Effect of Non-Steroidal Anti-Inflammatory Drugs on Bone Healing
Abstract
:1. Prostaglandins, Cyclooxygenases, and Bone Metabolism
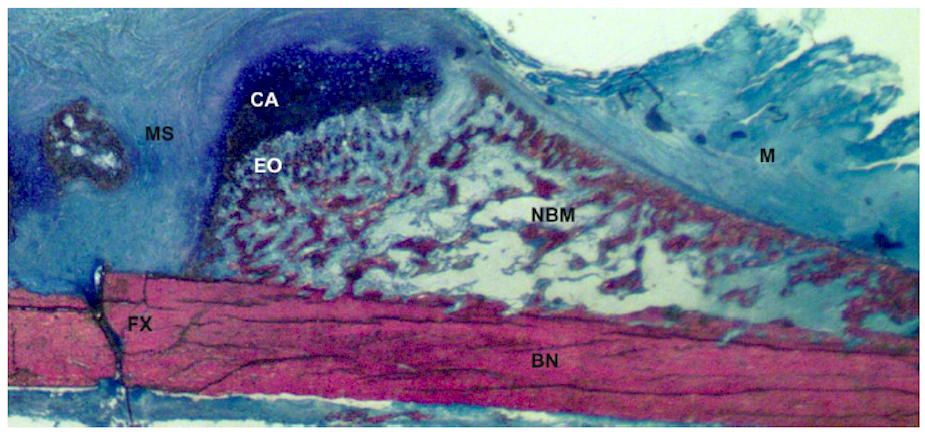
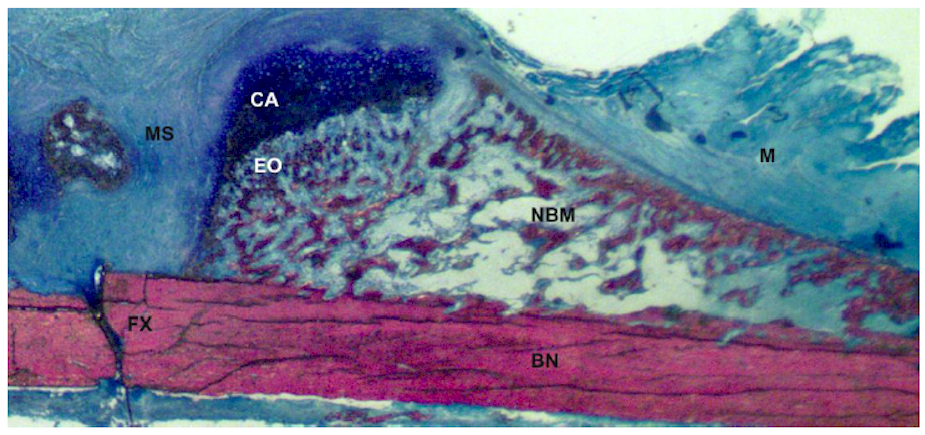
2. Types of Bone Healing
3. Cyclooxygenase Inhibitors
4. NSAID Effects on Experimental Models of Bone Healing
4.1. Effects of Traditional NSAIDs on Bone Healing

{kind=link}
| NSAID | Animal; Sex, Age, Weight | Bone Healing Model | Dose(s) mg/kg/day | Drug Administration (Post-Procedure) | Assay(s) | Longest Time Point | Outcome | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Aspirin | Rat, Male, 45 days | Closed, Unstable, Radius and ulna fractures | 100, 200, 300 | PO for 21 days | Histology | 22 days | Inhibition at highest dose | [23] | |
| Aspirin | Rabbit, N/A | Bone ingrowth, femur | 17, 34 | SQ injection | Histology | 8 weeks | Inhibition at high dose | Porous-coated chrome-cobalt implants; 4.5 mm diameter, 7 mm length | [38] |
| Celecoxib | Mice, 8–10 wks, ≈25 g | Closed, Stable, Tibia fracture | 10, 50 | In mice chow as a peanut butter pellet, PO daily till endpoint | Histology, mechanics | 12 weeks | No effect | [52] | |
| Celecoxib | Rat, Male, 6–9months, 584±62 g | Closed, Stable, Femur fracture | 4 | PO daily till endpoint | Histology, Radiography, Mechanics | 8 weeks | Inhibition | [25] | |
| Celecoxib | Rat, Male, 300 g | Closed, Stable, Femur fracture | 3 | Diet daily till endpoint | Radiography, histology mechanics | 12 weeks | No effect | Drug was given in chocolate | [51] |
| Celecoxib | Rat, Female, 281±20 g | Closed, Stable, Femur fracture | 3, 6 | PO daily till endpoint | Radiography, histology, mechanics | 8 weeks | Inhibition | [73] | |
| Celecoxib | Rat, Female, 272±7 g | Closed, Stable, Femur fracture | 2 , 4, 8 | 5-day before PO, PO daily till endpoint, 7 to 28 days daily, 14 to 28 days daily | Radiography, mechanics | 8 weeks | Inhibition, all doses, over time course, pre 5 day dose had no effect | [64] | |
| Celecoxib | Rat, Female, 250–300 g | Closed, Stable, Femur fracture | 4 | PO BID daily till endpoint | Radiography, histology, mechanics | 5 weeks | Inhibition | [98] | |
| Celecoxib | Rabbit, Male, 4.3–5.4 kg | Spinal fusion | 10 | PO daily till endpoint | Radiography, histology | 8 weeks | No effect | [41] | |
| Diclofenac | Rat, Male, 4–8 months 220–300 g | Closed, Stable, Tibia fracture | 1, 2 | PO daily for 10 days | Radiography, histology | 6 weeks | No effect | [60] | |
| Diclofenac | Rat, Male, 300–350 g | Open, Stable, Tibia fracture | 5 | PO daily for 7 or 21 days | Radiography, mechanics, CT scan | 3 weeks | Inhibition | [42] | |
| Diclofenac | Rat, Male, 30–350 g | Open, Stable, Tibia Osteotomy | 5 | PO daily for 7 days or 21 days | Histology | 3 weeks | 3 week dose inhibited callus maturation | [48] | |
| Etodolac | Rat, Female, 250–300 g | Closed, Stable, Femur fracture | 20 | PO daily till endpoint | Radiography, Mechanics | 3 weeks | Inhibition | [24] | |
| Etodolac | Rat, 12 wks, 250–300 g | Closed, Stable, Femur fracture | 20 | PO daily for 1 week, 3 weeks or during week 3 only | Radiography, mechanics | 3 weeks | Inhibition, when administered for 1 week or 3 weeks | [26] | |
| Flunixin | Rabbit, Female, 2.6–3.0 kg | Closed, Unstable, Tibia fracture | 1.1 | PO daily till endpoint | Mechanics, | 3 weeks | No effect | [58] | |
| Ibuprofen | Mice, Male, 8–10 wks, ≈25 g | Closed, Stable, Tibia fracture | 30 | In mice chow as a peanut butter pellet, PO daily till endpoint | Histology, mechanics | 12 weeks | No effect | [52] | |
| Ibuprofen | Rat, Male, 430–530 g | Closed, Unstable, Tibia fracture | 30–35 | Beginning 1 week prior to surgery and PO for 5 days a week | Callus size, calcium activity | 9 weeks | Ibuprofen activates calcium metabolism & decreases bone mass and composition | [31] | |
| Ibuprofen | Rat, Male, 430–530 g | Closed, Unstable, Tibia fracture | 30–35 | Beginning 1 week prior to surgery and PO for 5 days a week | Histology, mechanics | 9 weeks | Inhibition | [32] | |
| Ibuprofen | Rat, Male, 440–500 g | Closed, Stable, Tibia fracture | 30 | PO for 5 days/week, beginning 3 days post-fracture till endpoint | Histology, mechanics | 12 weeks | No effect | [59] | |
| Ibuprofen | Rat, Female, 375–450 g | Closed, Stable, Femur fracture | 30 | Diet for 4 or 12 weeks | Histology, mechanics | 12 weeks | Inhibition | [49] | |
| Ibuprofen | Rat, Male, 300 g | Closed, Stable, Femur fracture | 30 | PO daily till endpoint | Radiography, | 4 weeks | Inhibition | [45] | |
| Ibuprofen | Rabbit, Male & Female, 4 months, 2.1–3.5 kg | Open, Unstable, Femur osteotomy | 7.5 | PO daily till endpoint | Mechanics | 8 weeks | Inhibition | [34] | |
