Building Cell Selectivity into CPP-Mediated Strategies


Abstract
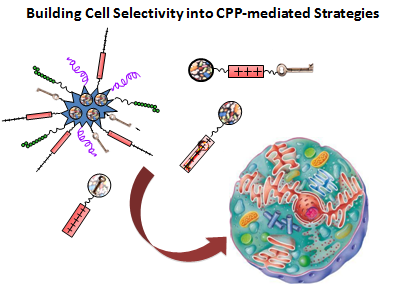
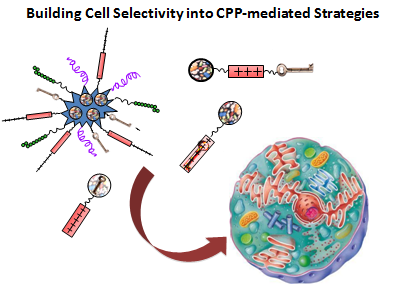
:
1. Introduction
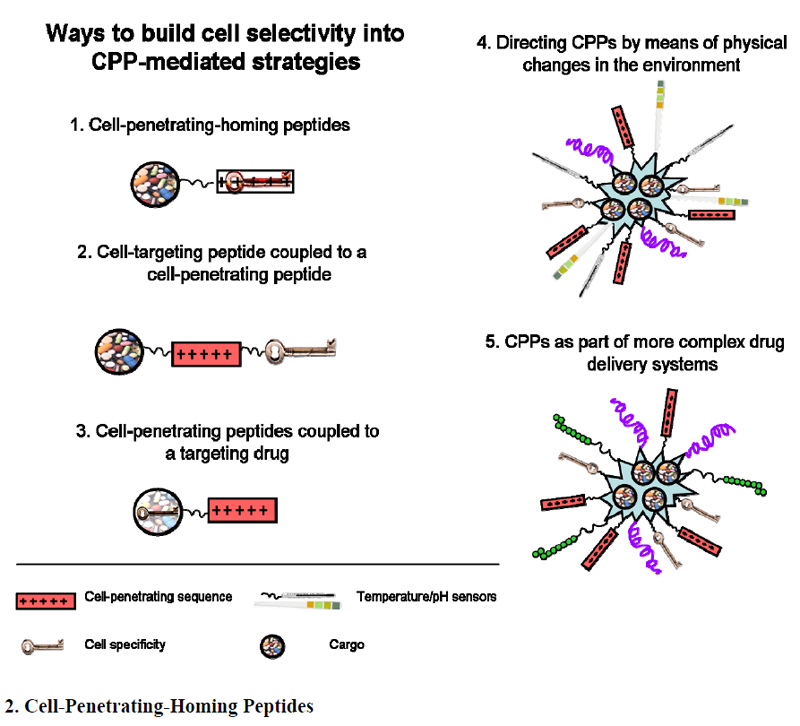
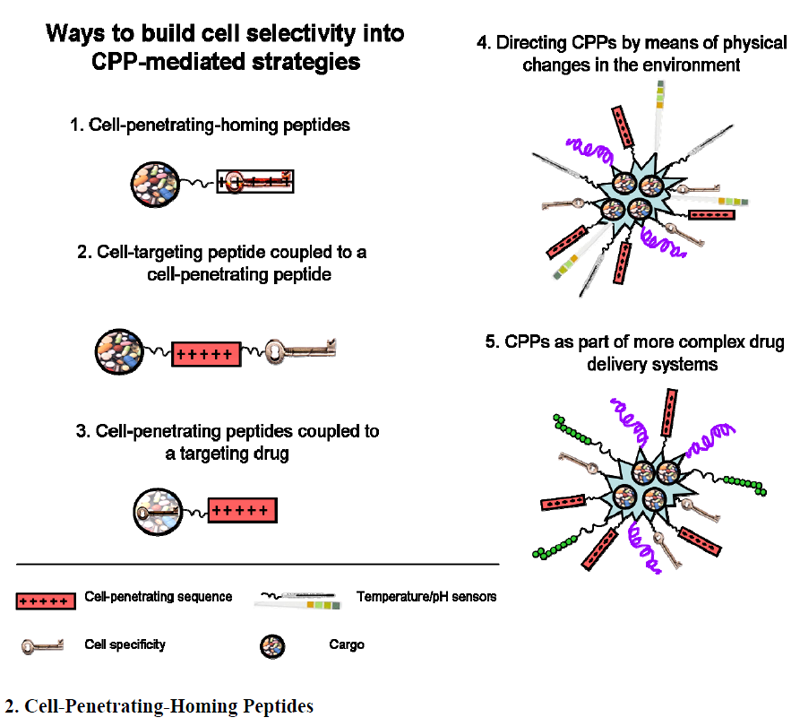
2. Cell-Penetrating-Homing Peptides

{kind=link}
{kind=link}
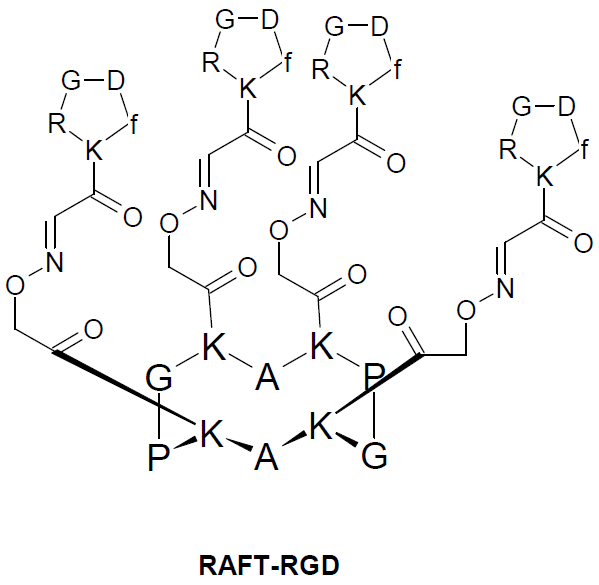
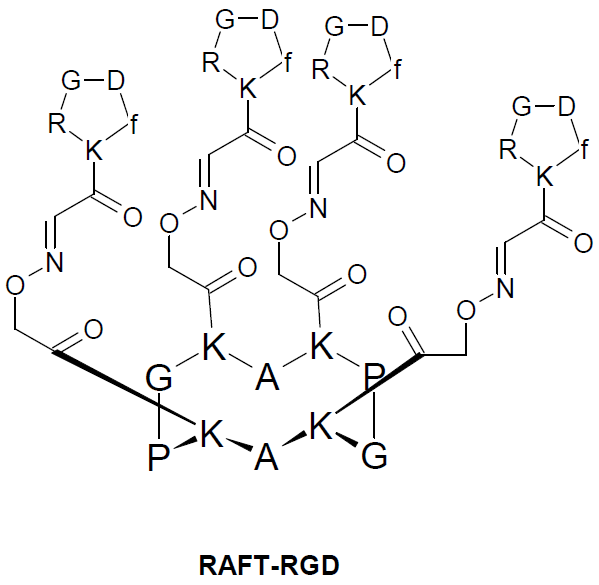{kind=link}
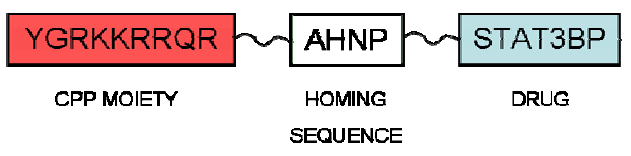{kind=link}
{kind=link}
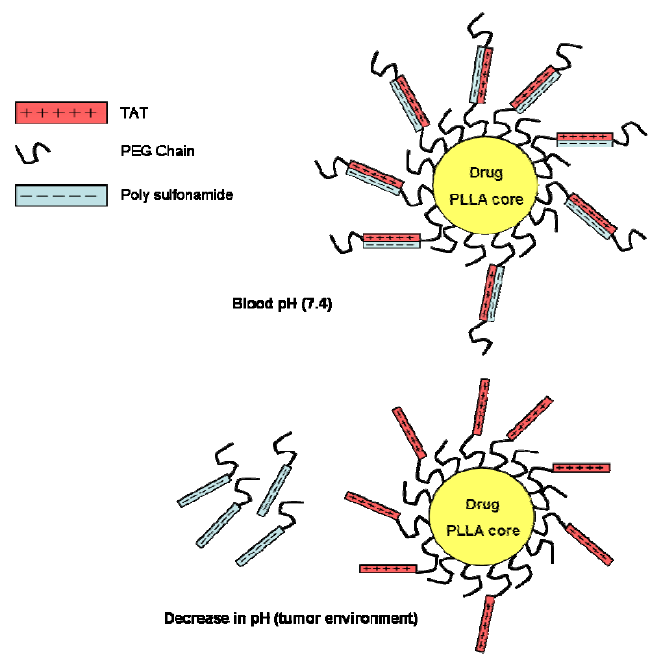{kind=link}
{kind=link}
| Peptide | Homes to |
|---|---|
| AGR (CAGRRSAYC) | TRAMP (prostate) |
| LyP-2 (CNRRTKAGC) | K14-HPV16 (skin) tumor |
| REA (CREAGRKAC) | TRAMP (prostate) |
| LSD (CLSDGKRKC) | C8161 (melanoma) |
3. Cell-Targeting Peptide Coupled to a CPP
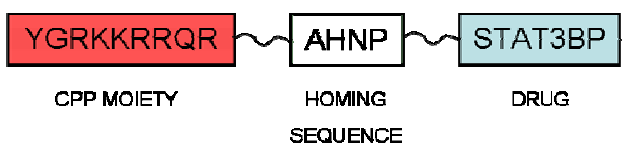
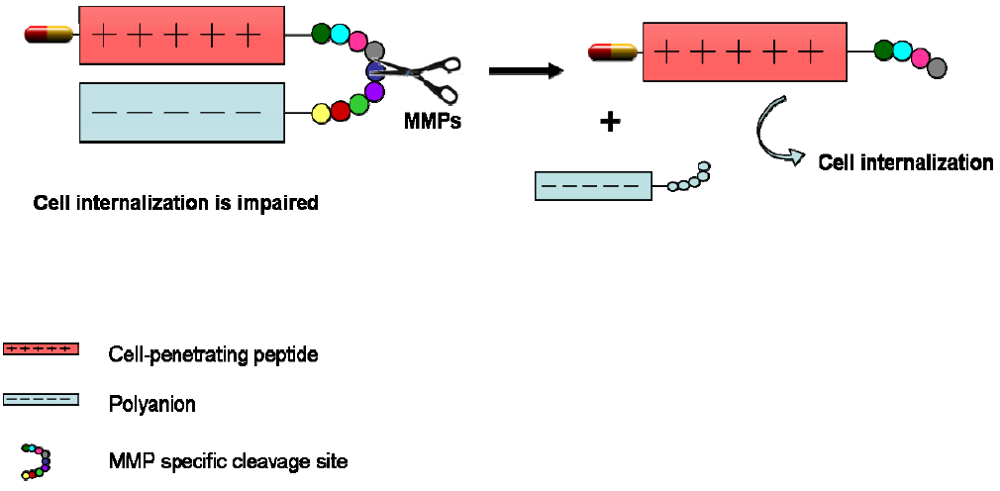
4. CPPs Coupled to a Targeting Drug
5. Directing CPPs by Means of Physical Changes in the Environment
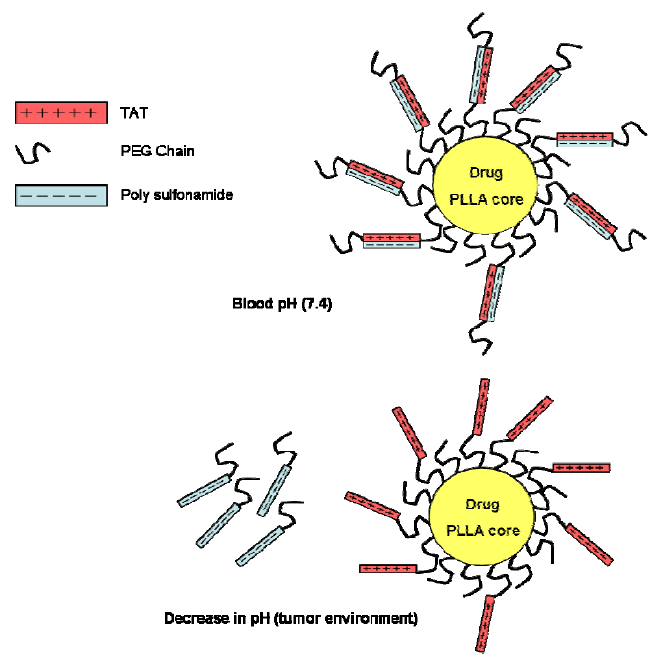
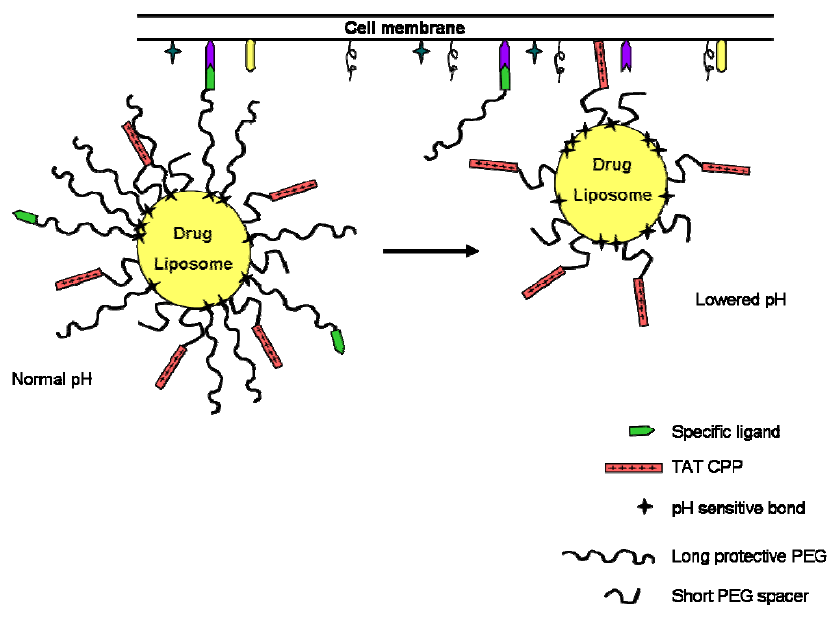
6. CPPs as Part of More Complex Drug Delivery Systems
7. Future Directions
Acknowledgments
References
- Hassane, F.S.; Saleh, A.; Abes, R.; Gait, M.; Lebleu, B. Cell-penetrating-peptides: Overview and applications to the delivery of oligonucleotides. Cell. Mol. Life Sci. 2009, 715–726. [Google Scholar]
- Andaloussi, S.E.; Guterstam, P.; Langel, U. Assessing the delivery efficacy and internalization route of cell-penetrating peptides. Nat. Protocols 2007, 2, 2043–2047. [Google Scholar]
- Pujals, S.; Fernandez-Carneado, J.; Kogan, M.J.; Martinez, J.; Cavelier, F.; Giralt, E. Replacement of a proline with silaproline causes a 20-fold increase in the cellular uptake of a pro-rich peptide. J. Am. Chem. Soc. 2006, 128, 8479–8483. [Google Scholar]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, A.T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.; Johansson, H.J.; Holm, T.; Langel, U. A Novel cell-penetrating peptide, M918, for efficient delivery of proteins and Peptide Nucleic Acids. Mol. Ther. 2007, 15, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Dowdy, S.F. TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Fernández-Carneado, J.; Ludevid, M.D.; Giralt, E. D-SAP: A new, noncytotoxic, and fully protease resistant cell-penetrating peptide. Chem. Med. Chem. 2008, 3, 296–301. [Google Scholar]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Delivery Rev. 2007, 60, 473–484. [Google Scholar] [CrossRef]
- Torchilin, V.P. Tatp-mediated intracellular delivery of pharmaceutical nanocarriers. Biochem. Soc. Trans. 2007, 35, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug Delivery Rev. 2009, 61, 704–709. [Google Scholar]
- Järver, P.; Langel, Ü. Cell-penetrating peptides-A brief introduction. Biochim. Biophys. Acta. 2006, 1758, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Enbäck, J.; Laakkonen, P. Tumour-homing peptides:Tools for targeting, imaging and destruction. Biochem. Soc. Trans. 2007, 035, 780–783. [Google Scholar]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.N.; Ohyama, C.; Lowitz, K.; Matsuo, O.; Pasqualini, R.; Ruoslahti, E.; Fukuda, M. A peptide mimic of E-selectin ligand inhibits Sialyl Lewis X-dependent lung colonization of tumor cells. Cancer Res. 2000, 60, 450–456. [Google Scholar] [PubMed]
- Wang, X.; Cao, B.B. Screening of specific internalization Fab fragment from Human Naive Phage Library by combinational bio-panning. Methods Mol. Biol. 2009, 525, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kolonin, M.; Pasqualini, R.; Arap, W. Molecular addresses in blood vessels as targets for therapy. Curr. Opin. Chem. Biol. 2001, 5, 308–313. [Google Scholar]
- Laakkonen, P.; Zhang, L.; Ruoslahti, E. Peptide targeting of tumor lymph vessels. Ann. N. Y. Acad. Sci. 2008, 1131, 37–43. [Google Scholar]
- Gehlsen, K.; Argraves, W.; Pierschbacher, M.; Ruoslahti, E. Inhibition of in vitro tumor cell invasion by Arg-Gly-Asp-containing synthetic peptides. J. Cell Biol. 1988, 106, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Russo, M.A.; Serra, M.; Colombo, L.; Schinelli, S. Small molecule integrin antagonists in cancer therapy. Mini-Rev. Med. Chem. 2009, 9, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Lucie, S.; Elisabeth, G.; Stephanie, F.; Guy, S.; Amandine, H.; Corinne, A.-R.; Didier, B.; Catherine, S.; Alexei, G.; Pascal, D.; Jean-Luc, C. Clustering and internalization of Integrin αvβ3 with a tetrameric RGD-synthetic peptide. Mol. Ther. 2009, 17, 837–843. [Google Scholar]
