain tumor growth and decreased tumor angiogenesis [95]. Mice lacking plasminogen, which is a precursor for angiostatin, showed spontaneous fibrin deposits with reduced fertility and survival indicating that plasminogen or plasmin are not essential for embryonic development [96]. The antiangiogenic functions of angiostatin were reported to be mediated by at least three different receptors that were identified on endothelial cell surface that include ATP synthase, angiomotin and integrin αVβ3, α4β1 and α9β1 [97-99]. The anticancer functions of angiostatin have been studied in several cancer types such as lung cancer, brain cancer, colon cancer, breast cancer, etc. With successful completion of PhaseI/II clinical trials of angiostatin for patients with progressive metastatic cancer and non-small-cell lung cancer, the results from phase III clinical trials of angiostatin are awaited in anticipation that the study would be completed by June 2012 as scheduled.
3.2. Prothrombin Kringle-2
The group led by Soung Soo Kim, have made vital studies in identification of prothrombin kringle-2 as an endogenous antiangiogenic molecule and made significant contributions to this area of research. Human prothrombin was digested with Factor Xa overnight and prothrombin fragments 1 and 2 are isolated as 30 kDa and 19 kDa proteins. The authors also identified that both the fragments inhibited bFGF induced endothelial cell growth in a dose dependent manner, and also inhibited in vivo angiogenesis through CAM assay [100]. Earlier the same group has reported the antiangiogenic functions of prothrombin kringle-2 domain from rabbits, which is a first report on its discovery [101]. Recombinant prothrombin kringle domains with antitumor properties, were studied using LLC tumor cells and reported that treatment with E. Coli expressed recombinant prothrombin kringle domain-2 not only inhibited tumor growth significantly but also prevented tumor metastasis [62]. The authors also detailed the mechanism by which prothrombin kringle domains are generated from prothrombin. Prothrombin is composed of 581 amino acids which when digested with Factor Xa results in two fragments 1–273 (amino terminal fragment) and 274–581 (active thrombin). Active thrombin then cleaves the amino terminal fragment and releases prothrombin kringle fragment-1 (1–155 amino acids) and prothrombin kringle fragment-2 (156–273 amino acids). Also the same group has identified two peptides NSA7 and NSA8 that were derived as C-terminal truncation products of NSA9 which was originally identified from prothrombin kringle-2 fragment. Although all the three peptides have significant antiangiogenic activity, NSA7 showed considerably higher effect than NSA8 and NSA9 as compared using cell proliferation inhibition assay, in vivo CAM angiogenesis assay, tube formation and migration of HUVEC cells. The peptide NSA7 was also found as an effective inhibitor for proliferation of B16F10, LLC and L929 tumor cells and gets internalized into endothelial and tumor cells more easily [102]. Endothelial cells when treated with prothrombin kringle-2 showed dose dependent inhibition of cellular migration and adhesion to ECM proteins especially using vitronectin matrix suggesting that αVβ3 could be a possible integrin receptor for prothrombim kringle-2 [103]. Mice treated with prothrombin kringle-2 showed resistance to melanoma pulmonary metastasis as they exhibited less metastatic colonies with small and isolated tumors, and also helped in restoring the acute lung injury associated with B16F10 melanoma metastasis to normal phenotype. Also, prothrombin kringle-2 was found to inhibit VEGF expression in type I and type II pneumocytes, endothelial cells and metastatic tumor cells with diminished CD31 expression which would have caused the inhibition of B16F10 melanoma metastasis associated with tumor neovascularization. Tumor cell derived MMP-2 or MMP-9 were reported to elicit secretion of soluble VEGF from the ECM [58,104]. Treatment with prothrombin kringle-2 decreased expression of MMP-2 and MMP-9 in the bronchiolar epithelial cells, pneumocytes, endothelial cells, and metastatic tumor cells of B16F10 melanoma, suggesting the possible mechanism of prothrombin kringle-2 antitumor actions [103]. Previously it was reported that activated microglia produces reactive oxygen species, resulting in oxidative damage and causes severe pathology in neurodegenerative diseases [105-108]. Prothrombin kringle-2 was shown to act as an endogenous microglial activator and exerts neurotoxicity in the cortex in vivo. Prothrombin kringle-2 induced up regulation of cytosolic protein p67phox co-localized within activated microglia in the cortex and activated microglial NADPH oxidase, enhanced reactive oxygen species production and protein oxidation which resulted in neurodegeneration in the cortex. However, prothrombin kringle-2 failed to cause neuronal loss in neuron enriched cortical cultures devoid of microglia suggesting that the activated microglia are required for prothrombin kringle-2 induced neurotoxicity. Supporting this observation, the authors also report that there is a concomitant increase in the level of nitrite formed from NO and TNF-α in cortical microglia cultures when treated with prothrombin kringle-2. Interestingly, the authors also identified that prothrombin kringle-2 treated cortex showed expression of iNOS and IL-1β in vivo [109,110].
3.3. Thrombospondins
Baenzinger in 1971 identified the presence of a high molecular weight thrombin sensitive protein when thrombin is added to intact platelets [111]. Later it was characterized as a high molecular weight glycoprotein isolated from human blood platelets and coined the term “thrombospondin” [112]. Thrombospondins are a family of extracellular matrix glycoproteins consisting of five members (TSP-1 to TSP-5) whose functions have been implicated in treating several cancer types. Among the five thrombospondins, TSP-1 and TSP-2 have equivalent domain structures and are widely studied. TSP-1 was reported as the first naturally occurring angiogenesis inhibitor to be identified for having angiostatic functions. The mechanisms by which TSPs exert their antiangiogenic functions include direct effects on inhibiting endothelial cell migration, apoptosis, or indirectly by inhibiting expression of growth factors, cytokines and proteases that regulate angiogenesis. TSP-1 and TSP-2 inhibit growth factors induced cell cycle progression by arresting the cells in the G0/G1 phase, and this inhibition is presumed to be independent of caspase activity. TSP-1 induces endothelial cell apoptosis by up regulating Bax and down regulating VEGF-mediated Bcl-2 expression [113]. CD47 (integrin-associated protein) was shown to impact angiogenesis to a large extent since binding of CD47 with TSP1 and other ligands inhibits VEGFR2 phosphorylation and angiogenesis. It was found that the C-terminal region of TSP-1 binds to CD47 and interacts with cell surface integrins [114]. TSP-1 was shown to bind to CD47 and regulates nitric oxide synthesis in both normal and pathological events [115]. Analysis of wound bed vascularity in TSP-1 and CD47 null mice showed increased angiogenesis indicating their essential role in antiangiogenesis [116]. Recently it was also reported that CD47 interacts with VEGFR2 receptor [117]. Thrombospondin type 1 repeats (TSRs) were found to be present in over 100 different proteins in the human genome, and the presence of these repeats has been correlated with the ability of these proteins to inhibit tumor angiogenesis and tumor growth [118,119]. The receptors for the TSR repeats in TSP-1 were identified as CD36, β1 integrins and TGF-β. The CD36 and TSP-1 interactions were reported to down regulate VEGF receptor-2 and p38 MAPK phosphorylation, inhibiting VEGF induced functions [120]. Also the interactions between TSRs and β1 integrins were shown to result in inhibition of endothelial cell migration [121]. The TSRs induced cell migration was also reported to be inhibited when treated with integrin α3, α5 and PI3 Kinase antagonists. The interaction between TSRs with CD36 was shown to result in endothelial cell apoptosis presumably mediated through Fyn and c-Jun N-terminal kinase (JNK) pathway [122,123]. The TSR sequence KRFKQDGGWSHWSPWSSC was reported to inhibit proliferation of both endothelial cells and tumor cells besides inhibiting angiogenesis in retinopathy and in solid tumors [124-126]. Also 18 functional peptides that were derived from type I thrombospondin repeat sequences were found to inhibit proliferation and migration of HUVECs when used up to 40 μg/mL concentrations. TSRs in TSP-1 were reported to bind to the type II fibronectin repeats of MMP-2 and inhibit its activation [127]. Also, in transgenic mice that over-express TSP-1 in the mammary glands, the levels of active MMP9 were found lowered in the developed tumors [128]. In TSP-2 null mice, implanted tumors showed increased tumor vascularization indicating its role in tumorangiogenesis [129]. Also overexpression of TSP-1 was shown to suppress tumor angiogenesis and metastasis but was reported to have no effect on lymphangiogenesis [130]. However, using CD36 null mice recently it was shown that TSP-1 inhibits lymphangiogenesis by binding to monocytes, and treatment of TSP-1 to macrophages resulted in suppression of lymphangiogenic factors VEGF-C and VEGF-D [131]. Signaling mechanisms of plasma derived endogenous angioinhibitors are shown in illustration in Figure 2.
4. Endogenous Angioinhibitors Identified from Other Sources
4.1. PEX Domain
MMPs are a family of zinc-dependent matrix-degrading enzymes with a wide variety of ECM proteins as substrates [132-134]. These MMPs were reported to have a prominent role in cellular invasion of several cell types, and an uncontrolled proteolysis of matrix by these MMPs would lead to severe pathological conditions [134]. However, a tight control was reported to exist in vivo that regulates this mechanism of matrix degradation during physiological angiogenesis. The noncatalytic C-terminal domain of MMP-2 (Hemopexin domain or PEX) was reported to interact with αVβ3 integrins on endothelial cell surface and prevents MMP-2 from binding to αVβ3 integrins which are key regulators of angiogenesis [135]. The authors also identified that PEX is a naturally occurring breakdown product of MMP-2 with detectable levels of PEX under normal physiological conditions and during retinal neovascularization and in in vivo tumors suggesting its vital role in angiogenesis and vasculogenesis, proving its antiangiogenic activity. It was also shown that αVβ3 integrins on CS-1 melanoma cells promotes collagenolytic activity that can be blocked using recombinant PEX domain, indicating that addition of PEX to αVβ3 expressing cells prevents binding of MMP-2 to the cell surface. Using lentiviral vectors, expression of PEX was shown to block bFGF induced MMP-2 activation, cell migration, proliferation, tube formation and CAM angiogenesis besides inhibiting human melanoma (M21L) tumor growth in nude mice [136]. The same group also showed that upon systemic administration of PEX, sustained inhibition of glioma tumors over a prolonged period of time, and the histological analysis showed decreased vascularity in tumors, with an increase in apoptosis [137]. The antitumor effects of PEX were evaluated by transfecting human neural stem cells and pTracer vector with PEX and reported inhibition of proliferation and migratory ability of PEX-producing cells in vitro [138]. It is interesting to see that neural stem cells were used here to deliver PEX into the xenograft tissue, and as reported, the transfected stem cells migrated to the tumor site and inhibited angiogenesis without inducing apoptosis or inhibiting cell motility. Recently, fusion of PEX domain with the N-terminal signal peptide of MMP-9 and stable transfection of SNB19 cells with the fusion construct showed secretion of PEX domain, and the cultured condition medium when treated to endothelial cells showed down regulation of MMP-9, VEGF and VEGFR2, induced cell cycle arrest and activated caspases-3, -8 and -9 besides PARP cleavage indicating onset of apoptosis and eventually leading to a significant reduction in tumor volume [139].