| Ibuprofen | Rabbit, N/A | Bone ingrowth, femur | 17, 34 | SQ injections | Histology | 8 weeks | Inhibition, both doses, with dose response | Porous-coated chrome-cobalt implants; 4.5 mm diameter, 7 mm length | [38] |
| Ibuprofen | Rabbit, Male, 3.5 kg | Open, Fibula, Osteoto my | 50 | PO three times daily for 28 days | Histology, mechanics | 12 weeks | Inhibition | [28] | |
| Indomethacin | Mice, Male, 8–10 wks, ≈25 g | Closed, Stable, Tibia fracture | 2 | In mice chow as a peanut butter pellet, PO daily till endpoint | Mechanics | 12 weeks | No effect | [52] | |
| Indomethacin | Rat, Male, 50 days, ≈160 g | Tooth extraction | 4 | PO, BID 2mg/kg/day for 5 days | Histology | 3 weeks | Inhibition | NSAID treated rats had delayed healing (1 week delay) | [29] |
| Indomethacin | Rat, Male, Adolesce nt | Closed, Unstable, Femur fracture | 2 | PO daily till endpoint | Radiography, histology, mechanics | 24 days | Inhibition | [20] | |
| Indomethacin | Rat, Male, 195±5 g | Closed, Unstable, Femur fracture | 2 | PO daily till endpoint | Callus weight, histology | 12 days | Indomethacin does not affect collagen metabolism | [55] | |
| Indomethacin | Rat, Male, 210–295 g | Closed, Unstable, Femur fracture | 2 | PO daily till endpoint | Radiography, manual assessment | 94 days | Inhibition | [22] | |
| Indomethacin | Rat, Male, 45 days | Closed, Unstable, | 1, 2, 4 | PO daily for 21 days | Histology | 22 days | Inhibition at all doses | [23] | |
| Indomethacin | Rat, Male, 2, 6–7, or 8–9 months | Fracture by drill hole in caudal vertebra | 4 | Beginning 1 week prior to surgery & daily till endpoint | Histology | 56 days | Inhibited | No effect if Rx was stopped day after lesion was induced | [33] |
| Indomethacin | Rat, Male, 4 wks | Drill hole in calvaria, 0.8mm | 2 | PO daily till 1, 2 or 4 weeks | Radiography, histology | 4 weeks | Inhibition | Dexamethasone was also tested and was found to inhibit bone wound healing more | [35] |
| Indomethacin | Rat, Male, 315–355 g or 329–479 g | Open, Stable, Femur fracture | 2 | 1 hr prior to surgery, PO daily for 3 days | Mechanics | 6 weeks | Inhibition | [107] | |
| Indomethacin | Rat, Male, 67–82 g or 52–64 g | Closed, Unstable, Femur fracture | 0.5, 2 | 2 mg/kg dose was given PO daily for 10 days; A single dose of 0.5 mg/kg was injected at fracture site | Radiography, | 20 days | Inhibition was found with oral and local treatment | Injection at fracture site was given in a poly-orthoester gel | [39] |
| Indomethacin | Rat, Female, 375–450 g | Closed, Stable, Femur fracture | 1 | Diet for 4 or 12 weeks | Histology, mechanics | 12 weeks | Inhibition | [49] | |
| Indomethacin | Rat, Male, 10–12 wks, 370–421 g | Spinal fusion | 3 | SQ injection, 6 days/week till endpoint | Manual assessment | 12 weeks | Inhibition | [79] | |
| Indomethacin | Rat, Female, 294±4 g | Mechanical loading | 0.02, 0.2, 2 | A single dose given 3 hours prior to loading | Histology | 12 days | Partially inhibited | [53] | |
| Indomethacin | Rat, Male, 335–345 g | Open, Stable, Femur osteotom y | 2 | IM for 3 days | Mechanics | 6 weeks | Inhibition | [54] | |
| Indomethacin | Rat, Male, 6–9 months, 584±62 g | Closed, Stable, Femur fracture | 1 | PO daily till endpoint | Radiography, histology, mechanics | 8 weeks | Inhibition | [25] | |
| Indomethacin | Rat, Male, ≈300 g | Closed, Stable, Femur fracture | 1 | PO daily till endpoint | Radiography, histology, mechanics | 12 weeks | No effect by 12 weeks | Indo delayed at 4 wks mechanically | [51] |
| Indomethacin | Rat, Female, ≈226 g | Closed, Stable, Tibia fracture | 0.625 | IP, prior to surgery, BID for 7 days | Radiography, histology, mechanics | 3 weeks | Inhibition | [50] | |
| Indomethacin | Rabbit, 4.5 months, 2.0–2.7 kg | Open, Unstable, Radius & ulna osteotomy | 10, 5 | Fed daily, 6 days a week; high dose for the first 2 weeks and then low dose for next 4 weeks | Radiography, histology | 43 days | Inhibition | [30] | |
| Indomethacin | Rabbit, Male & Female, 4 months, 2.1–3.5 kg | Open, Unstable, Femur osteotomy | 5 | PO daily till endpoint | Mechanics | 8 weeks | Inhibition | [34] | |
| Indomethacin | Rabbit, 4.3–6.0 kg | Open, Unstable, Tibia osteotomy | 50 | In drinking water, 4 days prior to surgery, PO daily till endpoint | Mechanics, bone mineral content | 6 weeks | Inhibition | [36] | |
| Indomethacin | Rabbit, Female, Juvenile | Open, Unstable, Femur osteotomy | 10 | SQ injections, | Radiography, | 6 weeks | Uncertain effect | [56] | |
| Indomethacin | Rabbit, N/A | Bone ingrowth, femur | 1, 2, 3 | SQ injections, daily | Histology | 8 weeks | Inhibition, all doses, with dose response | Porous-coated chrome-cobalt implants; 4.5 mm diameter, 7 mm length | [38] |