- Dumy, P.; Eggleston, I.M.; Cervigni, S.; Sila, U.; Sun, X.; Mutter, M. A convenient synthesis of cyclic peptides as regioselectively addressable functionalized templates (RAFT). Tetrahedron Lett. 1995, 36, 1255–1258. [Google Scholar]
- Koivunen, E.; Gay, D.A.; Ruoslahti, E. Selection of peptides binding to the alpha 5 beta 1 integrin from phage display library. J. Biol. Chem. 1993, 268, 20205–20210. [Google Scholar] [PubMed]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R. A.; Shapiro, L.H.; Arap, W.; Ruoslahti, E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727. [Google Scholar] [PubMed]
- Corti, A.; Curnis, F.; Arap, W.; Pasqualini, R. The neovasculature homing motif NGR: More than meets the eye. Blood 2008, 112, 2628–2635. [Google Scholar]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar]
- Pastorino, F.; Brignole, C.; Di Paolo, D.; Nico, B.; Pezzolo, A.; Marimpietri, D.; Pagnan, G.; Piccardi, F.; Cilli, M.; Longhi, R.; Ribatti, D.; Corti, A.; Allen, T.M.; Ponzoni, M. Targeting liposomal chemotherapy via both tumor cell-specific and tumor vasculature-specific ligands potentiates therapeutic efficacy. Cancer Res. 2006, 66, 10073–10082. [Google Scholar] [PubMed]
- Pastorino, F.; Brignole, C.; Marimpietri, D.; Cilli, M.; Gambini, C.; Ribatti, D.; Longhi, R.; Allen, T.M.; Corti, A.; Ponzoni, M. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003, 63, 7400–7409. [Google Scholar] [PubMed]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Rio, G.D.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Ruoslahti, E.; Bredesen, D.E.; Pasqualini, R. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038. [Google Scholar] [PubMed]
- Law, B.; Quinti, L.; Choi, Y.; Weissleder, R.; Tung, C.-H. A mitochondrial targeted fusion peptide exhibits remarkable cytotoxicity. Mol. Cancer Ther. 2006, 5, 1944–1949. [Google Scholar]
- Lemeshko, V.V. Potential-dependent membrane permeabilization and mitochondrial aggregation caused by anticancer polyarginine-KLA peptides. Arch. Biochem. Biophys. 2009, in press.. [Google Scholar]
- Sacchi, A.; Gasparri, A.; Curnis, F.; Bellone, M.; Corti, A. Crucial role for interferon γ in the synergism between tumor vasculature-targeted tumor necrosis factor-α (NGR-TNF) and doxorubicin. Cancer Res. 2004, 64, 7150–7155. [Google Scholar]
- Angelo, C.; Mirco, P. Tumor vascular targeting with Tumor Necrosis Factor α and chemotherapeutic drugs. Ann. N. Y. Acad. Sci. 2004, 1028, 104–112. [Google Scholar] [CrossRef]
- Curnis, F.; Sacchi, A.; Corti, A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J. Clin. Invest. 2002, 110, 475–482. [Google Scholar]
- Zarovni, N.; Monaco, L.; Corti, A. Inhibition of tumor growth by intramuscular injection of cDNA encoding Tumor Necrosis Factor α coupled to NGR and RGD tumor-homing peptides. Hum. Gene Ther. 2004, 15, 373–382. [Google Scholar]
- Sacchi, A.; Gasparri, A.; Gallo-Stampino, C.; Toma, S.; Curnis, F.; Corti, A. Synergistic antitumor activity of Cisplatin, Paclitaxel, and Gemcitabine with tumor vasculature-targeted Tumor Necrosis Factor α. Clin. Cancer Res. 2006, 12, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Yumi, Y.; Sundaram, R. Addition of an aminopeptidase N-binding sequence to human endostatin improves inhibition of ovarian carcinoma growth. Cancer 2005, 104, 321–331. [Google Scholar]
- Meng, J.; Ma, N.; Yan, Z.; Han, W.; Zhang, Y. NGR enhanced the anti-angiogenic activity of tum-5. J. Biochem. 2006, 140, 299–304. [Google Scholar]
- Meng, J.; Yan, Z.; Wu, J.; Li, L.; Xue, X.; Li, M.; Li, W.; Hao, Q.; Wan, Y.; Qin, X.; Zhang, C.; You, Y.; Han, W.; Zhang, Y. High-yield expression, purification and characterization of tumor-targeted IFN-α2a. Cytotherapy 2007, 9, 60–68. [Google Scholar]
- Curnis, F.; Gasparri, A.; Sacchi, A.; Cattaneo, A.; Magni, F.; Corti, A. Targeted delivery of IFN-γ to tumor vessels uncouples antitumor from counterregulatory mechanisms. Cancer Res. 2005, 65, 2906–2913. [Google Scholar]
- Bieker, R.; Kessler, T.; Schwoppe, C.; Padro, T.; Persigehl, T.; Bremer, C.; Dreischaluck, J.; Kolkmeyer, A.; Heindel, W.; Mesters, R.M.; Berdel, W.E. Infarction of tumor vessels by NGR-peptide-directed targeting of tissue factor: Experimental results and first-in-man experience. Blood 2009, 113, 5019–5027. [Google Scholar] [PubMed]
- Porkka, K.; Laakkonen, P.; Hoffman, J.A.; Bernasconi, M.; Ruoslahti, E. A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7444–7449. [Google Scholar]
- Bustin, M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 1999, 19, 5237–5246. [Google Scholar]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer. 2002, 2, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.D.; Clayman, G.L. Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Res. 2000, 60, 6551–6556. [Google Scholar] [PubMed]