4.2. Vasohibins
Vasohibin-1 (VASH1) was identified as a VEGF induced angiogenesis inhibitor whose expression was observed in endothelial cells both under physiological and pathological conditions associated with angiogenesis, and also in other cell types. Vasohibin-1 was reported to inhibit endothelial cell proliferation, migration and tube formation in vitro and angiogenesis in vivo [140]. Although expression of vasohibin-1 was observed in endothelial cells during the embryo development stages, because it is induced by VEGF and FGF, its expression was observed exclusively in newly formed blood vessels where angiogenesis terminates [140-142]. Steady state expression of vasohibin-1 was also reported in adult bone marrow derived haematopoietic stem cells[143]. Expression of vasohibin-1 in endothelial cells was also observed in various solid tumors, atherosclerotic lesions, age-dependent macular degeneration, diabetic retinopathy, and rheumatoid arthritis [144-151]. However, the molecular mechanisms of vasohibins remained largely unclear. Both human and murine vasohibin-1 proteins showed increased apoptosis in mouse fibroblasts, however only murine isoform was found to inhibit migration of human endothelial cells through scratch assay [152]. Recombinant vasohibin-1 protein when applied exogenously or when its murine isoform was overexpressed intracellular, strong inhibition of angiogenic sprouting of HUVEC spheroids was observed in the three-dimensional collagen gel [152]. Moreover, vasohibin-1 was demonstrated to be strongly upregulated by VEGF in the mouse retinopathy model in vivo. The effects of Vasohibin-1 as reported using corneal micro pocket assay showed its distinct antiangiogenic and antilymphangiogenic activity [153]. In addition, they also found that vasohibin-1 inhibits tumor lymphangiogenesis and lymph node metastasis. Moreover, tumor angiogenesis was reported to be inhibited when LLC cells were transfected with vasohibin-1. It was interesting to note that vasohibin-1 did not affect tyrosine phosphorylation of KDR or activation of ERK1/2 when HUVECs were stimulated with VEGF. However, hypoxia and TNF-α inhibited VEGF-stimulated induction of vasohibin-1 in endothelial cells. Vasohibin-2 shares over 52% similarity with vasohibin-1 and is expressed preferentially in mononuclear cells. In contrast to vasohibin-1, vasohibin-2 null mice showed increased angiogenesis [142]. The genetic organization of vasohibin-2 gene in the parasite Schistosoma mansoni, revealed identification of 14 different alternatively spliced variants that encode seven different protein isoforms [154]. Signaling mechanisms of angioinhibitor vasoinhibin are shown in Figure 3.
5. Conclusions
A new branch of cancer research emerged over forty years ago in the early 70s with the hypothesis of Judah Folkman from Harvard Medical School and with the discovery of thrombospondins. Since then, many different endogenous angioinhibitors have been discovered, with over 27 of them that have been identified for having antiangiogenic functions. The endogenous angioinhibitors described in the present review have been well characterized for their role in antiangiogenic functions. These molecules inhibit endothelial angiogenic functions that include cell migration, proliferation, and tube formation in vitro, besides inhibiting matrigel plug angiogenesis, CAM or tumor angiogenesis in vivo. These molecules typically mediate their antiangiogenic signaling by binding to cell surface integrins on endothelial cells and affect expression of kinases involved in cell survival pathways that include MAPK and PI3K, etc., which are induced by growth factors, especially VEGF. Besides they also affect expression of proangiogenic molecules such as MMPs, COX2 eNOS, etc., eventually regulating the cellular physiological processes and inhibiting cell growth. Also, some of these endogenous angioinhibitors were reported to induce apoptotic pathways in endothelial cells by upregulating caspases and the associated downstream signaling. As described before, some of these proteins not only exert their functions by interacting with integrins on cell surface, but they also get internalized and affect proangiogenic pathways. Also many of these proteins were reported to have direct effects on tumor cells as well, indicating the many diverse mechanisms of actions of these endogenous molecules in inhibiting the growth of new blood vessels. Although the mechanisms of action and the pharmacological and pharmacodynamic studies on these endogenous antiangiogenic molecules are yet to be carried out to fully understand their antiangiogenic and antitumor properties before being tested through clinical trials, in light of their multiple mechanisms of actions, these molecules are gaining the significance of having high therapeutic potential with their promising role in combination therapy for cancer treatment. The summary of the above matrix derived and plasma derived angioinhibitors and their receptors with possible mechanism of actions are stated in Table 1.
6. Future Perspectives
The very existence of these endogenous angioinhibitors in the normal healthy individuals indicates a new line of host defense system to maintain angiogenic balance, and this property could be potentially applied to prevent progression of angiogenesis related diseases. The striking feature that makes these endogenous angioinhibitors effective for use in cancer therapy is because of their little or no side effects and drug resistance. Several gene therapy options are available to apply antiangiogenic therapy that can be targeted to the disease locus. Since tumor recurrence is common to chemotherapeutic regimes, treating patients with these endogenous antiangiogenic agents in combination with chemotherapy or immunotherapy could prove to be an efficient means to prevent tumor recurrence. Use of these endogenous antiangiogenic agents in combination with chemotherapy is emerging as an effective option to address several angiogenesis related diseases. With about 27 of these endogenous antiangiogenic agents identified for their potential role in cancer treatment, and with many of them are presently under preclinical and clinical phase trials, the applicability of these endogenous molecules for the treatment of tumor angiogenesis appears very promising.

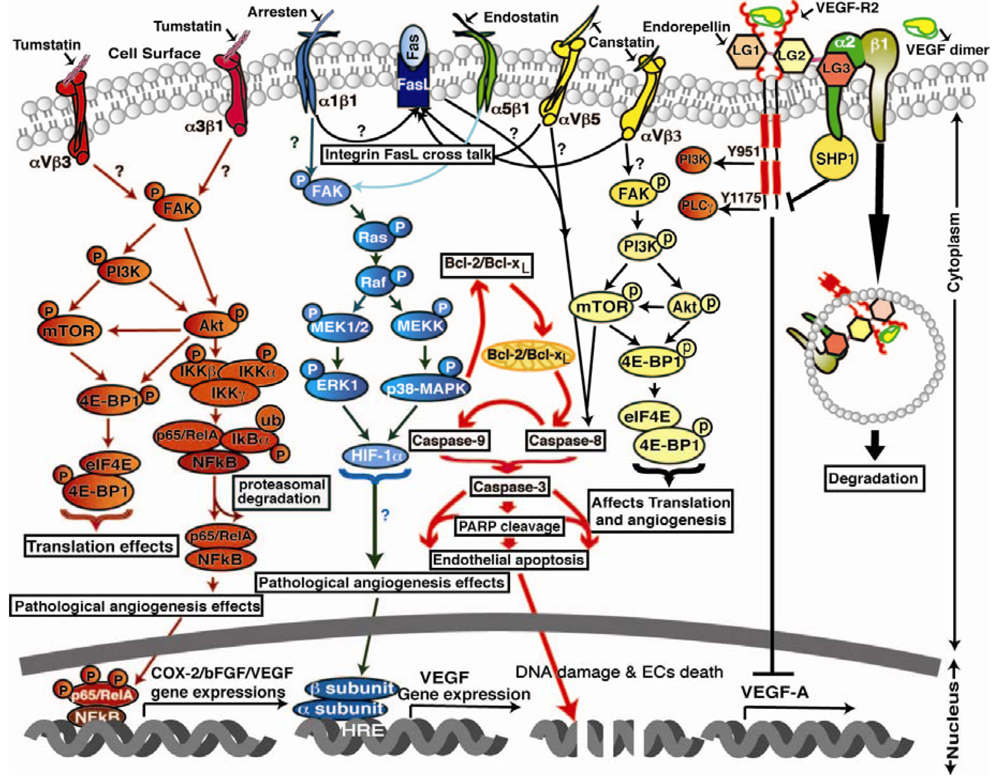
Tumstatin: It binds to αVβ3 and α3β1 integrins and inhibits FAK, Akt, PI3-K, mTOR, eIF4E and 4E-BP1 phosphorylation to decrease endothelial cell protein synthesis and proliferation. In addition tumstatin also inhibits transcription factor-NFκB mediated signaling in hypoxic conditions in endothelial cells leading to the inhibition of COX-2, bFGF and VEGF expressions, resulting in inhibition of tumor angiogenesis and LASER induced CNV in in-vivo mice models. Arresten: It binds to α1β1 integrin and inhibits FAK, Ras, Raf, ERK1 and p38-MAPK phosphorylation that leads to inhibition of HIF-1α and VEGF expression resulting in inhibition of endothelial cell proliferation, migration and tube formation. In addition arresten also initiates activation of caspase-9 and -8, leading to activation of caspase-3, PARP cleavage in two different ways: (i) arresten activates caspase-3 and caspase-9 directly through inhibition of FAK/p38-MAPK/Bcl-2/Bcl-xL in proliferating endothelial cells; (ii) possible, α1β1 integrin cross talk with Fas-Ligand through mitochondrial pathway leads to activation of caspase-8 and-3 in proliferating endothelial cells. Endostatin: It binds to α5β1 integrin and inhibits FAK phosphorylation that leads to of Ras, Raf, ERK1 and p38-MAPK phosphorylation inhibition that leads to inhibition of endothelial cell migration and promotes apoptosis in LASER induced CNV in in vivo mice models. Canstatin: It binds to integrins αVβ5/αVβ3 and inhibits two different apoptotic pathways, involving activation of caspase-9 and caspase-8 that leads to activation of caspase-3. Canstatin activates procaspase-9 not only through inhibition of the FAK/PI3K/Akt pathways but also by integrins cross talking mitochondrial pathway through Fas-Ligand dependent caspase-8 activation leads to endothelial cell apoptosis in LASER induced CNV models. Endorepellin: It binds to α2β1 integrins and VEGFR-2, activates SHP-1 mediated cAMP-PKA/FAK/p38-MAPK/Hsp27 signaling pathway.