| Indomethacin | Rabbit, Male | Bone ingrowth, femur | 10 | SQ injections, daily | Histology | 8 weeks | Inhibition | Radially drilled, cylindrical implants | [37] |
| Indomethacin | Rabbit, 3.5 kg | Open, Unstable, Tibia osteotomy | 12.5 | In drinking water, 4 days prior to surgery, PO daily till endpoint | Histology | 6 weeks | No effect | [61] | |
| Indomethacin | Rabbit, Male, 4.3–5.4 kg | Spinal fusion | 10 | PO daily till endpoint | Radiography, histology | 8 weeks | Inhibition | [41] | |
| Indomethacin | Rabbit, 5 kg | Spinal fusion | 10 | Started 2 or 4 weeks after PO, daily till endpoint | Manual assessment | 6 weeks | Inhibition when treatment was started at 2 weeks PO | [44] | |
| Indomethacin | Rabbit, Male, 3 months, 3.5 kg | Open, Unstable, Ulna fracture | 2 | PO daily till endpoint | Histology, mechanics | 6 weeks | Inhibition | [46] | |
| Indomethacin | Dog, 12–21 kg | Open, Stable, Transsecti on of the 3rd metacarpu s | 5 | PO BID daily for 8 days | Radiography, | 8 weeks | No effect | [57] | |
| Ketorolac | Mice, Male, 8–10 wks. ≈25 g | Closed, Stable, Tibia fracture | 2 | In mice chow as a peanut butter pellet, length not mentioned | Histology, mechanics | 12 weeks | Inhibition at 4 weeks | [52] | |
| Ketorolac | Rat, Male, 335–345 g | Open, Stable, Femur osteotomy | 1 | IM for 3 days | Mechanics | 6 weeks | Inhibition | [54] | |
| Ketorolac | Rat, Male, 425–600 g | Closed, Stable, Femur fracture | 4 | PO daily till endpoint | Histology, mechanics, gene expression | 35 days | Inhibition | [72] | |
| Ketorolac | Rabbit, Male, 3.0 kg | 2 cm defect, Ulna | 2, 4 | PO daily till endpoint | Radiography, histology | 6 weeks | No effect with low dose, high dose inhibition detected between 2nd & 4th week | DBM was added in conjunction to ketorolac | [76] |
| Ketorolac | Rabbit, 4.0–4.5 kg | Spinal fusion | 4 | Continuous infusion (sq pump) for 7 days | Palpation | 6 weeks | Inhibition | 75%, 35%, and 100% fusion in the saline, ketorolac, and ketorolac plus BMP-2 groups, respectively | [71] |
| Meloxicam | Rabbit, Male, 3 month 3.5 kg | Open, Unstable, Ulna osteotomy | 0.3 | PO daily till endpoint | Histology, mechanics | 6 weeks | Inhibition | [46] | |
| Naproxen | Rabbit, Male, 6–12 months, 3.5–4.2 kg | Bone-ingrowth chamber | 110 | Water for 4 weeks | Histology | 4 weeks | Inhibition | Proximal tibia site | [40] |
| NS-398 | Rat, Female, 294±4 g | Mechanical loading | 0.02, 0.2, 2 | A single dose given 3 hours prior to loading | Histology | 12 days | Inhibition | [53] | |
| Parecoxib | Rat, Male, 425–600 g | Closed, Stable, Femur fracture | 0.3, 1.5 | PO daily till endpoint | Histology, mechanics, gene expression | 35 days | Inhibition | [72] | |
| Parecoxib | Rat, Female, ≈226 g | Closed, Stable, Tibia fracture | 0.5 | IP, prior to surgery, BID for 7 days | Radiography, histology, mechanics | 3 weeks | Inhibition | [50] | |
| Piroxicam | Rabbit, Female, 2.6–3.0 kg | Closed, Unstable, Tibia fracture | 0.1, 0.2 | PO daily till endpoint | Mechanics, | 3 weeks | No effect | [58] | |
| Rofecoxib | Mice , Male, 8–10wks. ≈25 g | Closed, Stable, Tibia fracture | 1, 5 | In mice chow as a peanut butter pellet, PO daily till endpoint | Histology, mechanics | 12 weeks | Inhibition at 8 weeks | [52] | |
| Rofecoxib | Mice, Male, 4 months | Open, Stable, Femur osteotomy | 5 | PO daily till endpoint | Radiography, histology, mechanics, laser doppler flow | 32 days | Inhibition, affects blow flow across fracture gap | [75] | |
| Rofecoxib | Rat, Male, 6–9 months, 584±62 g | Closed, Stable, Femur fracture | 3 | PO daily till endpoint | Radiography, histology, mechanics | 8 weeks | Inhibition | [25] | |
| Rofecoxib | Rat, Male, 300 g | Closed, Stable, Femur fracture | 8 | PO BID daily until endpoint | Radiography, | 4 weeks | Inhibition | [45] | |
| Rofecoxib | Rabbit, Male | Closed, Stable, Femur fracturer | 3 | PO daily for 4 weeks | Histology | 4 weeks | Inhibition | Proximal tibia | [40] |
| Rofecoxib | Rabbit, Male, 6–12 months, 3.5–4.2 kg | Bone-ingrowth chamber | 3 | PO daily for 2 weeks, 6 weeks or last 2 weeks | Histology | 6 weeks | Inhibition when administered for 6 weeks, no effect found when treatment was given for 2 weeks | [74] | |
| Rofecoxib | Rabbit, Male, 3 months, 3.5 kg | Open, Unstable, Ulna fracture | 0.5 | PO daily till endpoint | Histology, mechanics | 6 weeks | Inhibition | [46] | |
| Rofecoxib | Rabbit, Male, 3.5 kg | Open, Fibula Osteotomy | 50 | PO 3x daily for 28 days | Histology, | 12 weeks | Inhibition | [28] | |
| Tenoxicam | Rat, Male, ≈100 g | Open, Unstable, Tibia Fracture | 10 | 1 week prior to PO, PO or 48hrs after PO, than daily till endpoint, IM injections | Histology | 4 weeks | Inhibition | [43] |