- Hoekman, K.; van der Vijgh, W.J.F.; Vermorken, J.B. Clinical and preclinical modulation of chemotherapy-induced toxicity in patients with cancer. Drugs 1999, 57, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lillo, A.M.; Steiniger, S.C.J.; Liu, Y.; Ballatore, C.; Anichini, A.; Mortarini, R.; Kaufmann, G.F.; Zhou, B.; Felding-Habermann, B.; Janda, K.D. Targeting Heat Shock Proteins on cancer cells: Selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand. Biochemistry 2006, 45, 9434–9444. [Google Scholar] [PubMed]
- Liu, Y.; Steiniger, S.C.J.; Kim, Y.; Kaufmann, G.F.; Felding-Habermann, B.; Janda, K.D. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol. Pharmaceutics 2007, 4, 435–447. [Google Scholar] [CrossRef]
- Lee, A.S. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, Y.; Jia, Z.; Li, Q.; Gong, W.; Wang, L.; Wei, D.; Yao, J.; Fang, S.; Xie, K. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metastasis 2006, 23, 401–410. [Google Scholar]
- Yoneda, Y.; Steiniger, S.C.J.; Čapková, K.; Mee, J.M.; Liu, Y.; Kaufmann, G.F.; Janda, K.D. A cell-penetrating peptidic GRP78 ligand for tumor cell-specific prodrug therapy. Bioorg. Med. Chem. Lett. 2008, 18, 1632–1636. [Google Scholar] [PubMed]
- Dubowchik, G.M.; Firestone, R.A. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg. Med. Chem. Lett. 1998, 8, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Haedicke, W.; Bernasconi, M.; Kain, R.; Rajotte, D.; Krajewski, S.; Ellerby, H.M.; Bredesen, D.E.; Pasqualini, R.; Ruoslahti, E. Targeting the prostate for destruction through a vascular address. Proc. Natl. Acad. Sci. USA 2002, 99, 1527–1531. [Google Scholar]
- Gingrich, J.R.; Barrios, R.J.; Morton, R.A.; Boyce, B.F.; DeMayo, F.J.; Finegold, M.J.; Angelopoulou, R.; Rosen, J.M.; Greenberg, N.M. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996, 56, 4096–4102. [Google Scholar] [PubMed]
- Tohru, Y.; Arsenio, M.F.; Vasu, P.; Laura, B.; Tapas, K.D.G.; Ananda, M.C. Internalization of bacterial redox protein azurin in mammalian cells: Entry domain and specificity. Cell. Microbiol. 2005, 7, 1418–1431. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; Das Gupta, T.K. Noncationic peptides obtained from Azurin preferentially enter cancer cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [PubMed]
- Chaudhari, A.; Mahfouz, M.; Fialho, A.M.; Yamada, T.; Granja, A.T.; Zhu, Y.; Hashimoto, W.; Schlarb-Ridley, B.; Cho, W.; Gupta, T.K.D.; Chakrabarty, A.M. Cupredoxin-cancer interrelationship: Azurin binding with EphB2, interference in EphB2 tyrosine phosphorylation, and inhibition of cancer growth. Biochemistry 2007, 46, 1799–1810. [Google Scholar] [PubMed]
- Yang, D.-S.; Miao, X.-D.; Ye, Z.-M.; Feng, J.; Xu, R.-Z.; Huang, X.; Ge, F.-F. Bacterial redox protein azurin induce apoptosis in human osteosarcoma U2OS cells. Pharmacol. Res. 2005, 52, 413–421. [Google Scholar]
- Yamada, T.; Hiraoka, Y.; Ikehata, M.; Kimbara, K.; Avner, B.S.; Das Gupta, T.K.; Chakrabarty, A.M. Apoptosis or growth arrest: Modulation of tumor suppressor p53's specificity by bacterial redox protein azurin. Proc. Natl. Acad. Sci. USA 2004, 101, 4770–4775. [Google Scholar]
- Laakkonen, P.; Åkerman, M.E.; Biliran, H.; Yang, M.; Ferrer, F.; Karpanen, T.; Hoffman, R.M.; Ruoslahti, E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9381–9386. [Google Scholar]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [PubMed]
- Kimberly, A.K.; Jones, D.A. Isolation of a colon tumor specific binding peptide using phage display selection. Neoplasia 2003, 5, 437–444. [Google Scholar] [PubMed][Green Version]
- Oi, J.; Terashima, T.; Kojima, H.; Fujimiya, M.; Maeda, K.; Arai, R.; Chan, L.; Yasuda, H.; Kashiwagi, A.; Kimura, H. Isolation of specific peptides that home to dorsal root ganglion neurons in mice. Neurosci. Lett. 2008, 434, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Sghirlanzoni, A.; Pareyson, D.; Lauria, G. Sensory neuron diseases. Lancet Neurol. 2005, 4, 349–361. [Google Scholar]
- Sadler, K.; Eom, K.D.; Yang, J.-L.; Dimitrova, Y.; Tam, J.P. Translocating proline-rich peptides from the antimicrobial peptide Bactenecin 7. Biochemistry 2002, 41, 14150–14157. [Google Scholar] [PubMed]
- Fillon, Y.A.; Anderson, J.P.; Chmielewski, J. Cell-penetrating agents based on a polyproline helix scaffold. J. Am. Chem. Soc. 2005, 127, 11798–11803. [Google Scholar] [PubMed]
- Geisler, I.; Chmielewsk, J. Probing length effects and mechanism of cell penetrating agents mounted on a polyproline helix scaffold. Bioorg. Med. Chem. Lett. 2007, 17, 2765–2768. [Google Scholar] [CrossRef] [PubMed]