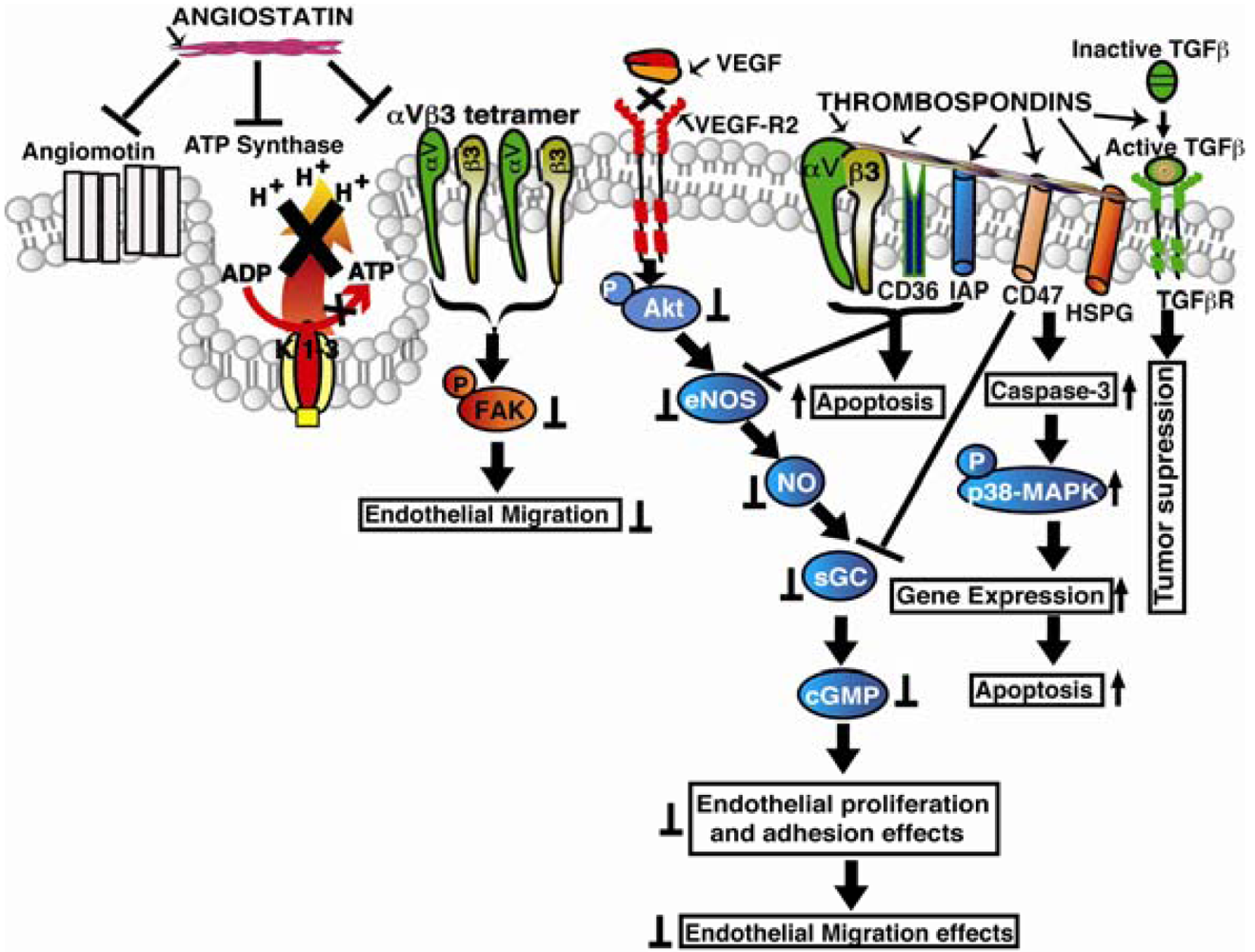
Angiostatin: It binds to αVβ3 integrin and inhibit FAK phosphorylation and ATP synthase in endothelial cells. Thrombospondins (TSPs): Bind to CD36 and integrin associated protein (IAP) promoting Src-family protein kinases/Caspase-3/p38-MAPK signaling, leading to activation of apoptosis In addition TSPs also binds to heparan sulfate proteoglycans (HSPGs), CD47, α3β1 and promotes TGF-β activation that leading to tumor cell death.
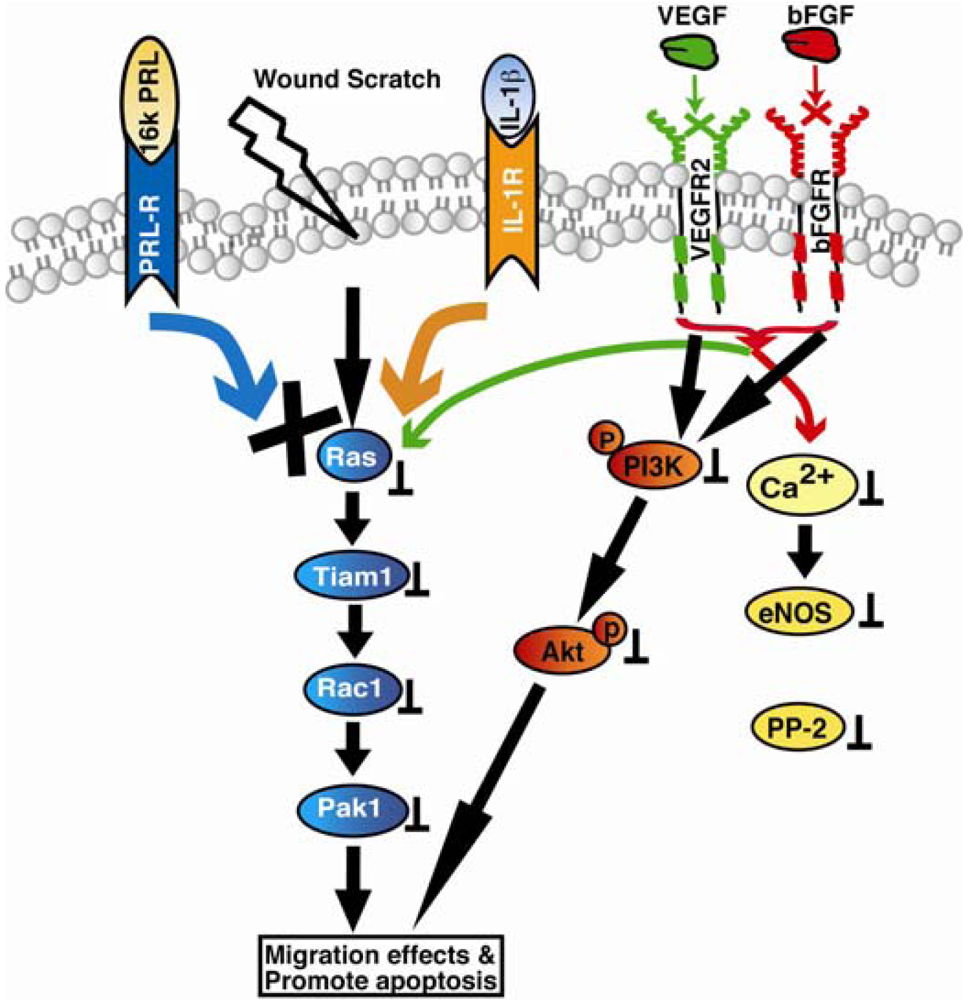
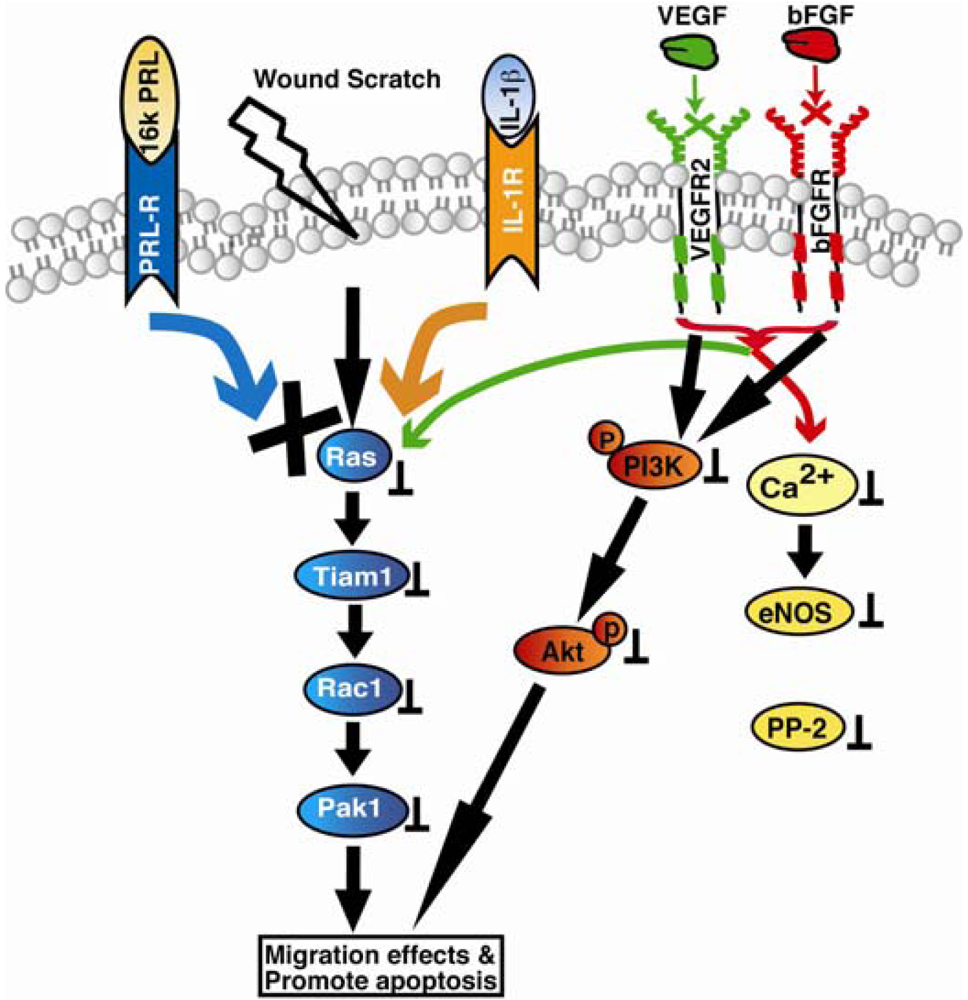
Vasoinhibin (16Kda PRL): It binds prolactin receptor (PRL-R) and interleukin-1 receptor (IL-1β) and promotes Ras/Tiam-1/Rac-1/Pak1 and Bcl-XL mediated apoptosis. In addition vasoinhibins also inhibit VEGF and bFGF mediated PI3K/Akt, and Ca2+/eNOS/protein phosphatase 2 signaling.