4.2. Effects of COX-2 Selective Inhibitors on Bone Healing.
4.3. Effects of NSAIDs on Spinal Fusion
5. Human Studies of NSAID Effects on Bone Healing and Formation
| Procedure or Injury | Number of Patients | Mean or Median age (yrs) | Follow-up (months) | Drug | Dose(s) | Results | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Fracture, Ankle-joint fracture dislocation | Exp:1 | Exp:64 | 10 weeks | Indomethacin | 25 mg, QID for 9 weeks | Inhibitory unhealed at 9 weeks | [84] | |
| Fracture, colles | Exp: 48 Con:50 | Exp: 62.9 Con: 58.7 | 12 | Flurbiprofen | 50 mg, 3–6 times a day for 14 days | Exp: 50% excellent functional result Con: 94% excellent functional result | [86] | |
| Fracture, colles | Exp:21 Con:21 | 63 | 12 weeks | Piroxicam | 20 mg/day for 8 weeks | Exp: 28% needed surgery Con: none needed surgery | [85] | |
| Acetabular fracture | Exp: 41 Con:34 | Exp: 43 Con:47 | (Range) Exp:12 Con:11.7 | Indomethacin | 25 mg, 3x/day for 6 weeks | No difference between grade distribution | Prospective study | [82] |
| Acetabular fracture | Exp: 72 Con: 16 | Exp: 41 Con: | Exp: 13 Con: | Indomethacin | 25 mg, QID for 6 weeks | Exp: 11.1% grade iii or iv ho Con: 37.5% grade iii or iv ho | Control patient group is small, study was designed to compare radiation vs. NSAID therapy for reduction of ho | [83] |
| Acetabular fracture | Exp: 74 Con:36 | Exp: 39.5 Con: 38.6 | 3–11 | Indomethacin | 25 mg, QID for 6 weeks | Inhibitory Exp: 29% non-union Con: 7% non-union | Also compared radiation | [88] |
| Fracture | Exp: 893 Con: 5781 | Exp: 74 Con:74 | 24 | NSAIDs | 5–7 times a week | No protective effect on subsequent risk of fractures | BMD & fracture risk assessment | [90] |
| Fracture | Exp: 32 Con:67 | Exp: 35 Con: 38 | 7+ | Ibuprofen or diclofenac | Varied, 1–21 weeks | Inhibitory Exp: 10.74 odds ratio | Retrospective study | [87] |
| Fracture | Exp1: 214,577 Exp2: 286,850 Con: 214,577 | Exp1: 55 Exp2:44 Con:55 | Exp1: 3.4 yrs Exp2: .7 years Con: 2.3 yrs | NSAIDs | Exp1: 3+ times per week Exp1: 1–2 times per week | Exp1: 1.47 Exp1 vs. Exp2: 1.04 | Retrospective study, fracture risk is similar between regular and incidental NSAID users | [91] |
| Fracture, humeral shaft | Exp: 1,032 Con:8,963 | Exp: 78 Con:77 | 3 | Traditional NSAID | Varied, at least 10 days of treatment in a 30 day period | Exposure to NSAID was associated with nonunion | Retrospective study | [89] |
| Acetabular fracture | Exp: 18 Con: 26 | Exp: 41 Con: 37 | 12+ | Indomethacin | 25 mg QID for 6 weeks | Exp: 1/18 grade ii ho, 0/18 grade iii or iv Con: 3/26 grade ii ho, 10/26 grade iii or iv ho | Heterotrophic bone ossification study | [80] |
| Spinal fusion | Exp: 167 Con: 121 | Exp: 43 Con: 45 | 24+ | Ketorolac | 60 mg IM, then 30 mg/IM very 6–8 h as needed | Inhibitory | Retrospective study; 4.25-fold increase in non-unions | [92] |
| Exp: 17% non-union Con: 4% non-union |
6. Role of COX-2 during Bone Healing
7. Summary
References
- O'Connor, J.P.; Lysz, T. Celecoxib, NSAIDs and the skeleton. Drugs 2008, 44, 693–709. [Google Scholar]
- Radi, Z.A.; Khan, N.K. Effects of cyclooxygenase inhibition on bone, tendon, and ligament healing. Inflamm. Res. 2005, 54, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Kolar, P.; Schmidt-Bleek, K.; Schell, H.; Gaber, T.; Toben, D.; Schmidmaier, G.; Perka, C.; Buttgereit, F.; Duda, G.N. The early fracture hematoma and its potential role in fracture healing. Tissue Eng. 2010. [Epub ahead of print]. [Google Scholar]
- Andrew, J.G.; Andrew, S.M.; Freemont, A.J.; Marsh, D.R. Inflammatory cells in normal human fracture healing. Acta Orthop. 1994, 65, 462–466. [Google Scholar]
- Oni, O.O. The early stages of the repair of adult human diaphyseal fractures. Injury 1997, 28, 521–525. [Google Scholar]
- Einhorn, T.A. The cell and molecular biology of fracture healing. Clin. Orthop. Relat. Res. 1998, 355, S7–S21. [Google Scholar]
- Greenbaum, M.A.; Kanat, I.O. Current concepts in bone healing. Review of the literature. J. Am. Podiatr. Med. Assoc. 1993, 83, 123–129. [Google Scholar] [PubMed]
- Glassman, S.D.; Carreon, L.; Djurasovic, M.; Campbell, M.J.; Puno, R.M.; Johnson, J.R.; Dimar, J.R. Posterolateral lumbar spine fusion with INFUSE bone graft. Spine J. 2007, 7, 44–49. [Google Scholar]
- Langford, R.M.; Mehta, V. Selective cyclooxygenase inhibition: Its role in pain and anaesthesia. Biomed. Pharmacother. 2006, 60, 323–328. [Google Scholar]
- Gonzalez, E.L.; Patrignani, P.; Tacconelli, S.; Rodriguez, L.A. Variability of risk of upper gastrointestinal bleeding among nonsteroidal anti-inflammatory drugs. Arthritis Rheum. 2010. [Epub ahead of print]. [Google Scholar]
- Lanas, A. A review of the gastrointestinal safety data—A gastroenterologist's perspective. Rheumatology 2010, 49 (Suppl. 2), ii3–ii10. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Levy, D.B.; Yannoni, Y.; Erikson, R.L. Identification of a phorbol ester-repressible v-src-inducible gene. Proc. Natl. Acad. Sci. USA 1989, 86, 1178–1182. [Google Scholar]
- O'Banion, M.K.; Sadowski, H.B.; Winn, V.; Young, D.A. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J. Biol. Chem. 1991, 266, 23261–23267. [Google Scholar]