- Geisler, I.; Chmielewski, J. Cationic amphiphilic polyproline helices: Side-chain variations and cell-specific internalization. Chem. Biol. Drug Des. 2009, 73, 39–45. [Google Scholar]
- Mäe, M.; Myrberg, H.; Andaloussi, S.E.; Langel, Ü. Design of a tumor homing cell-penetrating peptide for drug delivery. Int. J. Pept. Res. Ther. 2009, 15, 11–15. [Google Scholar] [CrossRef]
- Nakase, I.; Hirose, H.; Tanaka, G.; Tadokoro, A.; Kobayashi, S.; Takeuchi, T.; Futaki, S. Cell-surface accumulation of flock house virus-derived peptide leads to efficient internalization via macropinocytosis. Mol. Ther. 2009, 17, 1868–1876. [Google Scholar]
- Andreu, D.; Merrifield, R.B.; Steiner, H.; Boman, H.G. N-Terminal analogs of cecropin A: Synthesis, antibacterial activity, and conformational properties. Biochemistry 2002, 24, 1683–1688. [Google Scholar]
- Tan, M.; Lan, K.-H.; Yao, J.; Lu, C.-H.; Sun, M.; Neal, C.L.; Lu, J.; Yu, D. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of Transcription 3-Blocking Peptide. Cancer Res. 2006, 66, 3764–3772. [Google Scholar] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Dihua, Y.; Mien-Chie, H. Role of ErbB2 in breast cancer chemosensitivity. BioEssays 2000, 22, 673–680. [Google Scholar]
- Tan, M.; Jing, T.; Lan, K.-H.; Neal, C.L.; Li, P.; Lee, S.; Fang, D.; Nagata, Y.; Liu, J.; Arlinghaus, R.; Hung, M.-C.; Yu, D. Phosphorylation on Tyrosine-15 of p34Cdc2 by ErbB2 Inhibits p34Cdc2 Activation and is Involved in Resistance to Taxol-Induced Apoptosis. Mol. Cell 2002, 9, 993–1004. [Google Scholar]
- Bromberg, J.; Darnell, J.E. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Yang, H.; Stanley, J.T.; Holloway, P.H.; Moudgil, B.M.; Walter, G.; Mericle, R.A. Rapid and effective labeling of brain tissue using TAT-conjugated CdSMn/ZnS quantum dots. Chem. Commun. 2005, 3144–3146. [Google Scholar]
- Wadia, J.S.; Dowdy, S.F. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv. Drug Delivery Rev. 2005, 57, 579–596. [Google Scholar] [CrossRef]
- Melnick, A. Targeting aggressive B-cell lymphomas with cell-penetrating peptides. Biochem. Soc. Trans. 2007, 035, 802–806. [Google Scholar]
- Essler, M.; Ruoslahti, E. Molecular specialization of breast vasculature: A breast-homing phage-displayed peptide binds to Aminopeptidase P in breast vasculature. Proc. Natl. Acad. Sci. USA 2002, 99, 2252–2257. [Google Scholar]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, Ü. VE-Cadherin-derived cell-penetrating peptide, pVEC, with carrier functions. Exp. Cell Res. 2001, 269, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Myrberg, H.; Zhang, L.; Mäe, M.; Langel, Ü. Design of a tumor-homing cell-penetrating peptide. Bioconjugate Chem. 2007, 19, 70–75. [Google Scholar]
- Menard, S.; Pupa, S.M.; Campiglio, M.; Tagliabue, E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22, 6570–6578. [Google Scholar] [CrossRef] [PubMed]
- Dayanidhi, R.; Paige, J.B.; Yee Mon, T.; Ann, R. Role of chemokines in tumor growth. Cancer Lett. 2007, 256, 137–165. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.C.; Chang, H.K.; Eugene, C.B. Chemokines in the systemic organization of immunity. Immunol. Rev. 2003, 195, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer. 2004, 4, 540–550. [Google Scholar]
- Snyder, E.L.; Saenz, C.C.; Denicourt, C.; Meade, B.R.; Cui, X.-S.; Kaplan, I.M.; Dowdy, S.F. Enhanced targeting and killing of tumor cells expressing the CXC chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005, 65, 10646–10650. [Google Scholar] [PubMed]
- Zhou, N.; Luo, Z.; Luo, J.; Fan, X.; Cayabyab, M.; Hiraoka, M.; Liu, D.; Han, X.; Pesavento, J.; Dong, C.-Z.; Wang, Y.; An, J.; Kaji, H.; Sodroski, J.G.; Huang, Z. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with d-peptides derived from chemokines. J. Biol. Chem. 2002, 277, 17476–17485. [Google Scholar] [PubMed]
- Snyder, E.L.; Meade, B.R.; Saenz, C.C.; Dowdy, S.F. Treatment of terminal Peritoneal Carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004, 2, e36. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-N.P.; Sharma, S.K.; Ramsey, T.M.; Jiang, L.; Martin, M.S.; Baker, K.; Adams, P.D.; Bair, K.W.; Kaelin, W.G. Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc. Natl. Acad. Sci. USA 1999, 96, 4325–4329. [Google Scholar]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; Lopez, E.; Falcon, V.; Alonso, D.F. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the Protein Kinase 2 (Casein Kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [PubMed]
- Yaylim, I.; Isbir, T. Enhanced casein kinase II (CK II) activity in human lung tumours. Anticancer Res. 2002, 22, 215–218. [Google Scholar]
