The Renin-Angiotensin System in the Development of Salt-Sensitive Hypertension in Animal Models and Humans

Abstract
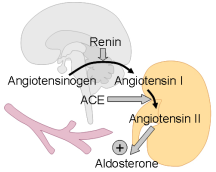
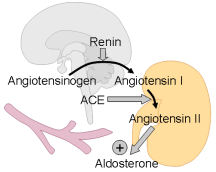
:
{kind=link}
1. Introduction
2. Pathogenetic Processes in the Development of Salt Sensitivity and Hypertension
2.1. Brain
2.2. Kidney
2.3. Vasculature and heart
3. Therapeutic Effects of RAS Antagonists
4. Conclusions
References
- Garcia, E.A.; Newhouse, S.; Caulfield, M.J.; Munroe, P.B. Genes and Hypertension. Curr. Pharm. Des. 2003, 9, 1679–1689. [Google Scholar] [CrossRef]
- Weinberger, M.H. Salt sensitivity of blood pressure in humans. Hypertension 1996, 27, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E.D.; Varagic, J. Sodium directly impairs target organ function in hypertension. Curr. Opin. Cardiol. 2005, 20, 424–429. [Google Scholar] [CrossRef]
- Calhoun, D.A.; Zhu, S.; Wyss, J.M.; Oparil, S. Diurnal blood pressure variation and dietary salt in spontaneously hypertensive rats. Hypertension 1994, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Leenen, F.H. Brain amiloride-sensitive Phe-Met-Arg-Phe-NH(2)--gated Na(+) channels and Na(+)-induced sympathoexcitation and hypertension. Hypertension 2002, 39, 557–561. [Google Scholar] [CrossRef]
- Morgan, T.O.; Aubert, J.F.; Wang, Q. Sodium, angiotensin II, blood pressure and cardiac hypertrophy. Kidney Int. 1998, 54, S213–S215. [Google Scholar] [CrossRef]
- Labat, C.; Lacolley, P.; Lajemi, M.; de Gasparo, M.; Safar, M.E.; Benetos, A. Effects of valsartan on mechanical properties of the carotid artery in spontaneously hypertensive rats under high-salt diet. Hypertension 2001, 38, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Varagic, J.; Slama, M.; Susic, D.; Frohlich, E.D. Cardiac structural and functional responses to salt loading in SHR. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H767–H772. [Google Scholar] [CrossRef]
- Varagic, J.; Frohlich, E.D.; Díez, J.; Susic, D.; Ahn, J.; González, A.; López, B. Myocardial fibrosis, impaired coronary hemodynamics, and biventricular dysfunction in salt-loaded SHR. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1503–H1509. [Google Scholar] [CrossRef]
- Sasaki, Y.; Fujimura, M.; Furukawa, M.; Kubo, T. Sensitivity of pressor responses to central hypertonic saline is greatly enhanced even in pre-hypertensive spontaneously hypertensive rats. Neurosci. Lett. 2006, 399, 255–258. [Google Scholar] [CrossRef]
- Ziegelhöffer-Mihalovicova, B.; Arnold, N.; Marx, G.; Tannapfel, A. Effects of salt loading and various therapies on cardiac hypertrophy and fibrosis in young spontaneously hypertensive rats. Life Sci. 2006, 79, 838–846. [Google Scholar] [CrossRef]
- Beeks, E.; Kessels, A.G.; Kroon, A.A.; van der Klauw, M.M.; de Leeuw, P.W. Genetic predisposition to salt-sensitivity: a systematic review. J. Hypertens. 2004, 22, 1243–1249. [Google Scholar] [CrossRef]
- Iwai, N.; Tsujita, Y.; Kinoshita, M. Isolation of a chromosome 1 region that contributes to high blood pressure and salt sensitivity. Hypertension 1998, 32, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Ely, D.; Turner, M.; Milsted, A. Review of the Y chromosome and hypertension. Braz. J. Med. Biol. Res. 2000, 33, 679–691. [Google Scholar] [CrossRef]
- Aneas, I.; Rodrigues, M.V.; Pauletti, B.A.; Silva, G.J.; Carmona, R.; Cardoso, L.; Kwitek, A.E.; Jacob, H.J.; Soler, J.M.; Krieger, J.E. Congenic strains provide evidence that four mapped loci in chromosomes 2, 4, and 16 influence hypertension in the SHR. Physiol. Genomics 2009, 37, 52–57. [Google Scholar] [CrossRef]
- Johnson, M.D.; He, L.; Herman, D.; Wakimoto, H.; Wallace, C.A.; Zidek, V.; Mlejnek, P.; Musilova, A.; Simakova, M.; Vorlicek, J.; Kren, V.; Viklicky, O.; Qi, N.R.; Wang, J.; Seidman, C.E. Dissection of chromosome 18 blood pressure and salt-sensitivity quantitative trait loci in the spontaneously hypertensive rat. Hypertension 2009, 54, 639–645. [Google Scholar] [CrossRef]