- Fletcher, B.S.; Kujubu, D.A.; Perrin, D.M.; Herschman, H.R. Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J. Biol. Chem. 1992, 267, 4338–4344. [Google Scholar]
- Salvemini, D.; Misko, T.P.; Masferrer, J.L.; Seibert, K.; Currie, M.G.; Needleman, P. Nitric oxide activates cyclooxygenase enzymes. Proc. Natl. Acad. Sci. USA 1993, 90, 7240–7244. [Google Scholar]
- Zhang, Y.; Shaffer, A.; Portanova, J.; Seibert, K.; Isakson, P.C. Inhibition of cyclooxygenase-2 rapidly reverses inflammatory hyperalgesia and prostaglandin E2 production. J. Pharmacol. Exp. Ther. 1997, 283, 1069–1075. [Google Scholar]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar]
- FitzGerald, G.A.; Patrono, C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 2001, 345, 433–442. [Google Scholar]
- Fitzgerald, G.A. Coxibs and cardiovascular disease. N. Engl. J. Med. 2004, 351, 1709–1711. [Google Scholar]
- Ro, J.; Sudmann, E.; Marton, P.F. Effect of indomethacin on fracture healing in rats. Acta Orthop. 1976, 47, 588–599. [Google Scholar]
- Baratieri, A.; Deli, R. The effect on bone repair of aspirin cones placed in extraction sockets in dogs: A histopathologic study. J. Oral Pathol. 1979, 8, 198–206. [Google Scholar]
- Sudmann, E.; Dregelid, E.; Bessesen, A.; Morland, J. Inhibition of fracture healing by indomethacin in rats. Eur. J. Clin. Invest. 1979, 9, 333–339. [Google Scholar]
- Allen, H.L.; Wase, A.; Bear, W.T. Indomethacin and aspirin: Effect of nonsteroidal anti-inflammatory agents on the rate of fracture repair in the rat. Acta Orthop. 1980, 51, 595–600. [Google Scholar]
- Endo, K.; Sairyo, K.; Kornatsurbara, S.; Sasa, T.; Egawa, H.; Yonekura, D.; Adachi, K.; Ogawa, T. Cyclooxygenase-2 Inhibitor Inhibits the Fracture Healing. J. Physiol. Anthropol. Appl. Hum. Sci. 2002, 21, 235–238. [Google Scholar]
- Simon, A.M.; Manigrasso, M.B.; O'Connor, J.P. Cyclo-oxygenase 2 function is essential for bone fracture healing. J. Bone Miner. Res. 2002, 17, 963–976. [Google Scholar]
- Endo, K.; Sairyo, K.; Komatsubara, S.; Sasa, T.; Egawa, H.; Ogawa, T.; Yonekura, D.; Murakami, R.; Yasui, N. Cyclooxygenase-2 inhibitor delays fracture healing in rats. Acta Orthop. 2005, 76, 470–474. [Google Scholar]
- Dimmen, S.; Engebretsen, L.; Nordsletten, L.; Madsen, J.E. Negative effects of parecoxib and indomethacin on tendon healing: An experimental study in rats. Knee Surg. Sports Traumatol. Arthrosc. 2009, 17, 835–839. [Google Scholar]
- O'Connor, J.P.; Capo, J.T.; Tan, V.; Cottrell, J.A.; Manigrasso, M.B.; Bontempo, N.; Parsons, J.R. A comparison of the effects of ibuprofen and rofecoxib on rabbit fibula osteotomy healing. Acta Orthop. 2009, 80, 597–605. [Google Scholar]
- Huusko, P.J.; Nieminen, L.H.; Nieminen, L.S. The effect of indomethacin on tooth extraction wound healing in rats. Experientia 1975, 31, 1056–1058. [Google Scholar]
- Sudmann, E.; Bang, G. Indomethacin-induced inhibition of haversian remodelling in rabbits. Acta Orthop. 1979, 50, 621–627. [Google Scholar]
- Tornkvist, H.; Lindholm, T.S. Effect of ibuprofen on mass and composition of fracture callus and bone—An experimental study on adult rat. Scand. J. Rheumatol. 1980, 9, 167–171. [Google Scholar]
- Lindholm, T.S.; Tornkvist, H. Inhibitory effect on bone formation and calcification exerted by the anti-inflammatory drug ibuprofen—An experimental study on adult rat with fracture. Scand. J. Rheumatol. 1981, 10, 38–42. [Google Scholar]
- Elves, M.W.; Bayley, I.; Roylance, P.J. The effect of indomethacin upon experimental fractures in the rat. Acta Orthop. Scand. 1982, 53, 35–41. [Google Scholar]
- Tornkvist, H.; Lindholm, T.S.; Netz, P.; Stromberg, L.; Lindholm, T.C. Effect of ibuprofen and indomethacin on bone metabolism reflected in bone strength. Clin. Orthop. Relat. Res. 1984, 187, 255–259. [Google Scholar]
- Sato, S.; Kim, T.; Arai, T.; Maruyama, S.; Tajima, M.; Utsumi, N. Comparison between the effects of dexamethasone and indomethacin on bone wound healing. Jpn. J. Pharmacol. 1986, 42, 71–78. [Google Scholar]
- Keller, J.; Bunger, C.; Andreassen, T.T.; Bak, B.; Lucht, U. Bone repair inhibited by indomethacin. Effects on bone metabolism and strength of rabbit osteotomies. Acta Orthop. 1987, 58, 379–383. [Google Scholar]
- Keller, J.C.; Trancik, T.M.; Young, F.A.; St Mary, E. Effects of indomethacin on bone ingrowth. J. Orthop. Res. 1989, 7, 28–34. [Google Scholar]
- Trancik, T.; Mills, W.; Vinson, N. The effect of indomethacin, aspirin, and ibuprofen on bone ingrowth into a porous-coated implant. Clin. Orthop. Relat. Res. 1989, 249, 113–121. [Google Scholar] [PubMed]
- Engesaeter, L.B.; Sudmann, B.; Sudmann, E. Fracture healing in rats inhibited by locally administered indomethacin. Acta Orthop. Scand. 1992, 63, 330–333. [Google Scholar]
- Goodman, S.; Ma, T.; Trindade, M.; Ikenoue, T.; Matsuura, I.; Wong, N.; Fox, N.; Genovese, M.; Regula, D.; Smith, R.L. COX-2 selective NSAID decreases bone ingrowth in vivo. J. Orthop. Res. 2002, 20, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Lewis, S.; Kuklo, T.; Zhu, Y.; Riew, K.D. The effect of cyclooxygenase-2 inhibitors on spinal fusion. J. Bone Joint Surg. Am. 2002, 84A, 1763–1768. [Google Scholar]