- Wang, H.; Davis, A.; Yu, S.; Ahmed, K. Response of cancer cells to molecular interruption of the CK2 signal. Mol. Cell. Biochem. 2001, 227, 167–174. [Google Scholar]
- Ruzzene, M.; Penzo, D.; Pinna, L.A. Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem. J. 2002, 364, 41–47. [Google Scholar] [PubMed]
- Mueller, J.; Gaertner, F.C.; Blechert, B.; Janssen, K.-P.; Essler, M. Targeting of tumor blood vessels: A phage-displayed tumor-homing peptide specifically binds to matrix metalloproteinase-2-processed collagen IV and blocks angiogenesis in vivo. Mol. Cancer Res. 2009, 7, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar]
- Roy, R.; Yang, J.; Moses, M.A. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J. Clin. Oncol. 2009, 27, 5287–5297. [Google Scholar] [PubMed]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. Biol. Cell. 2008, 100, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Friend, S. p53: A glimpse at the puppet behind the shadow play. Science 1994, 265, 334–335. [Google Scholar]
- Abarzua, P.; Losardo, J.E.; Gubler, M.L.; Spathis, R.; Lu, Y.-A.; Felix, A.; Nerri, A. Restoration of the transcription activation function to mutant p53 in human cancer cells. Oncogene 1996, 13, 2477–2248. [Google Scholar] [PubMed]
- Selivanova, G.; Iotsova, V.; Okan, I.; Fritsche, M.; Strom, M.; Groner, B.; Grafstrom, R.C.; Wiman, K.G. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 1997, 3, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Senatus, P.B.; Li, Y.; Mandigo, C.; Nichols, G.; Moise, G.; Mao, Y.; Brown, M.D.; Anderson, R.C.; Parsa, A.T.; Brandt-Rauf, P.W.; Bruce, J.N.; Fine, R.L. Restoration of p53 function for selective Fas-mediated apoptosis in human and rat glioma cells in vitro and in vivo by a p53 COOH-terminal peptide. Mol. Cancer Ther. 2006, 5, 20–28. [Google Scholar] [PubMed][Green Version]
- Li, Y.; Mao, Y.; Rosal, R.V.; Dinnen, R.D.; Williams, A.C.; Brandt-Rauf, P.W.; Fine, R.L. Selective induction of apoptosis through the FADD/caspase-8 pathway by a p53 c-terminal peptide in human pre-malignant and malignant cells. Int. J. Cancer 2005, 115, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rosal, R.V.; Brandt-Rauf, P.W.; Fine, R.L. Correlation between hydrophobic properties and efficiency of carrier-mediated membrane transduction and apoptosis of a p53 C-terminal peptide. Biochem. Biophys. Res. Commun. 2002, 298, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Vocero-Akbani, A.M.; Heyden, N.V.; Lissy, N.A.; Ratner, L.; Dowdy, S.F. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 1999, 5, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondoh, S.; Tanaka, S.; Hiraoka, M. Imaging and targeting of the hypoxia-inducible factor 1-active microenvironment. Toxicol. Pathol. 2009, 22, 93–100. [Google Scholar]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572. [Google Scholar] [PubMed]
- Hahn, J.-S. The Hsp90 chaperone machinery: From structure to drug development. BMB Rep. 2009, 42, 623–630. [Google Scholar]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Fortugno, P.; Beltrami, E.; Plescia, J.; Fontana, J.; Pradhan, D.; Marchisio, P.C.; Sessa, W.C.; Altieri, D.C. Regulation of survivin function by Hsp90. Proc. Natl. Acad. Sci. USA 2003, 100, 13791–13796. [Google Scholar]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; Gabrilovich, D.I.; Colombo, G.; Altieri, D.C. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B. Prion protein conversions: insight into mechanisms, TSE transmission barriers and strains. Br. Med. Bull. 2003, 66, 109–120. [Google Scholar] [Green Version]
- Lundberg, P.; Magzoub, M.; Lindberg, M.; Hällbrink, M.; Jarvet, J.; Eriksson, L.E.G.; Langel, Ü.; Gräslund, A. Cell membrane translocation of the N-terminal (1-28) part of the prion protein. Biochem. Biophys. Res. Commun. 2002, 299, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Oglęcka, K.; Pramanik, A.; Eriksson, L.E.G.; Gräslund, A. Membrane perturbation effects of peptides derived from the N-termini of unprocessed prion proteins. Biochim. Biophys. Acta Biomembr. 2005, 1716, 126–136. [Google Scholar] [CrossRef]
- Lofgren, K.; Wahlstrom, A.; Lundberg, P.; Langel, U.; Graslund, A.; Bedecs, K. Antiprion properties of prion protein-derived cell-penetrating peptides. FASEB J. 2008, 22, 2177–2184. [Google Scholar]
- Patel, L.; Zaro, J.; Shen, W.-C. Cell-penetrating-peptides: Intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007, 24, 1977–1992. [Google Scholar]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Meade, B.R.; Dowdy, S.F. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv. Drug Delivery Rev. 2008, 60, 530–536. [Google Scholar] [CrossRef]