{kind=link}
{kind=link}
{kind=link}
| Angioinhibitor | Parent molecule | Receptors | Mode of action |
|---|---|---|---|
| Angiostatin | Plasminogen | ATP synthases, αVβ3 integrin, angiomotin | αVβ3 integrin mediated apoptosis in endothelial cells, ATP synthase α1β1 integrin dependent endothelial |
| Arresten | Type IV Collagen α1 NC1 domain | α1β1 integrin, HSPG | Raf/MEK/ERK1/2/p38-MAPK, HIF-1 inhibition and integrin independent MMP-2 activation inhibition |
| Canstatin | Type IV Collagen α2 NC1 domain | αVβ3, αVβ5 integrins and cross talk with, Fas Ligand | Integrins dependent inhibition of Akt/FAK/mToR, eIF-4EBP-1 activation, inhibition of caspase-8 and -9 activation and Ribosomal S6-kinase |
| Endorepellin | Perlecan | α2β1 integrins, lipid rafts, caveolin | Inhibition of cAMP-PKA/FAK/p38-MAPK/Hsp27, SHP-1, Ca2+ signaling |
| Endostatin | Type XVIII Collagen NC1 domain | αVβ1/α5β1 integrins, HSP, glypican, caveolin-1 | Inhibition of Ras/Raf/KDR/Flk-1/ERK/p38-MAPK/p125 FAK/HIF-1α/Ephrin/TNFα/ NF-κβ, Wnt signaling |
| PEX domain | MMP-2 | αVβ3 integrin | Interacts with αVβ3 integrins and prevents MMP-2 binding to αVβ3 integrins |
| Prothrombin Kringle-2 | Prothrombin | αVβ3 integrin | Inhibits VEGF, MMP-2 and 9 expression, affects EC growth. |
| Thrombospondins | TSP | α3β1, CD47, HSPG, CD36, IAP | Inhibition of Src-family kinases/ Caspase-3/p38 MAPK, TGF-β signaling |
| Tumstatin | Type IV Collagen α3 NC1 domain | αVβ3, α3β1, α6β1 integrins, CD47/IAP | Inhibition of FAK/Akt/PI3K/mTOR/eIF-4EBP1 signaling; NFκB, COX-2 dependent tumor angiogenesis inhibition signaling |
| Vasoinhibins | Prolactin, growth hormone | Not known | Sos/Ras/MAPK or eNOS/Raf/MAPK, Ca2+/eNOS/protein phosphatase 2, Ras/ Tiam-1/Rac1/Pak1, Bcl-XL, NF-κβ, caspases |
Akt: protein kinase B, Bcl-XL: B-cell lymphoma-extra large, bFGF: basic fibroblast growth factor, CD47 Integrin Associated Protein, COX-2: cyclooxygenase-2, eIF-4EBP-1: eukaryotic translation initiation factor-4E binding protein-1, eNOS: endothelial nitric oxide synthase, ECs: endothelial cells, ERK1/2: extracellular signal-regulated kinase1/2, FAK: focal adhesion kinase, HIF-1a: hypoxia inducible factor-1a, Hsp: heat shock protein, HSPG: Heparan sulfate proteoglycan, IAP: integrin associated protein, IKK: IκB kinase, KDR: kinase insert domain receptor, MAPK: Mitogen activated protein kinase, MEK: MAPK-ERK kinase, MIP: macrophage inflammatory protein-1/-2, MMPs: matrix metallo proteinases, mTOR: mammalian target of rapamycin, NF-κβ: nuclear factor kappa β, PEX: noncatalytic carboxy-terminal hemopexin-like domain of MMP, PI3K: phosphatidyl inositol 3-kinase, Rac: Ras-related C3 botulinin toxin susbtrate 1, Raf: Ras activated factor, Ras: Rat sarcoma, SHP: Src homology region 2 domain-containing phopshatase, Src: Schmidt-Ruppin A-2 sarcoma viral oncogene homolog, TIAM: T-lymphoma invasion and metastasis-inducing protein, TGF-β: transforming growth factor β, TNFα: tumor necrosis factor α, TSP: thrombospondin.
Acknowledgments
We would like to apologize to those of our colleagues whose work we were unable to cite in this review due to space concerns. Research related to this work in the authors' laboratory is supported by Flight Attendant Medical Research Institute Young Clinical Scientist Award Grant FAMRI-062558, NIH/NCI Grant RO1CA143128, Dobleman Head and Neck Cancer Institute Grant DHNCI-61905 and startup research funds of Cell Signaling, Retinal and Tumor Angiogenesis Laboratory at Boys Town National Research Hospital to YAS.
Conflict of Interest
The authors declare no conflict of interest.
References
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Abdollahi, A.; Lipson, K.E.; Sckell, A.; Zieher, H.; Klenke, F.; Poerschke, D.; Roth, A.; Han, X.; Krix, M.; Bischof, M.; et al. Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res. 2003, 63, 8890–8898. [Google Scholar]
- Filleur, S.; Volpert, O.V.; Degeorges, A.; Voland, C.; Reiher, F.; Clezardin, P.; Bouck, N.; Cabon, F. In vivo mechanisms by which tumors producing thrombospondin-1 bypass its inhibitory effects. Genes Dev. 2001, 15, 1373–1382. [Google Scholar]
- Filleur, S.; Courtin, A.; Ait-Si-Ali, S.; Guglielmi, J.; Merle, C.; Harel-Bellan, A.; Clezardin, P.; Cabon, F. siRNA-mediated inhibition of vascular endothelial growth factor severely limits tumor resistance to antiangiogenic thrombospondin-1 and slows tumor vascularization and growth. Cancer Res. 2003, 63, 3919–3922. [Google Scholar]
- Fernando, N.T.; Koch, M.; Rothrock, C.; Gollogly, L.K.; D'Amore, P.A.; Ryeom, S.; Yoon, S.S. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin. Cancer Res. 2008, 14, 1529–1539. [Google Scholar]
- Colorado, P.C.; Torre, A.; Kamphaus, G.; Maeshima, Y.; Hopfer, H.; Takahashi, K.; Volk, R.; Zamborsky, E.D.; Herman, S.; Sarkar, P.K.; et al. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000, 60, 2520–2526. [Google Scholar]
- Sudhakar, A.; Nyberg, P.; Keshamouni, V.G.; Mannam, A.P.; Li, J.; Sugimoto, H.; Cosgrove, D.; Kalluri, R. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by α1β1 integrin. J. Clin. Invest. 2005, 115, 2801–2810. [Google Scholar]
- Nyberg, P.; Xie, L.; Sugimoto, H.; Colorado, P.; Sund, M.; Holthaus, K.; Sudhakar, A.; Salo, T.; Kalluri, R. Characterization of the anti-angiogenic properties of arresten, an α1β1 integrin-dependent collagen-derived tumor suppressor. Exp. Cell Res. 2008, 314, 3292–3305. [Google Scholar]
- Boosani, C.S.; Nalabothula, N.; Sheibani, N.; Sudhakar, A. Inhibitory effects of arresten on bFGF-induced proliferation, migration, and matrix metalloproteinase-2 activation in mouse retinal endothelial cells. Curr. Eye Res. 2010, 35, 45–55. [Google Scholar]
- Boosani, C.S.; Nalabothula, N.; Munugalavadla, V.; Cosgrove, D.; Keshamoun, V.G.; Sheibani, N.; Sudhakar, A. Fak and p38-MAP kinase-dependent activation of apoptosis and caspase-3 in retinal endothelial cells by α1(IV)NC1. Invest. Ophthalmol. Vis. Sci. 2009, 50, 4567–4575. [Google Scholar]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar]
- Magnon, C.; Galaup, A.; Mullan, B.; Rouffiac, V.; Bouquet, C.; Bidart, J.M.; Griscelli, F.; Opolon, P.; Perricaudet, M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with αVβ3 and αVβ5 integrins. Cancer Res. 2005, 65, 4353–4361. [Google Scholar]
- Panka, D.J.; Mier, J.W. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J. Biol. Chem. 2003, 278, 37632–37636. [Google Scholar]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular flip. Nature 1997, 388, 190–195. [Google Scholar]
- He, G.A.; Luo, J.X.; Zhang, T.Y.; Wang, F.Y.; Li, R.F. Canstatin-N fragment inhibits in vitro endothelial cell proliferation and suppresses in vivo tumor growth. Biochem. Biophys. Res. Commun. 2003, 312, 801–805. [Google Scholar]
- He, G.A.; Luo, J.X.; Zhang, T.Y.; Hu, Z.S.; Wang, F.Y. The C-terminal domain of canstatin suppresses in vivo tumor growth associated with proliferation of endothelial cells. Biochem. Biophys. Res. Commun. 2004, 318, 354–360. [Google Scholar]
- Magnon, C.; Opolon, P.; Ricard, M.; Connault, E.; Ardouin, P.; Galaup, A.; Metivier, D.; Bidart, J.M.; Germain, S.; Perricaudet, M. Radiation and inhibition of angiogenesis by canstatin synergize to induce HIF-1α-mediated tumor apoptotic switch. J. Clin. Invest. 2007, 117, 1844–1855. [Google Scholar]
- Hwang-Bo, J.; Yoo, K.H.; Jeong, H.S.; Chung, I.S. Recombinant canstatin inhibits tumor growth in an orthotopic AT-84 oral squamous cell carcinoma model. Biotechnol. Lett. 2010, 32, 189–194. [Google Scholar]
- He, X.P.; Su, C.Q.; Wang, X.H.; Pan, X.; Tu, Z.X.; Gong, Y.F.; Gao, J.; Liao, Z.; Jin, J.; Wu, H.Y.; et al. E1B-55kd-deleted oncolytic adenovirus armed with canstatin gene yields an enhanced anti-tumor efficacy on pancreatic cancer. Cancer Lett. 2009, 285, 89–98. [Google Scholar]