- Beck, A.; Krischak, G.; Sorg, T.; Augat, P.; Farker, K.; Merkel, U.; Kinzl, L.; Claes, L. Influence of diclofenac (group of nonsteroidal anti-inflammatory drugs) on fracture healing. Arch. Orthop. Trauma Surg. 2003, 123, 327–332. [Google Scholar]
- Giordano, V.; Giordano, M.; Knackfuss, I.G.; Apfel, M.I.; Gomes, R.D. Effect of tenoxicam on fracture healing in rat tibiae. Injury 2003, 34, 85–94. [Google Scholar]
- Riew, K.D.; Long, J.; Rhee, J.; Lewis, S.; Kuklo, T.; Kim, Y.J.; Yukawa, Y.; Zhu, Y. Time-dependent inhibitory effects of indomethacin on spinal fusion. J. Bone Joint Surg. Am. 2003, 85A, 632–634. [Google Scholar]
- Leonelli, S.M.; Goldberg, B.A.; Safanda, J.; Bagwe, M.R.; Sethuratnam, S.; King, S.J. Effects of a cyclooxygenase-2 inhibitor (rofecoxib) on bone healing. Am. J. Orthop. 2006, 35, 79–84. [Google Scholar]
- Karachalios, T.; Boursinos, L.; Poultsides, L.; Khaldi, L.; Malizos, K.N. The effects of the short-term administration of low therapeutic doses of anti-COX-2 agents on the healing of fractures. An experimental study in rabbits. J. Bone Joint Surg. Br. 2007, 89, 1253–1260. [Google Scholar] [PubMed]
- Krischak, G.D.; Augat, P.; Blakytny, R.; Claes, L.; Kinzl, L.; Beck, A. The non-steroidal anti-inflammatory drug diclofenac reduces appearance of osteoblasts in bone defect healing in rats. Arch. Orthop. Trauma Surg. 2007, 127, 453–458. [Google Scholar]
- Krischak, G.D.; Augat, P.; Sorg, T.; Blakytny, R.; Kinzl, L.; Claes, L.; Beck, A. Effects of diclofenac on periosteal callus maturation in osteotomy healing in an animal model. Arch. Orthop. Trauma Surg. 2007, 127, 3–9. [Google Scholar]
- Altman, R.D.; Latta, L.L.; Keer, R.; Renfree, K.; Hornicek, F.J.; Banovac, K. Effect of nonsteroidal antiinflammatory drugs on fracture healing: A laboratory study in rats. J. Orthop. Trauma 1995, 9, 392–400. [Google Scholar]
- Dimmen, S.; Nordsletten, L.; Madsen, J.E. Parecoxib and indomethacin delay early fracture healing: A study in rats. Clin. Orthop. Relat. Res. 2009, 467, 1992–1999. [Google Scholar]
- Brown, K.M.; Saunders, M.M.; Kirsch, T.; Donahue, H.J.; Reid, J.S. Effect of COX-2-specific inhibition on fracture-healing in the rat femur. J. Bone Joint Surg. Am. 2004, 86A, 116–123. [Google Scholar]
- Mullis, B.H.; Copland, S.T.; Weinhold, P.S.; Miclau, T.; Lester, G.E.; Bos, G.D. Effect of COX-2 inhibitors and non-steroidal anti-inflammatory drugs on a mouse fracture model. Injury 2006, 37, 827–837. [Google Scholar]
- Forwood, M.R. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J. Bone Miner. Res. 1996, 11, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Reikeraas, O.; Engebretsen, L. Effects of ketoralac tromethamine and indomethacin on primary and secondary bone healing—An experimental study in rats. Arch. Orthop. Trauma Surg. 1998, 118, 50–52. [Google Scholar]
- Ro, J.; Langeland, N.; Sander, J. Effect of indomethacin on collagen metabolism of rat fracture callus in vitro. Acta Orthop. 1978, 49, 323–328. [Google Scholar] [CrossRef]
- Shindell, R.; Lippiello, L.; Connolly, J.F. Uncertain effect of indomethacin on physeal growth injury—Experiments in rabbits. Acta Orthop. 1988, 59, 46–49. [Google Scholar]
- Mbugua, S.W.; Skoglund, L.A.; Lokken, P. Effects of phenylbutazone and indomethacin on the post-operative course following experimental orthopaedic surgery in dogs. Acta Vet Scand. 1989, 30, 27–35. [Google Scholar]
- More, R.C.; Kody, M.H.; Kabo, J.M.; Dorey, F.J.; Meals, R.A. The effects of two nonsteroidal antiinflammatory drugs on limb swelling, joint stiffness, and bone torsional strength following fracture in a rabbit model. Clin. Orthop. Relat. Res. 1989, 247, 306–312. [Google Scholar] [PubMed]
- Huo, M.H.; Troiano, N.W.; Pelker, R.R.; Gundberg, C.M.; Friedlaender, G.E. The influence of ibuprofen on fracture repair: Biomechanical, biochemical, histologic, and histomorphometric parameters in rats. J. Orthop. Res. 1991, 9, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Akman, S.; Gogus, A.; Sener, N.; Bilgic, B.; Aksoy, B.; Seckin, F. Effect of diclofenac sodium on union of tibial fractures in rats. Adv. Ther. 2002, 19, 119–125. [Google Scholar]
- Keller, J.; Kjaersgaard-Andersen, P.; Bayer-Kristensen, I.; Melsen, F. Indomethacin and bone trauma. Effects on remodeling of rabbit bone. Acta Orthop. 1990, 61, 66–69. [Google Scholar]
- Blackwell, K.A.; Raisz, L.G.; Pilbeam, C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010, 21, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Dekel, S.; Lenthall, G.; Francis, M.J. Release of prostaglandins from bone and muscle after tibial fracture—An experimental study in rabbits. J. Bone Joint Surg. Br. 1981, 63B, 185–189. [Google Scholar]
- Simon, A.M.; O'Connor, J.P. Dose and time-dependent effects of cyclooxygenase-2 inhibition on fracture-healing. J. Bone Joint Surg. Am. 2007, 89, 500–511. [Google Scholar]
- Perazella, M.A.; Buller, G.K. NSAID nephrotoxicity revisited: Acute renal failure due to parenteral ketorolac. South Med. J. 1993, 86, 1421–1424. [Google Scholar]
- Roth, S.H.; Tindall, E.A.; Jain, A.K.; McMahon, F.G.; April, P.A.; Bockow, B.I.; Cohen, S.B.; Fleischmann, R.M. A controlled study comparing the effects of nabumetone, ibuprofen, and ibuprofen plus misoprostol on the upper gastrointestinal tract mucosa. Arch. Intern. Med. 1993, 153, 2565–2571. [Google Scholar] [PubMed]