- Moschos, S.A.; Jones, S.W.; Perry, M.M.; Williams, A.E.; Erjefalt, J.S.; Turner, J.J.; Barnes, P.J.; Sproat, B.S.; Gait, M.J.; Lindsay, M.A. Lung selivery studies using siRNA conjugated to TAT(48-60) and Penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug. Chem. 2007, 18, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Delivery Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef]
- Sebbage, V. Cell-penetrating peptides and their therapeutic applications. Biosci. Horiz. 2009, 2, 64–72. [Google Scholar]
- Turner, J.J.; Jones, S.; Fabani, M.M.; Ivanova, G.; Arzumanov, A.A.; Gait, M.J. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol. Dis. 2007, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2008, 17, 95–103. [Google Scholar] [PubMed]
- Eguchi, A.; Meade, B.R.; Chang, Y.-C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat. Biotech. 2009, 27, 567–571. [Google Scholar]
- El-Andaloussi, S.; Johansson, H.J.; Lundberg, P.; Langel, Ü. Induction of splice correction by cell-penetrating peptide nucleic acids. J. Gen. Med. 2006, 8, 1262–1273. [Google Scholar] [CrossRef]
- Hassane, F.S.; Ivanova, G.D.; Bolewska-Pedyczak, E.; Abes, R.; Arzumanov, A.A.; Gait, M.J.; Lebleu, B.; Garièpy, J. A peptide-based dendrimer that enhances the splice-redirecting activity of PNA conjugates in cells. Bioconjugate Chem. 2009, 20, 1523–1530. [Google Scholar]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.A.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucl. Acids Res. 2008, 36, 6418–6428. [Google Scholar] [CrossRef]
- Moulton, H.M.; Fletcher, S.; Neuman, B.W.; McClorey, G.; Stein, D.A.; Abes, S.; Wilton, S.D.; Buchmeier, M.J.; Lebleu, B.; Iversen, P.L. Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem. Soc. Trans. 2007, 35, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Wust, P.; Hildebrandt, B.; Sreenivasa, G.; Rau, B.; Gellermann, J.; Riess, H.; Felix, R.; Schlag, P.M. Hyperthermia in combined treatment of cancer. Lancet Oncol. 2002, 3, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lebleu, B.; Moulton, H.M.; Abes, R.; Ivanova, G.D.; Abes, S.; Stein, D.A.; Iversen, P.L.; Arzumanov, A.A.; Gait, M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 2008, 60, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Abes, R.; Moulton, H.M.; Clair, P.; Yang, S.-T.; Abes, S.; Melikov, K.; Prevot, P.; Youngblood, D.S.; Iversen, P.L.; Chernomordik, L.V.; Lebleu, B. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: Structure-activity studies. Nucl. Acids Res. 2008, 36, 6343–6354. [Google Scholar]
- Sethuraman, V.A.; Na, K.; Bae, Y.H. pH-responsive Sulfonamide/PEI system for tumor specific gene delivery: An in vitro study. Biomacromolecules 2005, 7, 64–70. [Google Scholar] [CrossRef]
- Sethuraman, V.A.; Bae, Y.H. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Control. Release 2007, 118, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Rotin, D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989, 49, 4373–4384. [Google Scholar] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar]
- Semenza, G.L.; Artemov, D.; Bedi, A.; Bhujwalla, Z.; Chiles, K.; Feldser, D.; Laughner, E.; Ravi, R.; Simons, J.; Taghavi, P.; Zhong, H. The metabolism of tumours: 70 years later. In the Tumour Microenvironment: Causes and Consequences of Hypoxia and Acidity; Goode, J.A., Chadwick, D.J., Eds.; John Wiley & Sons: Chichester, UK, 2001; pp. 251–264. [Google Scholar]
- Sawant, R.M.; Hurley, J.P.; Salmaso, S.; Kale, A.; Tolcheva, E.; Levchenko, T.S.; Torchilin, V.P. "SMART" drug delivery systems: Double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjugate Chem. 2006, 17, 943–949. [Google Scholar] [CrossRef]
- Kale, A.A.; Torchilin, V.P. Enhanced transfection of tumor cells in vivo using “Smart” pH-sensitive TAT-modified pegylated liposomes. J. Drug Targeting 2007, 15, 538–545. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Viglianti, B.L.; Lora-Michiels, M.; Hanson, M.; Hoopes, P.J. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int. J. Hyperthermia 2003, 19, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.E.; Shin, B.C.; Kong, G.A.; Dewhirst, M.W.; Chilkoti, A. Drug targeting using thermally responsive polymers and local hyperthermia. J. Control. Release 2001, 74, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Mart, R.J.; Osborne, R.D.; Stevens, M.M.; Ulijn, R.V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. [Google Scholar] [CrossRef]