- Johnson, A.G.; Nguyen, T.V.; Davis, D. Blood pressure is linked to salt intake and modulated by the angiotensinogen gene in normotensive and hypertensive elderly subjects. J. Hypertens. 2001, 19, 1053–1060. [Google Scholar] [CrossRef]
- Hiraga, H.; Oshima, T.; Watanabe, M.; Ishida, M.; Ishida, T.; Shingu, T.; Kambe, M.; Matsuura, H.; Kajiyama, G. Angiotensin I-converting enzyme gene polymorphism and salt sensitivity in essential hypertension. Hypertension 1996, 27, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Poch, E.; González, D.; Giner, V.; Bragulat, E.; Coca, A.; de La Sierra, A. Molecular basis of salt sensitivity in human hypertension. Evaluation of renin-angiotensin-aldosterone system gene polymorphisms. Hypertension 2001, 38, 1204–1209. [Google Scholar] [CrossRef]
- Shimoike, H.; Iwai, N.; Kinoshita, M. Genetic analysis of renin gene expression in the central nervous system of spontaneously hypertensive rats. Neurosci. Lett. 1997, 221, 133–136. [Google Scholar] [CrossRef]
- Pamies-Andreu, E.; Ramirez-Lorca, R.; Stiefel García-Junco, P.; Muñiz-Grijalbo, O.; Vallejo-Maroto, I.; Garcia Morillo, S.; Miranda-Guisado, M.L.; Ortíz, J.V.; Carneado de la Fuente, J. Renin-angiotensin-aldosterone system and G-protein beta-3 subunit gene polymorphisms in salt-sensitive essential hypertension. J. Hum. Hypertens. 2003, 17, 187–191. [Google Scholar] [CrossRef]
- Jose, P.A.; Eisner, G.M.; Felder, R.A. Dopamine and the kidney: a role in hypertension? Curr. Opin. Nephrol. Hypertens. 2003, 12, 189–194. [Google Scholar] [CrossRef]
- Cusi, D.; Barlassina, C.; Taglietti, M.V. Genetics of human arterial hypertension. J. Nephrol. 2003, 16, 609–615. [Google Scholar] [PubMed]
- Manunta, P.; Ferrandi, M.; Messaggio, E.; Ferrari, P. A new antihypertensive agent that antagonizes the prohypertensive effect of endogenous ouabain and adducin. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 61–66. [Google Scholar] [CrossRef]
- Takata, Y.; Yamashita, Y.; Takishita, S.; Tsuchihashi, T.; Tomita, Y.; Kimura, Y.; Koga, T.; Fujishima, M. Central and peripheral mechanisms of the enhanced hypertension following long-term salt loading in spontaneously hypertensive rats. Jpn. Circ. J. 1988, 52, 1317–1322. [Google Scholar] [CrossRef]
- Huang, B.S.; Leenen, F.H. Brain "ouabain" and angiotensin II in salt-sensitive hypertension in spontaneously hypertensive rats. Hypertension 1996, 28, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Budzikowski, A.S.; Leenen, F.H. ANG II in median preoptic nucleus and pressor responses to CSF sodium and high sodium intake in SHR. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1210–H1216. [Google Scholar] [PubMed]
- Fang, Z.; Carlson, S.H.; Peng, N.; Wyss, J.M. Circadian rhythm of plasma sodium is disrupted in spontaneously hypertensive rats fed a high-NaCl diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1490–R1495. [Google Scholar] [PubMed]
- Huang, B.S.; van Vliet, B.N.; Leenen, F.H. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1160–H1166. [Google Scholar] [CrossRef]
- Huang, B.S.; Amin, M.S.; Leenen, F.H. The central role of the brain in salt-sensitive hypertension. Curr. Opin. Cardiol. 2006, 21, 295–304. [Google Scholar] [CrossRef]
- Amin, M.S.; Reza, E.; Wang, H.; Leenen, F.H. Sodium transport in the choroid plexus and salt-sensitive hypertension. Hypertension 2009, 54, 860–867. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef]
- van Huysse, J.W. Endogenous brain Na pumps, brain ouabain-like substance and the alpha2 isoform in salt-dependent hypertension. Pathophysiology 2007, 14, 213–220. [Google Scholar] [CrossRef] [PubMed]
- van Huysse, J.W.; Hou, X. Pressor response to CSF sodium in mice: mediation by a ouabain-like substance and renin-angiotensin system in the brain. Brain Res. 2004, 1021, 219–231. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Agalakova, N.I.; Talan, M.I.; Lakatta, E.G.; Bagrov, A.Y. Brain ouabain stimulates peripheral marinobufagenin via angiotensin II signalling in NaCl-loaded Dahl-S rats. J. Hypertens. 2005, 23, 1515–1523. [Google Scholar] [CrossRef]