- Wang, W.B.; Zhou, Y.L.; Heng, D.F.; Miao, C.H.; Cao, Y.L. Combination of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and canstatin gene suppression therapy on breast tumor xenograft growth in mice. Breast Cancer Res. Treat. 2008, 110, 283–295. [Google Scholar]
- Wang, Y.; Yin, H.; Chen, P.; Xie, L. Inhibitory effect of canstatin in alkali burn-induced corneal neovascularization. Ophthalmic Res. 2011, 46, 66–72. [Google Scholar]
- Maeshima, Y.; Colorado, P.C.; Torre, A.; Holthaus, K.A.; Grunkemeyer, J.A.; Ericksen, M.B.; Hopfer, H.; Xiao, Y.; Stillman, I.E.; Kalluri, R. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J. Biol. Chem. 2000, 275, 21340–21348. [Google Scholar]
- Monboisse, J.C.; Garnotel, R.; Bellon, G.; Ohno, N.; Perreau, C.; Borel, J.P.; Kefalides, N.A. The α3 chain of type IV collagen prevents activation of human polymorphonuclear leukocytes. J. Biol. Chem. 1994, 269, 25475–25482. [Google Scholar]
- Maeshima, Y.; Manfredi, M.; Reimer, C.; Holthaus, K.A.; Hopfer, H.; Chandamuri, B.R.; Kharbanda, S.; Kalluri, R. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J. Biol. Chem. 2001, 276, 15240–15248. [Google Scholar]
- Maeshima, Y.; Colorado, P.C.; Kalluri, R. Two RGD-independent αVβ3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J. Biol. Chem. 2000, 275, 23745–23750. [Google Scholar]
- Maeshima, Y.; Sudhakar, A.; Lively, J.C.; Ueki, K.; Kharbanda, S.; Kahn, C.R.; Sonenberg, N.; Hynes, R.O.; Kalluri, R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science 2002, 295, 140–143. [Google Scholar]
- Han, J.; Ohno, N.; Pasco, S.; Monboisse, J.C.; Borel, J.P.; Kefalides, N.A. A cell binding domain from the α3 chain of type IV collagen inhibits proliferation of melanoma cells. J. Biol. Chem. 1997, 272, 20395–20401. [Google Scholar]
- Kawaguchi, T.; Yamashita, Y.; Kanamori, M.; Endersby, R.; Bankiewicz, K.S.; Baker, S.J.; Bergers, G.; Pieper, R.O. The PTEN/Akt pathway dictates the direct αVβ3-dependent growth-inhibitory action of an active fragment of tumstatin in glioma cells in vitro and in vivo. Cancer Res. 2006, 66, 11331–11340. [Google Scholar]
- Xie, L.; Duncan, M.B.; Pahler, J.; Sugimoto, H.; Martino, M.; Lively, J.; Mundel, T.; Soubasakos, M.; Rubin, K.; Takeda, T.; et al. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc. Natl. Acad. Sci. USA 2011, 108, 9939–9944. [Google Scholar]
- Goto, T.; Ishikawa, H.; Matsumoto, K.; Nishimura, D.; Kusaba, M.; Taura, N.; Shibata, H.; Miyaaki, H.; Ichikawa, T.; Hamasaki, K.; et al. Tum-1, a tumstatin fragment, gene delivery into hepatocellular carcinoma suppresses tumor growth through inhibiting angiogenesis. Int. J. Oncol. 2008, 33, 33–40. [Google Scholar]
- Yan, Y.; Xu, W.; Qian, H.; Zhu, W.; Mao, F.; Zhang, X. Tumstatin45-132-TNFα suppresses tumour growth through anti-angiogenic effects and cytotoxicity. Biotechnol. Appl. Biochem. 2010, 56, 119–127. [Google Scholar]
- Chung, I.S.; Son, Y.I.; Ko, Y.J.; Baek, C.H.; Cho, J.K.; Jeong, H.S. Peritumor injections of purified tumstatin delay tumor growth and lymphatic metastasis in an orthotopic oral squamous cell carcinoma model. Oral Oncol. 2008, 44, 1118–1126. [Google Scholar]
- Petitclerc, E.; Boutaud, A.; Prestayko, A.; Xu, J.; Sado, Y.; Ninomiya, Y.; Sarras, M.P., Jr; Hudson, B.G.; Brooks, P.C. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 2000, 275, 8051–8061. [Google Scholar]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by αVβ3 and α5β1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar]
- Boosani, C.S.; Mannam, A.P.; Cosgrove, D.; Silva, R.; Hodivala-Dilke, K.M.; Keshamouni, V.G.; Sudhakar, A. Regulation of COX-2 mediated signaling by α3 type IV noncollagenous domain in tumor angiogenesis. Blood 2007, 110, 1168–1177. [Google Scholar]
- Eikesdal, H.P.; Sugimoto, H.; Birrane, G.; Maeshima, Y.; Cooke, V.G.; Kieran, M.; Kalluri, R. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor activity. Proc. Natl. Acad. Sci. USA 2008, 105, 15040–15045. [Google Scholar]
- Thevenard, J.; Ramont, L.; Devy, J.; Brassart, B.; Dupont-Deshorgue, A.; Floquet, N.; Schneider, L.; Ouchani, F.; Terryn, C.; Maquart, F.X.; et al. The YSNSG cyclopeptide derived from tumstatin inhibits tumor angiogenesis by down-regulating endothelial cell migration. Int. J. Cancer 2010, 126, 1055–1066. [Google Scholar]
- Zhang, X.; Xu, W.; Qian, H.; Zhu, W.; Zhang, R. Mesenchymal stem cells modified to express lentivirus TNFα tumstatin(45-132) inhibit the growth of prostate cancer. J. Cell. Mol. Med. 2011, 15, 433–444. [Google Scholar]
- Liu, Y.; Li, J.; Xu, H.; Zhang, Y.; Liu, X. Mitochondria-mediated tumstatin peptide-induced HepG-2 cell apoptosis. Int. J. Mol. Med. 2009, 24, 653–659. [Google Scholar]
- Mundel, T.M.; Yliniemi, A.M.; Maeshima, Y.; Sugimoto, H.; Kieran, M.; Kalluri, R. Type IV collagen α6 chain-derived noncollagenous domain 1 (α6(IV)NC1) inhibits angiogenesis and tumor growth. Int. J. Cancer 2008, 122, 1738–1744. [Google Scholar]
- O'Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar]
- Sertie, A.L.; Sossi, V.; Camargo, A.A.; Zatz, M.; Brahe, C.; Passos-Bueno, M.R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 2000, 9, 2051–2058. [Google Scholar]
- Suzuki, O.T.; Sertie, A.L.; Der Kaloustian, V.M.; Kok, F.; Carpenter, M.; Murray, J.; Czeizel, A.E.; Kliemann, S.E.; Rosemberg, S.; Monteiro, M.; et al. Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in knobloch syndrome. Am. J. Hum. Genet. 2002, 71, 1320–1329. [Google Scholar]
- Hefler, L.; Tempfer, C.; Kainz, C.; Obermair, A. Serum concentrations of endostatin in patients with vulvar cancer. Gynecol. Oncol. 1999, 74, 151–152. [Google Scholar]
- Zorick, T.S.; Mustacchi, Z.; Bando, S.Y.; Zatz, M.; Moreira-Filho, C.A.; Olsen, B.; Passos-Bueno, M.R. High serum endostatin levels in down syndrome: Implications for improved treatment and prevention of solid tumours. Eur. J. Hum. Genet. 2001, 9, 811–814. [Google Scholar]
- Kim, H.S.; Lim, S.J.; Park, Y.K. Anti-angiogenic factor endostatin in osteosarcoma. APMIS 2009, 117, 716–723. [Google Scholar]
- Iizasa, T.; Chang, H.; Suzuki, M.; Otsuji, M.; Yokoi, S.; Chiyo, M.; Motohashi, S.; Yasufuku, K.; Sekine, Y.; Iyoda, A.; et al. Overexpression of collagen XVIII is associated with poor outcome and elevated levels of circulating serum endostatin in non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 5361–5366. [Google Scholar]
- Hu, T.H.; Huang, C.C.; Wu, C.L.; Lin, P.R.; Liu, S.Y.; Lin, J.W.; Chuang, J.H.; Tai, M.H. Increased endostatin/collagen XVIII expression correlates with elevated VEGF level and poor prognosis in hepatocellular carcinoma. Mod. Pathol. 2005, 18, 663–672. [Google Scholar]
- Hata, K.; Dhar, D.K.; Kanasaki, H.; Nakayama, K.; Fujiwaki, R.; Katabuchi, H.; Okamura, H.; Nagasue, N.; Miyazaki, K. Serum endostatin levels in patients with epithelial ovarian cancer. Anticancer Res. 2003, 23, 1907–1912. [Google Scholar]
- Guan, K.P.; Ye, H.Y.; Yan, Z.; Wang, Y.; Hou, S.K. Serum levels of endostatin and matrix metalloproteinase-9 associated with high stage and grade primary transitional cell carcinoma of the bladder. Urology 2003, 61, 719–723. [Google Scholar]
- Homer, J.J.; Greenman, J.; Stafford, N.D. Circulating angiogenic cytokines as tumour markers and prognostic factors in head and neck squamous cell carcinoma. Clin. Otolaryngol. Allied Sci. 2002, 27, 32–37. [Google Scholar]
- Feldman, A.L.; Alexander, H.R., Jr; Yang, J.C.; Linehan, W.M.; Eyler, R.A.; Miller, M.S.; Steinberg, S.M.; Libutti, S.K. Prospective analysis of circulating endostatin levels in patients with renal cell carcinoma. Cancer 2002, 95, 1637–1643. [Google Scholar]