- Hart, D.S.; Gehrke, S.H. Thermally associating polypeptides designed for drug delivery produced by genetically engineered cells. J. Pharm. Sci. 2007, 96, 484–516. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.E.; Kong, G.A.; Dewhirst, M.W.; Zalutsky, M.R.; Chilkoti, A. Targeting a genetically engineered elastin-like polypeptide to solid tumors by local hyperthermia. Cancer Res. 2001, 61, 1548–1554. [Google Scholar] [PubMed]
- Bidwell, G.L.; Raucher, D. Application of thermally responsive polypeptides directed against c-Myc transcriptional function for cancer therapy. Mol. Cancer Ther. 2005, 4, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Massodi, I.; Bidwell III, G.L.; Raucher, D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J. Control. Release 2005, 108, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Manceur, A.P.; Driscoll, B.D.; Sun, W.; Audet, J. Selective enhancement of the uptake and bioactivity of a TAT-conjugated peptide inhibitor of Glycogen Synthase Kinase-3. Mol. Ther. 2008, 17, 500–507. [Google Scholar] [PubMed]
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar] [PubMed]
- Koo, J. A randomized, double-blind study comparing the efficacy, safety and optimal dose of two formulations of Cyclosporin, Neoral and Sandimmun, in patients with severe psoriasis. Br. J. Dermatol. 1998, 139, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Naeyaert, J.M.; Lachapelle, J.M.; Degreef, H.; de la Brassinne, M.; Heenen, M.; Lambert, J. Cyclosporin in Atopic Dermatitis. Dermatology 1999, 198, 145–152. [Google Scholar] [CrossRef] [PubMed]
- McCusker, C.T.; Wang, Y.; Shan, J.; Kinyanjui, M.W.; Villeneuve, A.; Michael, H.; Fixman, E.D. Inhibition of experimental allergic airways disease by local application of a cell-penetrating dominant-negative STAT-6 peptide. J. Immunol. 2007, 179, 2556–2564. [Google Scholar] [PubMed]
- Kuperman, D.A.; Huang, X.; Koth, L.L.; Chang, G.H.; Dolganov, G.M.; Zhu, Z.; Elias, J.A.; Sheppard, D.; Erle, D.J. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 2002, 8, 885–889. [Google Scholar] [PubMed]
- Heckl, S.; Pipkorn, R.; Waldeck, W.; Spring, H.; Jenne, J.; von der Lieth, C.-W.; Corban-Wilhelm, H.; Debus, J.; Braun, K. Intracellular visualization of prostate cancer using magnetic resonance imaging. Cancer Res. 2003, 63, 4766–4772. [Google Scholar]
- Louie, A.Y.; Huber, M.M.; Ahrens, E.T.; Rothbacher, U.; Moats, R.; Jacobs, R.E.; Fraser, S.E.; Meade, T.J. In vivo visualization of gene expression using magnetic resonance imaging. Nat. Biotech. 2000, 18, 321–325. [Google Scholar]
- Lewin, M.; Carlesso, N.; Tung, C.-H.; Tang, X.-W.; Cory, D.; Scadden, D.T.; Weissleder, R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotech. 2000, 18, 410–414. [Google Scholar]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar]
- Tan, M.L.; Choong, P.F.M.; Dass, C.R. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides 2010, 184–193. [Google Scholar] [PubMed]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Fanciullino, R.; Ciccolini, J. Liposome-encapsulated anticancer drugs: Still waiting for the magic bullet? Curr. Med. Chem. 2009, 16, 4361–4373. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.R.; Hersh, E.M. Cationic lipid-mediated gene transfer: current concepts. Curr. Opin. Mol. Ther. 1999, 1, 158–176. [Google Scholar] [PubMed]
- Ko, Y.T.; Hartner, W.C.; Kale, A.; Torchilin, V.P. Gene delivery into ischemic myocardium by double-targeted lipoplexes with anti-myosin antibody and TAT peptide. Gene Ther. 2008, 16, 52–59. [Google Scholar] [PubMed]
- Sheikh, F.; Sontag, D.P.; Fandrich, R.R.; Kardami, E.; Cattini, P.A. Overexpression of FGF-2 increases cardiac myocyte viability after injury in isolated mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1039–H1050. [Google Scholar] [PubMed]
- Jayakumar, J.; Suzuki, K.; Sammut, I.A.; Smolenski, R.T.; Khan, M.; Latif, N.; Abunasra, H.; Murtuza, B.; Amrani, M.; Yacoub, M.H. Heat Shock Protein 70 gene transfection protects mitochondrial and ventricular function against ischemia-reperfusion injury. Circulation 2001, 104, I303–I307. [Google Scholar] [PubMed]
- Mack, C.A.; Patel, S.R.; Schwarz, E.A.; Zanzonico, P.; Hahn, R.T.; Ilercil, A.; Devereux, R.B.; Goldsmith, S.J.; Christian, T.F.; Sanborn, T.A.; Kovesdi, I.; Hackett, N.; Isom, O.W.; Crystal, R.G.; Rosengart, T.K. Biologic bypass with the use of adenovirus-mediated gene transfer of the complementary deoxyribonucleic acid for vascular endothelial growth factor 121 improves myocardial perfusion and function in the ischemic porcine heart. J. Thorac. Cardiovasc. Surg. 1998, 115, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Keiji, I.; Yoshiki, S.; Satoru, K.-S.; Naomasa, K.; Nariaki, M.; Toshikazu, N.; Hikaru, M. Gene transfection of hepatocyte growth factor attenuates the progression of cardiac remodeling in the hypertrophied heart. J. Thorac. Cardiovasc. Surg. 2005, 130, 719–725. [Google Scholar] [Green Version]