- Splinter, W.M.; Rhine, E.J.; Roberts, D.W.; Reid, C.W.; MacNeill, H.B. Preoperative ketorolac increases bleeding after tonsillectomy in children. Can. J. Anaesth. 1996, 43, 560–563. [Google Scholar]
- Langman, M.J.; Weil, J.; Wainwright, P.; Lawson, D.H.; Rawlins, M.D.; Logan, R.F.; Murphy, M.; Vessey, M.P.; Colin-Jones, D.G. Risks of bleeding peptic ulcer associated with individual non-steroidal anti-inflammatory drugs. Lancet 1994, 343, 1075–1078. [Google Scholar]
- Lanas, A.; Perez-Aisa, M.A.; Feu, F.; Ponce, J.; Saperas, E.; Santolaria, S.; Rodrigo, L.; Balanzo, J.; Bajador, E.; Almela, P.; Navarro, J.M.; Carballo, F.; Castro, M.; Quintero, E. A nationwide study of mortality associated with hospital admission due to severe gastrointestinal events and those associated with nonsteroidal antiinflammatory drug use. Am. J. Gastroenterol. 2005, 100, 1685–1693. [Google Scholar]
- Dubois, R.W.; Melmed, G.Y.; Henning, J.M.; Bernal, M. Risk of Upper Gastrointestinal Injury and Events in Patients Treated With Cyclooxygenase (COX)-1/COX-2 Nonsteroidal Antiinflammatory Drugs (NSAIDs), COX-2 Selective NSAIDs, and Gastroprotective Cotherapy: An Appraisal of the Literature. J. Clin. Rheumatol. 2004, 10, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.J., Jr.; Boden, S.D.; Titus, L. Recombinant human bone morphogenetic protein-2 overcomes the inhibitory effect of ketorolac, a nonsteroidal anti-inflammatory drug (NSAID), on posterolateral lumbar intertransverse process spine fusion. Spine 1999, 24, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Gerstenfeld, L.C.; Thiede, M.; Seibert, K.; Mielke, C.; Phippard, D.; Svagr, B.; Cullinane, D.; Einhorn, T.A. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J. Orthop. Res. 2003, 21, 670–675. [Google Scholar]
- Bergenstock, M.; Min, W.; Simon, A.M.; Sabatino, C.; O'Connor, J.P. A comparison between the effects of acetaminophen and celecoxib on bone fracture healing in rats. J. Orthop. Trauma 2005, 19, 717–723. [Google Scholar]
- Goodman, S.B.; Ma, T.; Mitsunaga, L.; Miyanishi, K.; Genovese, M.C.; Smith, R.L. Temporal effects of a COX-2-selective NSAID on bone ingrowth. J. Biomed. Mater. Res. 2005, 72, 279–287. [Google Scholar]
- Murnaghan, M.; Li, G.; Marsh, D.R. Nonsteroidal anti-inflammatory drug-induced fracture nonunion: An inhibition of angiogenesis? J. Bone Joint Surg. Am. 2006, 88 Suppl. 3, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.L.; Chang, J.K.; Wang, G.J. Effects of ketorolac on bone repair: A radiographic study in modeled demineralized bone matrix grafted rabbits. Pharmacology 1998, 57, 148–159. [Google Scholar]
- Paulson, S.K.; Zhang, J.Y.; Breau, A.P.; Hribar, J.D.; Liu, N.W.; Jessen, S.M.; Lawal, Y.M.; Cogburn, J.N.; Gresk, C.J.; Markos, C.S.; Maziasz, T.J.; Schoenhard, G.L.; Burton, E.G. Pharmacokinetics, tissue distribution, metabolism, and excretion of celecoxib in rats. Drug Metab. Dispos. 2000, 28, 514–521. [Google Scholar] [PubMed]
- Virchenko, O.; Skoglund, B.; Aspenberg, P. Parecoxib impairs early tendon repair but improves later remodeling. Am. J. Sports Med. 2004, 32, 1743–1747. [Google Scholar]
- Dimar, J.R., 2nd.; Ante, W.A.; Zhang, Y.P.; Glassman, S.D. The effects of nonsteroidal anti-inflammatory drugs on posterior spinal fusions in the rat. Spine 1996, 21, 1870–1876. [Google Scholar] [PubMed]
- McLaren, A.C. Prophylaxis with indomethacin for heterotopic bone—After open reduction of fractures of the acetabulum. J. Bone Joint Surg. Am. 1990, 72, 245–247. [Google Scholar]
- Gebuhr, P.; Wilbek, H.; Soelberg, M. Naproxen for 8 days can prevent heterotopic ossification after hip arthroplasty. Clin. Orthop. Relat. Res. 1995, 166–169. [Google Scholar]
- Moore, K.D.; Goss, K.; Anglen, J.O. Indomethacin versus radiation therapy for prophylaxis against heterotopic ossification in acetabular fractures: A randomised, prospective study. J. Bone Joint Surg. Br. 1998, 80, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Burd, T.A.; Lowry, K.J.; Anglen, J.O. Indomethacin compared with localized irradiation for the prevention of heterotopic ossification following surgical treatment of acetabular fractures. J. Bone Joint Surg. Am. 2001, 83A, 1783–1788. [Google Scholar]
- Sudmann, E.; Hagen, T. Indomethacin-induced delayed fracture healing. Arch. Orthop. Traum Surg. 1976, 85, 151–154. [Google Scholar]
- Adolphson, P.; Abbaszadegan, H.; Jonsson, U.; Dalen, N.; Sjoberg, H.E.; Kalen, S. No effects of piroxicam on osteopenia and recovery after Colles' fracture—A randomized, double-blind, placebo-controlled, prospective trial. Arch. Orthop. Trauma Surg. 1993, 112, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.R.; Ackroyd, C.E. Non-steroidal anti-inflammatory agents in the management of Colles' fractures. Br. J.Clin. Pract. 1988, 42, 184–189. [Google Scholar]
- Giannoudis, P.V.; MacDonald, D.A.; Matthews, S.J.; Smith, R.M.; Furlong, A.J.; De Boer, P. Nonunion of the femoral diaphysis—The influence of reaming and non-steroidal anti-inflammatory drugs. J. Bone Joint Surg. Br. 2000, 82, 655–658. [Google Scholar]
- Burd, T.A.; Hughes, M.S.; Anglen, J.O. Heterotopic ossification prophylaxis with indomethacin increases the risk of long-bone nonunion. J. Bone Joint Surg. Br. 2003, 85B, 700–705. [Google Scholar]