- Feldman, A.L.; Pak, H.; Yang, J.C.; Alexander, H.R., Jr; Libutti, S.K. Serum endostatin levels are elevated in patients with soft tissue sarcoma. Cancer 2001, 91, 1525–1529. [Google Scholar]
- Wrobel, T.; Mazur, G.; Kapelko, K.; Kuliczkowski, K. Endostatin serum level in acute myeloid leukemia. Neoplasma 2005, 52, 182–184. [Google Scholar]
- Feldman, A.L.; Alexander, H.R., Jr; Bartlett, D.L.; Kranda, K.C.; Miller, M.S.; Costouros, N.G.; Choyke, P.L.; Libutti, S.K. A prospective analysis of plasma endostatin levels in colorectal cancer patients with liver metastases. Ann. Surg. Oncol. 2001, 8, 741–745. [Google Scholar]
- Felbor, U.; Dreier, L.; Bryant, R.A.; Ploegh, H.L.; Olsen, B.R.; Mothes, W. Secreted cathepsin L generates endostatin from collagen XVIII. EMBO J. 2000, 19, 1187–1194. [Google Scholar]
- Fang, J.; Shing, Y.; Wiederschain, D.; Yan, L.; Butterfield, C.; Jackson, G.; Harper, J.; Tamvakopoulos, G.; Moses, M.A. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc. Natl. Acad. Sci. USA 2000, 97, 3884–3889. [Google Scholar]
- Itoh, Y.; Ito, A.; Iwata, K.; Tanzawa, K.; Mori, Y.; Nagase, H. Plasma membrane-bound tissue inhibitor of metalloproteinases (TIMP)-2 specifically inhibits matrix metalloproteinase 2 (gelatinase a) activated on the cell surface. J. Biol. Chem. 1998, 273, 24360–24367. [Google Scholar]
- Kim, Y.M.; Jang, J.W.; Lee, O.H.; Yeon, J.; Choi, E.Y.; Kim, K.W.; Lee, S.T.; Kwon, Y.G. Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res. 2000, 60, 5410–5413. [Google Scholar]
- Hajitou, A.; Grignet, C.; Devy, L.; Berndt, S.; Blacher, S.; Deroanne, C.F.; Bajou, K.; Fong, T.; Chiang, Y.; Foidart, J.M.; et al. The antitumoral effect of endostatin and angiostatin is associated with a down-regulation of vascular endothelial growth factor expression in tumor cells. FASEB J. 2002, 16, 1802–1804. [Google Scholar]
- Kim, T.H.; Kim, E.; Yoon, D.; Kim, J.; Rhim, T.Y.; Kim, S.S. Recombinant human prothrombin kringles have potent anti-angiogenic activities and inhibit lewis lung carcinoma tumor growth and metastases. Angiogenesis 2002, 5, 191–201. [Google Scholar]
- Skovseth, D.K.; Veuger, M.J.; Sorensen, D.R.; de Angelis, P.M.; Haraldsen, G. Endostatin dramatically inhibits endothelial cell migration, vascular morphogenesis, and perivascular cell recruitment in vivo. Blood 2005, 105, 1044–1051. [Google Scholar]
- Abdollahi, A.; Hahnfeldt, P.; Maercker, C.; Grone, H.J.; Debus, J.; Ansorge, W.; Folkman, J.; Hlatky, L.; Huber, P.E. Endostatin's antiangiogenic signaling network. Mol. Cell 2004, 13, 649–663. [Google Scholar]
- Shi, H.; Huang, Y.; Zhou, H.; Song, X.; Yuan, S.; Fu, Y.; Luo, Y. Nucleolin is a receptor that mediates antiangiogenic and antitumor activity of endostatin. Blood 2007, 110, 2899–2906. [Google Scholar]
- Ginisty, H.; Amalric, F.; Bouvet, P. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J. 1998, 17, 1476–1486. [Google Scholar]
- Erard, M.S.; Belenguer, P.; Caizergues-Ferrer, M.; Pantaloni, A.; Amalric, F. A major nucleolar protein, nucleolin, induces chromatin decondensation by binding to histone H1. Eur. J. Biochem. 1988, 175, 525–530. [Google Scholar]
- Kharrat, A.; Derancourt, J.; Doree, M.; Amalric, F.; Erard, M. Synergistic effect of histone H1 and nucleolin on chromatin condensation in mitosis: Role of a phosphorylated heteromer. Biochemistry 1991, 30, 10329–10336. [Google Scholar]
- Huang, Y.; Shi, H.; Zhou, H.; Song, X.; Yuan, S.; Luo, Y. The angiogenic function of nucleolin is mediated by vascular endothelial growth factor and nonmuscle myosin. Blood 2006, 107, 3564–3571. [Google Scholar]
- Dhanabal, M.; Ramchandran, R.; Waterman, M.J.; Lu, H.; Knebelmann, B.; Segal, M.; Sukhatme, V.P. Endostatin induces endothelial cell apoptosis. J. Biol. Chem. 1999, 274, 11721–11726. [Google Scholar]
- Dhanabal, M.; Volk, R.; Ramchandran, R.; Simons, M.; Sukhatme, V.P. Cloning, expression, and in vitro activity of human endostatin. Biochem. Biophys. Res. Commun. 1999, 258, 345–352. [Google Scholar]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar]
- MacDonald, N.J.; Shivers, W.Y.; Narum, D.L.; Plum, S.M.; Wingard, J.N.; Fuhrmann, S.R.; Liang, H.; Holland-Linn, J.; Chen, D.H.; Sim, B.K. Endostatin binds tropomyosin. A potential modulator of the antitumor activity of endostatin. J. Biol. Chem. 2001, 276, 25190–25196. [Google Scholar]
- Sund, M.; Hamano, Y.; Sugimoto, H.; Sudhakar, A.; Soubasakos, M.; Yerramalla, U.; Benjamin, L.E.; Lawler, J.; Kieran, M.; Shah, A.; et al. Function of endogenous inhibitors of angiogenesis as endothelium-specific tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 2934–2939. [Google Scholar]
- Mongiat, M.; Sweeney, S.M.; San Antonio, J.D.; Fu, J.; Iozzo, R.V. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 2003, 278, 4238–4249. [Google Scholar]
- Gonzalez, E.M.; Reed, C.C.; Bix, G.; Fu, J.; Zhang, Y.; Gopalakrishnan, B.; Greenspan, D.S.; Iozzo, R.V. BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 2005, 280, 7080–7087. [Google Scholar]
- Cailhier, J.F.; Sirois, I.; Laplante, P.; Lepage, S.; Raymond, M.A.; Brassard, N.; Prat, A.; Iozzo, R.V.; Pshezhetsky, A.V.; Hebert, M.J. Caspase-3 activation triggers extracellular cathepsin L release and endorepellin proteolysis. J. Biol. Chem. 2008, 283, 27220–27229. [Google Scholar]
- Woodall, B.P.; Nystrom, A.; Iozzo, R.A.; Eble, J.A.; Niland, S.; Krieg, T.; Eckes, B.; Pozzi, A.; Iozzo, R.V. Integrin α2β1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 2008, 283, 2335–2343. [Google Scholar]
- Bix, G.; Fu, J.; Gonzalez, E.M.; Macro, L.; Barker, A.; Campbell, S.; Zutter, M.M.; Santoro, S.A.; Kim, J.K.; Hook, M.; et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through α2β1 integrin. J. Cell Biol. 2004, 166, 97–109. [Google Scholar]
- Laplante, P.; Raymond, M.A.; Labelle, A.; Abe, J.; Iozzo, R.V.; Hebert, M.J. Perlecan proteolysis induces an α2β1 integrin- and Src family kinase-dependent anti-apoptotic pathway in fibroblasts in the absence of focal adhesion kinase activation. J. Biol. Chem. 2006, 281, 30383–30392. [Google Scholar]
- Bix, G.; Iozzo, R.A.; Woodall, B.; Burrows, M.; McQuillan, A.; Campbell, S.; Fields, G.B.; Iozzo, R.V. Endorepellin, the C-terminal angiostatic module of perlecan, enhances collagen-platelet responses via the α2β1-integrin receptor. Blood 2007, 109, 3745–3748. [Google Scholar]
- Nystrom, A.; Shaik, Z.P.; Gullberg, D.; Krieg, T.; Eckes, B.; Zent, R.; Pozzi, A.; Iozzo, R.V. Role of tyrosine phosphatase SHP-1 in the mechanism of endorepellin angiostatic activity. Blood 2009, 114, 4897–4906. [Google Scholar]
- Seo, D.W.; Li, H.; Guedez, L.; Wingfield, P.T.; Diaz, T.; Salloum, R.; Wei, B.Y.; Stetler-Stevenson, W.G. TIMP-2 mediated inhibition of angiogenesis: An MMP-independent mechanism. Cell 2003, 114, 171–180. [Google Scholar]
- Seo, D.W.; Kim, S.H.; Eom, S.H.; Yoon, H.J.; Cho, Y.R.; Kim, P.H.; Kim, Y.K.; Han, J.W.; Diaz, T.; Wei, B.Y.; et al. TIMP-2 disrupts FGF-2-induced downstream signaling pathways. Microvasc. Res. 2008, 76, 145–151. [Google Scholar]
- Hoegy, S.E.; Oh, H.R.; Corcoran, M.L.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J. Biol. Chem. 2001, 276, 3203–3214. [Google Scholar]
- Corcoran, M.L.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinase-2 stimulates fibroblast proliferation via a CAMP-dependent mechanism. J. Biol. Chem. 1995, 270, 13453–13459. [Google Scholar]
- Goyal, A.; Pal, N.; Concannon, M.; Paul, M.; Doran, M.; Poluzzi, C.; Sekiguchi, K.; Whitelock, J.M.; Neill, T.; Iozzo, R.V. Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEFGR2). A dual receptor antagonism. J. Biol. Chem. 2011, 286, 25947–25962. [Google Scholar]