- Khaw, B.A.; Mattis, J.A.; Melincoff, G.; Strauss, H.W.; Gold, H.K.; Haber, E. Monoclonal antibody to cardiac myosin: Imaging of experimental myocardial infarction. Hybridoma 1984, 3, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Dass, C.R.; Choong, P.F.M. Selective gene delivery for cancer therapy using cationic liposomes: In vivo proof of applicability. J. Control. Release 2009, 113, 155–163. [Google Scholar]
- Leonie, B.; Klaas, P.; Grietje, M.; Dirk, K.F.M. Targeting of sugar- and charge-modified albumins to fibrotic rat livers: The accessibility of hepatic cells after chronic bile duct ligation. J. Hepatol. 1998, 29, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Tuan Giam Chuang, V.; Kragh-Hansen, U.; Otagiri, M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharm. Res. 2002, 19, 569–577. [Google Scholar]
- Zoltán, S.; Alisa, E.K. Vascular endothelium and immune responses: Implications for inflammation and angiogenesis. Rheum. Dis. Clin. North Am. 2004, 30, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Temming, K.; Lacombe, M.; van der Hoeven, P.; Prakash, J.; Gonzalo, T.; Dijkers, E.C.F.; Orfi, L.; Keri, G.; Poelstra, K.; Molema, G.; Kok, R.J. Delivery of the p38 MAPkinase inhibitor SB202190 to angiogenic endothelial cells: Development of novel RGD-Equipped and PEGylated drug-albumin conjugates using platinum(II)-based drug linker technology. Bioconjugate Chem. 2006, 17, 1246–1255. [Google Scholar] [CrossRef]
- Temming, K.; Meyer, D.L.; Zabinski, R.; Senter, P.D.; Poelstra, K.; Molema, G.; Kok, R.J. Improved efficacy of αvβ3-targeted albumin conjugates by conjugation of a novel Auristatin derivative. Mol. Pharmaceutics 2007, 4, 686–694. [Google Scholar] [CrossRef]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar]
- Singh, R.; Kostarelos, K. Designer adenoviruses for nanomedicine and nanodiagnostics. Trends Biotechnol. 2009, 27, 220–229. [Google Scholar]
- Zochowska, M.; Paca, A.; Schoehn, G.; Andrieu, J.-P.; Chroboczek, J.; Dublet, B.; Szolajska, E. Adenovirus Dodecahedron, as a drug delivery vector. PLoS One 2009, 4, e5569. [Google Scholar]
- Ogris, M.; Wagner, E. Targeting tumors with non-viral gene delivery systems. Drug Discov. Today 2002, 7, 479–485. [Google Scholar]
- Järver, P.; Langel, Ü. The use of cell-penetrating peptides as a tool for gene regulation. Drug Discov. Today 2004, 9, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2008, 109, 259–302. [Google Scholar]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug. Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Boeckle, S.; Wagner, E. Optimizing targeted gene delivery: Chemical modification of viral vectors and synthesis of artificial virus vector systems. AAPS J. 2006, 8, 731–742. [Google Scholar]
- Kreppel, F.; Gackowski, J.; Schmidt, E.; Stefan, K. Combined genetic and chemical capsid modifications enable flexible and efficient de- and retargeting of Adenovirus vectors. Mol. Ther. 2005, 12, 107–117. [Google Scholar]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Harrison, S.D. Cell-penetrating peptides in drug development: Enabling intracellular targets. Biochem. Soc. Trans. 2007, 35, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, M.J. Antisense morpholino oligomers and theirs peptide conjugates. In Therapeutic Oligonucleotide; Kurreck, J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2008; pp. 43–79. [Google Scholar]
- Juliano, R.L.; Alam, R.; Dixit, V.; Kang, H.M. Cell-targeting and cell-penetrating peptides for delivery of therapeutic and imaging agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 324–335. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martín, I.; Teixidó, M.; Giralt, E. Building Cell Selectivity into CPP-Mediated Strategies. Pharmaceuticals 2010, 3, 1456-1490. https://doi.org/10.3390/ph3051456
Martín I, Teixidó M, Giralt E. Building Cell Selectivity into CPP-Mediated Strategies. Pharmaceuticals. 2010; 3(5):1456-1490. https://doi.org/10.3390/ph3051456
Chicago/Turabian StyleMartín, Irene, Meritxell Teixidó, and Ernest Giralt. 2010. "Building Cell Selectivity into CPP-Mediated Strategies" Pharmaceuticals 3, no. 5: 1456-1490. https://doi.org/10.3390/ph3051456
APA StyleMartín, I., Teixidó, M., & Giralt, E. (2010). Building Cell Selectivity into CPP-Mediated Strategies. Pharmaceuticals, 3(5), 1456-1490. https://doi.org/10.3390/ph3051456