- Bhattacharyya, T.; Levin, R.; Vrahas, M.S.; Solomon, D.H. Nonsteroidal antiinflammatory drugs and nonunion of humeral shaft fractures. Arthritis Rheum. 2005, 53, 364–367. [Google Scholar]
- Bauer, D.C.; Orwoll, E.S.; Fox, K.M.; Vogt, T.M.; Lane, N.E.; Hochberg, M.C.; Stone, K.; Nevitt, M.C. Aspirin and NSAID use in older women: effect on bone mineral density and fracture risk—Study of Osteoporotic Fractures Research Group. J. Bone Miner. Res. 1996, 11, 29–35. [Google Scholar]
- van Staa, T.P.; Leufkens, H.G.; Cooper, C. Use of nonsteroidal anti-inflammatory drugs and risk of fractures. Bone 2000, 27, 563–568. [Google Scholar]
- Glassman, S.D.; Rose, S.M.; Dimar, J.R.; Puno, R.M.; Campbell, M.J.; Johnson, J.R. The effect of postoperative nonsteroidal anti-inflammatory drug administration on spinal fusion. Spine 1998, 23, 834–838. [Google Scholar]
- Zhang, X.; Schwarz, E.M.; Young, D.A.; Puzas, J.E.; Rosier, R.N.; O'Keefe, R.J. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Invest. 2002, 109, 1405–1415. [Google Scholar]
- Chikazu, D.; Li, X.; Kawaguchi, H.; Sakuma, Y.; Voznesensky, O.S.; Adams, D.J.; Xu, M.; Hoshio, K.; Katavic, V.; Herschman, H.R.; Raisz, L.G.; Pilbeam, C.C. Bone morphogenetic protein 2 induces cyclo-oxygenase 2 in osteoblasts via a Cbfal binding site: Role in effects of bone morphogenetic protein 2 in vitro and in vivo. J. Bone Miner. Res. 2002, 17, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.A.; Meyenhofer, M.; Medicherla, S.; Higgins, L.; O'Connor, J.P. Analgesic effects of p38 kinase inhibitor treatment on bone fracture healing. Pain 2009, 142, 116–126. [Google Scholar]
- Einhorn, T.A.; Majeska, R.J.; Rush, E.B.; Levine, P.M.; Horowitz, M.C. The expression of cytokine activity by fracture callus. J. Bone Miner. Res. 1995, 10, 1272–1281. [Google Scholar]
- Hausman, M.R.; Schaffler, M.B.; Majeska, R.J. Prevention of fracture healing in rats by an inhibitor of angiogenesis. Bone 2001, 29, 560–564. [Google Scholar]
- Katoh, H.; Hosono, K.; Ito, Y.; Suzuki, T.; Ogawa, Y.; Kubo, H.; Kamata, H.; Mishima, T.; Tamaki, H.; Sakagami, H.; Sugimoto, Y.; Narumiya, S.; Watanabe, M.; Majima, M. COX-2 and prostaglandin EP3/EP4 signaling regulate the tumor stromal proangiogenic microenvironment via CXCL12-CXCR4 chemokine systems. Am. J. Pathol. 2010, 176, 1469–1483. [Google Scholar]
- Xie, C.; Liang, B.; Xue, M.; Lin, A.S.; Loiselle, A.; Schwarz, E.M.; Guldberg, R.E.; O'Keefe, R.J.; Zhang, X. Rescue of impaired fracture healing in COX-2-/- mice via activation of prostaglandin E2 receptor subtype 4. Am. J. Pathol. 2009, 175, 772–785. [Google Scholar]
- Cottrell, J.A.; O'Connor, J.P. Pharmacological inhibition of 5-lipoxygenase accelerates and enhances fracture-healing. J. Bone Joint Surg. Am. 2009, 91, 2653–2665. [Google Scholar]
- Ren, W.; Dziak, R. Effects of leukotrienes on osteoblastic cell proliferation. Calcified Tissue Int. 1991, 49, 197–201. [Google Scholar]
- Gallwitz, W.E.; Mundy, G.R.; Lee, C.H.; Qiao, M.; Roodman, G.D.; Raftery, M.; Gaskell, S.J.; Bonewald, L.F. 5-Lipoxygenase metabolites of arachidonic acid stimulate isolated osteoclasts to resorb calcified matrices. J. Biol. Chem. 1993, 268, 10087–10094. [Google Scholar]
- Garcia, C.; Boyce, B.F.; Gilles, J.; Dallas, M.; Qiao, M.; Mundy, G.R.; Bonewald, L.F. Leukotriene B4 stimulates osteoclastic bone resorption both in vitro and in vivo. J. Bone Miner. Res. 1996, 11, 1619–1627. [Google Scholar] [PubMed]
- Bonewald, L.F.; Flynn, M.; Qiao, M.; Dallas, M.R.; Mundy, G.R.; Boyce, B.F. Mice lacking 5-lipoxygenase have increased cortical bone thickness. Adv. Exp. Med. Biol. 1997, 433, 299–302. [Google Scholar]
- Traianedes, K.; Dallas, M.R.; Garrett, I.R.; Mundy, G.R.; Bonewald, L.F. 5-Lipoxygenase metabolites inhibit bone formation in vitro. Endocrinology 1998, 139, 3178–3184. [Google Scholar] [PubMed]
- Maxis, K.; Delalandre, A.; Martel-Pelletier, J.; Pelletier, J.P.; Duval, N.; Lajeunesse, D. The shunt from the cyclooxygenase to lipoxygenase pathway in human osteoarthritic subchondral osteoblasts is linked with a variable expression of the 5-lipoxygenase-activating protein. Arthritis Res. Ther. 2006, 8, R181. [Google Scholar]
- Hogevold, H.E.; Grogaard, B.; Reikeras, O. Effects of short-term treatment with corticosteroids and indomethacin on bone healing—A mechanical study of osteotomies in rats. Acta Orthop. 1992, 63, 607–611. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cottrell, J.; O’Connor, J.P. Effect of Non-Steroidal Anti-Inflammatory Drugs on Bone Healing. Pharmaceuticals 2010, 3, 1668-1693. https://doi.org/10.3390/ph3051668
Cottrell J, O’Connor JP. Effect of Non-Steroidal Anti-Inflammatory Drugs on Bone Healing. Pharmaceuticals. 2010; 3(5):1668-1693. https://doi.org/10.3390/ph3051668
Chicago/Turabian StyleCottrell, Jessica, and J. Patrick O’Connor. 2010. "Effect of Non-Steroidal Anti-Inflammatory Drugs on Bone Healing" Pharmaceuticals 3, no. 5: 1668-1693. https://doi.org/10.3390/ph3051668
APA StyleCottrell, J., & O’Connor, J. P. (2010). Effect of Non-Steroidal Anti-Inflammatory Drugs on Bone Healing. Pharmaceuticals, 3(5), 1668-1693. https://doi.org/10.3390/ph3051668