- Bix, G.; Castello, R.; Burrows, M.; Zoeller, J.J.; Weech, M.; Iozzo, R.A.; Cardi, C.; Thakur, M.L.; Barker, C.A.; Camphausen, K.; et al. Endorepellin in vivo: Targeting the tumor vasculature and retarding cancer growth and metabolism. J. Natl. Cancer Inst. 2006, 98, 1634–1646. [Google Scholar]
- Chang, J.W.; Kang, U.B.; Kim, D.H.; Yi, J.K.; Lee, J.W.; Noh, D.Y.; Lee, C.; Yu, M.H. Identification of circulating endorepellin IG3 fragment: Potential use as a serological biomarker for breast cancer. Proteomics Clin. Appl. 2008, 2, 23–32. [Google Scholar]
- O'Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar]
- Gately, S.; Twardowski, P.; Stack, M.S.; Cundiff, D.L.; Grella, D.; Castellino, F.J.; Enghild, J.; Kwaan, H.C.; Lee, F.; Kramer, R.A.; et al. The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Proc. Natl. Acad. Sci. USA 1997, 94, 10868–10872. [Google Scholar]
- Gately, S.; Twardowski, P.; Stack, M.S.; Patrick, M.; Boggio, L.; Cundiff, D.L.; Schnaper, H.W.; Madison, L.; Volpert, O.; Bouck, N.; et al. Human prostate carcinoma cells express enzymatic activity that converts human plasminogen to the angiogenesis inhibitor, angiostatin. Cancer Res. 1996, 56, 4887–4890. [Google Scholar]
- Claesson-Welsh, L.; Welsh, M.; Ito, N.; Anand-Apte, B.; Soker, S.; Zetter, B.; O'Reilly, M.; Folkman, J. Angiostatin induces endothelial cell apoptosis and activation of focal adhesion kinase independently of the integrin-binding motif RGD. Proc. Natl. Acad. Sci. USA 1998, 95, 5579–5583. [Google Scholar]
- Redlitz, A.; Daum, G.; Sage, E.H. Angiostatin diminishes activation of the mitogen-activated protein kinases ERK-1 and ERK-2 in human dermal microvascular endothelial cells. J. Vasc. Res. 1999, 36, 28–34. [Google Scholar]
- Meneses, P.I.; Abrey, L.E.; Hajjar, K.A.; Gultekin, S.H.; Duvoisin, R.M.; Berns, K.I.; Rosenfeld, M.R. Simplified production of a recombinant human angiostatin derivative that suppresses intracerebral glial tumor growth. Clin. Cancer Res. 1999, 5, 3689–3694. [Google Scholar]
- Lijnen, H.R. Pathophysiology of the plasminogen/plasmin system. Int. J. Clin. Lab. Res. 1996, 26, 1–6. [Google Scholar]
- Moser, T.L.; Stack, M.S.; Asplin, I.; Enghild, J.J.; Hojrup, P.; Everitt, L.; Hubchak, S.; Schnaper, H.W.; Pizzo, S.V. Angiostatin binds ATP synthase on the surface of human endothelial cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2811–2816. [Google Scholar]
- Troyanovsky, B.; Levchenko, T.; Mansson, G.; Matvijenko, O.; Holmgren, L. Angiomotin: An angiostatin binding protein that regulates endothelial cell migration and tube formation. J. Cell Biol. 2001, 152, 1247–1254. [Google Scholar]
- Tarui, T.; Miles, L.A.; Takada, Y. Specific interaction of angiostatin with integrin αVβ3 in endothelial cells. J. Biol. Chem. 2001, 276, 39562–39568. [Google Scholar]
- Rhim, T.Y.; Park, C.S.; Kim, E.; Kim, S.S. Human prothrombin fragment 1 and 2 inhibit bFGF-induced BCE cell growth. Biochem. Biophys. Res. Commun. 1998, 252, 513–516. [Google Scholar]
- Lee, T.H.; Rhim, T.; Kim, S.S. Prothrombin kringle-2 domain has a growth inhibitory activity against basic fibroblast growth factor-stimulated capillary endothelial cells. J. Biol. Chem. 1998, 273, 28805–28812. [Google Scholar]
- Hwang, H.S.; Kim, D.W.; Kim, S.S. Structure-activity relationships of the human prothrombin kringle-2 peptide derivative NSA9: Anti-proliferative activity and cellular internalization. Biochem. J. 2006, 395, 165–172. [Google Scholar]
- Kim, T.H.; Ahn, S.; Kim, J.; Kim, I.; Yang, M.Z.; Lee, J.E.; Kim, S.S. Recombinant human prothrombin kringle-2 inhibits B16F10 melanoma metastasis through inhibition of neovascularization and reduction of matrix metalloproteinase expression. Clin. Exp. Metastasis 2006, 23, 391–399. [Google Scholar]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar]
- Block, M.L.; Li, G.; Qin, L.; Wu, X.; Pei, Z.; Wang, T.; Wilson, B.; Yang, J.; Hong, J.S. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: Substance P vs. Dynorphin. FASEB J. 2006, 20, 251–258. [Google Scholar]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar]
- Gao, H.M.; Liu, B.; Hong, J.S. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci. 2003, 23, 6181–6187. [Google Scholar]
- Moisse, K.; Strong, M.J. Innate immunity in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1083–1093. [Google Scholar]
- Won, S.Y.; Choi, S.H.; Jin, B.K. Prothrombin kringle-2-induced oxidative stress contributes to the death of cortical neurons in vivo and in vitro: Role of microglial NADPH oxidase. J. Neuroimmunol. 2009, 214, 83–92. [Google Scholar]
- Kim, S.R.; Chung, E.S.; Bok, E.; Baik, H.H.; Chung, Y.C.; Won, S.Y.; Joe, E.; Kim, T.H.; Kim, S.S.; Jin, M.Y.; et al. Prothrombin kringle-2 induces death of mesencephalic dopaminergic neurons in vivo and in vitro via microglial activation. J. Neurosci. Res. 2010, 88, 1537–1548. [Google Scholar]
- Baenziger, N.L.; Brodie, G.N.; Majerus, P.W. A thrombin-sensitive protein of human platelet membranes. Proc. Natl. Acad. Sci. USA 1971, 68, 240–243. [Google Scholar]
- Lawler, J.W.; Slayter, H.S.; Coligan, J.E. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J. Biol. Chem. 1978, 253, 8609–8616. [Google Scholar]
- Nor, J.E.; Mitra, R.S.; Sutorik, M.M.; Mooney, D.J.; Castle, V.P.; Polverini, P.J. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J. Vasc. Res. 2000, 37, 209–218. [Google Scholar]
- Calzada, M.J.; Sipes, J.M.; Krutzsch, H.C.; Yurchenco, P.D.; Annis, D.S.; Mosher, D.F.; Roberts, D.D. Recognition of the N-terminal modules of thrombospondin-1 and thrombospondin-2 by α6β1 integrin. J. Biol. Chem. 2003, 278, 40679–40687. [Google Scholar]
- Isenberg, J.S.; Ridnour, L.A.; Dimitry, J.; Frazier, W.A.; Wink, D.A.; Roberts, D.D. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J. Biol. Chem. 2006, 281, 26069–26080. [Google Scholar]
- Isenberg, J.S.; Pappan, L.K.; Romeo, M.J.; Abu-Asab, M.; Tsokos, M.; Wink, D.A.; Frazier, W.A.; Roberts, D.D. Blockade of thrombospondin-1-CD47 interactions prevents necrosis of full thickness skin grafts. Ann. Surg. 2008, 247, 180–190. [Google Scholar]
- Kaur, S.; Martin-Manso, G.; Pendrak, M.L.; Garfield, S.H.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem. 2010, 285, 38923–38932. [Google Scholar]
- Tucker, R.P. The thrombospondin type 1 repeat superfamily. Int. J. Biochem. Cell Biol. 2004, 36, 969–974. [Google Scholar]
- Lawler, J.; Detmar, M. Tumor progression: The effects of thrombospondin-1 and -2. Int. J. Biochem. Cell Biol. 2004, 36, 1038–1045. [Google Scholar]
- Primo, L.; Ferrandi, C.; Roca, C.; Marchio, S.; di Blasio, L.; Alessio, M.; Bussolino, F. Identification of CD36 molecular features required for its in vitro angiostatic activity. FASEB J. 2005, 19, 1713–1715. [Google Scholar]
- Short, S.M.; Derrien, A.; Narsimhan, R.P.; Lawler, J.; Ingber, D.E.; Zetter, B.R. Inhibition of endothelial cell migration by thrombospondin-1 type-1 repeats is mediated by β1 integrins. J. Cell Biol. 2005, 168, 643–653. [Google Scholar]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar]
- Jimenez, B.; Volpert, O.V.; Reiher, F.; Chang, L.; Munoz, A.; Karin, M.; Bouck, N. c-jun N-terminal kinase activation is required for the inhibition of neovascularization by thrombospondin-1. Oncogene 2001, 20, 3443–3448. [Google Scholar]
- Bogdanov, A., Jr; Marecos, E.; Cheng, H.C.; Chandrasekaran, L.; Krutzsch, H.C.; Roberts, D.D.; Weissleder, R. Treatment of experimental brain tumors with trombospondin-1 derived peptides: An in vivo imaging study. Neoplasia 1999, 1, 438–445. [Google Scholar]
- Guo, N.; Krutzsch, H.C.; Inman, J.K.; Roberts, D.D. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res. 1997, 57, 1735–1742. [Google Scholar]
- Shafiee, A.; Penn, J.S.; Krutzsch, H.C.; Inman, J.K.; Roberts, D.D.; Blake, D.A. Inhibition of retinal angiogenesis by peptides derived from thrombospondin-1. Invest. Ophthalmol. Vis. Sci. 2000, 41, 2378–2388. [Google Scholar]
- Bein, K.; Simons, M. Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J. Biol. Chem. 2000, 275, 32167–32173. [Google Scholar]
- Rodriguez-Manzaneque, J.C.; Lane, T.F.; Ortega, M.A.; Hynes, R.O.; Lawler, J.; Iruela-Arispe, M.L. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc. Natl. Acad. Sci. USA 2001, 98, 12485–12490. [Google Scholar]
- Hawighorst, T.; Velasco, P.; Streit, M.; Hong, Y.K.; Kyriakides, T.R.; Brown, L.F.; Bornstein, P.; Detmar, M. Thrombospondin-2 plays a protective role in multistep carcinogenesis: A novel host anti-tumor defense mechanism. EMBO J. 2001, 20, 2631–2640. [Google Scholar]
- Hawighorst, T.; Oura, H.; Streit, M.; Janes, L.; Nguyen, L.; Brown, L.F.; Oliver, G.; Jackson, D.G.; Detmar, M. Thrombospondin-1 selectively inhibits early-stage carcinogenesis and angiogenesis but not tumor lymphangiogenesis and lymphatic metastasis in transgenic mice. Oncogene 2002, 21, 7945–7956. [Google Scholar]
- Cursiefen, C.; Maruyama, K.; Bock, F.; Saban, D.; Sadrai, Z.; Lawler, J.; Dana, R.; Masli, S. Thrombospondin 1 inhibits inflammatory lymphangiogenesis by CD36 ligation on monocytes. J. Exp. Med. 2011, 208, 1083–1092. [Google Scholar]
- Mignatti, P.; Rifkin, D.B. Biology and biochemistry of proteinases in tumor invasion. Physiol. Rev. 1993, 73, 161–195. [Google Scholar]
- DeClerck, Y.A.; Laug, W.E. Cooperation between matrix metalloproteinases and the plasminogen activator-plasmin system in tumor progression. Enzyme Protein 1996, 49, 72–84. [Google Scholar]
- Stetler-Stevenson, W.G.; Hewitt, R.; Corcoran, M. Matrix metalloproteinases and tumor invasion: From correlation and causality to the clinic. Semin. Cancer Biol. 1996, 7, 147–154. [Google Scholar]
- Brooks, P.C.; Silletti, S.; von Schalscha, T.L.; Friedlander, M.; Cheresh, D.A. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell 1998, 92, 391–400. [Google Scholar]
- Pfeifer, A.; Kessler, T.; Silletti, S.; Cheresh, D.A.; Verma, I.M. Suppression of angiogenesis by lentiviral delivery of PEX, a noncatalytic fragment of matrix metalloproteinase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12227–12232. [Google Scholar]
- Pluderi, M.; Lucini, V.; Caronzolo, D.; Pannacci, M.; Costa, F.; Carrabba, G.; Giussani, C.; Grosso, S.; Colleoni, F.; Scaglione, F.; Villani, R.; Bikfalvi, A.; Bello, L. Long-term inhibition of glioma growth by systemic administration of human PEX. J. Neurosurg. Sci. 2003, 47, 69–78. [Google Scholar]
- Kim, S.K.; Cargioli, T.G.; Machluf, M.; Yang, W.; Sun, Y.; Al-Hashem, R.; Kim, S.U.; Black, P.M.; Carroll, R.S. PEX-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin. Cancer Res. 2005, 11, 5965–5970. [Google Scholar]
- Ezhilarasan, R.; Jadhav, U.; Mohanam, I.; Rao, J.S.; Gujrati, M.; Mohanam, S. The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int. J. Cancer 2009, 124, 306–315. [Google Scholar]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Invest. 2004, 114, 898–907. [Google Scholar]
- Shimizu, K.; Watanabe, K.; Yamashita, H.; Abe, M.; Yoshimatsu, H.; Ohta, H.; Sonoda, H.; Sato, Y. Gene regulation of a novel angiogenesis inhibitor, vasohibin, in endothelial cells. Biochem. Biophys. Res. Commun. 2005, 327, 700–706. [Google Scholar]
- Kimura, H.; Miyashita, H.; Suzuki, Y.; Kobayashi, M.; Watanabe, K.; Sonoda, H.; Ohta, H.; Fujiwara, T.; Shimosegawa, T.; Sato, Y. Distinctive localization and opposed roles of vasohibin-1 and vasohibin-2 in the regulation of angiogenesis. Blood 2009, 113, 4810–4818. [Google Scholar]
- Naito, H.; Kidoya, H.; Sato, Y.; Takakura, N. Induction and expression of anti-angiogenic vasohibins in the hematopoietic stem/progenitor cell population. J. Biochem. 2009, 145, 653–659. [Google Scholar]
- Yamashita, H.; Abe, M.; Watanabe, K.; Shimizu, K.; Moriya, T.; Sato, A.; Satomi, S.; Ohta, H.; Sonoda, H.; Sato, Y. Vasohibin prevents arterial neointimal formation through angiogenesis inhibition. Biochem. Biophys. Res. Commun. 2006, 345, 919–925. [Google Scholar]
- Yoshinaga, K.; Ito, K.; Moriya, T.; Nagase, S.; Takano, T.; Niikura, H.; Yaegashi, N.; Sato, Y. Expression of vasohibin as a novel endothelium-derived angiogenesis inhibitor in endometrial cancer. Cancer Sci. 2008, 99, 914–919. [Google Scholar]
- Wakusawa, R.; Abe, T.; Sato, H.; Yoshida, M.; Kunikata, H.; Sato, Y.; Nishida, K. Expression of vasohibin, an antiangiogenic factor, in human choroidal neovascular membranes. Am. J. Ophthalmol. 2008, 146, 235–243. [Google Scholar]
- Tamaki, K.; Moriya, T.; Sato, Y.; Ishida, T.; Maruo, Y.; Yoshinaga, K.; Ohuchi, N.; Sasano, H. Vasohibin-1 in human breast carcinoma: A potential negative feedback regulator of angiogenesis. Cancer Sci. 2009, 100, 88–94. [Google Scholar]
- Sato, H.; Abe, T.; Wakusawa, R.; Asai, N.; Kunikata, H.; Ohta, H.; Sonoda, H.; Sato, Y.; Nishida, K. Vitreous levels of vasohibin-1 and vascular endothelial growth factor in patients with proliferative diabetic retinopathy. Diabetologia 2009, 52, 359–361. [Google Scholar]
- Sato, Y. Delta-like 4 and vasohibin 1: Two endothelium-produced negative regulators of angiogenesis with distinctive roles. Eur. Cytokine Netw. 2009, 20, 220–224. [Google Scholar]
- Hosaka, T.; Kimura, H.; Heishi, T.; Suzuki, Y.; Miyashita, H.; Ohta, H.; Sonoda, H.; Moriya, T.; Suzuki, S.; Kondo, T.; et al. Vasohibin-1 expression in endothelium of tumor blood vessels regulates angiogenesis. Am. J. Pathol. 2009, 175, 430–439. [Google Scholar]
- Miyake, K.; Nishida, K.; Kadota, Y.; Yamasaki, H.; Nasu, T.; Saitou, D.; Tanabe, K.; Sonoda, H.; Sato, Y.; Maeshima, Y.; et al. Inflammatory cytokine-induced expression of vasohibin-1 by rheumatoid synovial fibroblasts. Acta Med. Okayama 2009, 63, 349–358. [Google Scholar]
- Kern, J.; Steurer, M.; Gastl, G.; Gunsilius, E.; Untergasser, G. Vasohibin inhibits angiogenic sprouting in vitro and supports vascular maturation processes in vivo. BMC Cancer 2009, 9, 284. [Google Scholar]
- Heishi, T.; Hosaka, T.; Suzuki, Y.; Miyashita, H.; Oike, Y.; Takahashi, T.; Nakamura, T.; Arioka, S.; Mitsuda, Y.; Takakura, T.; et al. Endogenous angiogenesis inhibitor vasohibin1 exhibits broad-spectrum antilymphangiogenic activity and suppresses lymph node metastasis. Am. J. Pathol. 2010, 176, 1950–1958. [Google Scholar]
- Venancio, T.M.; DeMarco, R.; Almeida, G.T.; Oliveira, K.C.; Setubal, J.C.; Verjovski-Almeida, S. Analysis of schistosoma mansoni genes shared with deuterostomia and with possible roles in host interactions. BMC Genomics 2007, 8, 407. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boosani, C.S.; Sudhakar, Y.A. Proteolytically Derived Endogenous Angioinhibitors Originating from the Extracellular Matrix. Pharmaceuticals 2011, 4, 1551-1577. https://doi.org/10.3390/ph4121551
Boosani CS, Sudhakar YA. Proteolytically Derived Endogenous Angioinhibitors Originating from the Extracellular Matrix. Pharmaceuticals. 2011; 4(12):1551-1577. https://doi.org/10.3390/ph4121551
Chicago/Turabian StyleBoosani, Chandra Shekhar, and Yakkanti A. Sudhakar. 2011. "Proteolytically Derived Endogenous Angioinhibitors Originating from the Extracellular Matrix" Pharmaceuticals 4, no. 12: 1551-1577. https://doi.org/10.3390/ph4121551
APA StyleBoosani, C. S., & Sudhakar, Y. A. (2011). Proteolytically Derived Endogenous Angioinhibitors Originating from the Extracellular Matrix. Pharmaceuticals, 4(12), 1551-1577. https://doi.org/10.3390/ph4121551