- Takata, Y.; Yamashita, Y.; Takishita, S.; Kimura, Y.; Fujishima, M. Brain renin angiotensin system contributes to the salt-induced enhancement of hypertension in SHR. Clin. Exp. Hypertens. A. 1986, 8, 1149–1170. [Google Scholar] [CrossRef]
- Schelling, P.; Meyer, D.; Loos, H.E.; Speck, G.; Phillips, M.I.; Johnson, A.K.; Ganten, D. A micromethod for the measurement of renin in brain nuclei: its application in spontaneously hypertensive rats. Neuropharmacology 1982, 21, 455–463. [Google Scholar] [CrossRef]
- Ganten, D.; Hermann, K.; Bayer, C.; Unger, T.; Lang, R.E. Angiotensin synthesis in the brain and increased turnover in hypertensive rats. Science 1983, 221, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.I.; Kimura, B. Converting enzyme inhibitors and brain angiotensin. J. Cardiovasc. Pharmacol. 1986, 8, S82–S90. [Google Scholar] [CrossRef]
- Huang, B.S.; White, R.A.; Jeng, A.Y.; Leenen, F.H. Role of central nervous system aldosterone synthase and mineralocorticoid receptors in salt-induced hypertension in Dahl salt-sensitive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R994–R1000. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; White, R.A.; Ahmad, M.; Jeng, A.Y.; Leenen, F.H. Central infusion of aldosterone synthase inhibitor prevents sympathetic hyperactivity and hypertension by central Na+ in Wistar rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R166–R172. [Google Scholar] [CrossRef] [PubMed]
- Gehlert, D.R.; Speth, R.C.; Wamsley, J.K. Quantitative autoradiography of angiotensin II receptors in the SHR brain. Peptides 1986, 7, 1021–1027. [Google Scholar] [CrossRef]
- Gutkind, J.S.; Kurihara, M.; Castren, E.; Saavedra, J.M. Increased concentration of angiotensin II binding sites in selected brain areas of spontaneously hypertensive rats. J. Hypertens. 1988, 6, 79–84. [Google Scholar] [CrossRef]
- Wright, J.W.; Sullivan, M.J.; Quirk, W.S.; Batt, C.M.; Harding, J.W. Heightened pressor effect and dipsogenicity to intracerebroventricularly applied angiotensin II and III in spontaneously hypertensive rats. J. Hypertens. Suppl. 1986, 4, S408–S411. [Google Scholar] [PubMed]
- Kubo, T.; Hagiwara, Y. Angiotensin II sensitivity of anterior hypothalamic area neurons is enhanced in both spontaneously hypertensive rats and Dahl salt-sensitive rats. Neurosci. Lett. 2006, 397, 297–300. [Google Scholar] [CrossRef]
- Kubo, T.; Yamaguchi, H.; Tsujimura, M.; Hagiwara, Y.; Fukumori, R. An angiotensin system in the anterior hypothalamic area anterior is involved in the maintenance of hypertension in spontaneously hypertensive rats. Brain Res. Bull. 2000, 52, 291–296. [Google Scholar] [CrossRef]
- Huang, B.S.; Wang, H.; Leenen, F.H. Chronic central infusion of aldosterone leads to sympathetic hyperreactivity and hypertension in Dahl S but not Dahl R rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H517–H524. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.P.; Fort, C.; Thwaites, D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am. J. Physiol. Endocrinol. Metab. 1992, 262, E96–E99. [Google Scholar]
- Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Effect of central infusion of benzamil on Dahl S rat hypertension. Am. J. Physiol. Heart Circ. Physiol. 1995, 269, H1044–H1047. [Google Scholar]
- Lon, S.; Szczepanska-Sadowska, E.; Szczypaczewska, M. Evidence that centrally released arginine vasopressin is involved in central pressor action of angiotensin II. Am. J. Physiol. Heart Circ. Physiol. 1996, 270, H167–H173. [Google Scholar]
- Wyss, J.M.; Mozaffari, M.S.; Roysommuti, S. Contribution of the sympathetic nervous system to salt-sensitivity in lifetime captopril-treated spontaneously hypertensive rats. J. Hypertens. 1995, 13, 1037–1042. [Google Scholar] [CrossRef]
- Budzikowski, A.S.; Leenen, F.H. Brain 'ouabain' in the median preoptic nucleus mediates sodium-sensitive hypertension in spontaneously hypertensive rats. Hypertension 1997, 29, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Budzikowski, A.S.; Huang, B.S.; Leenen, F.H. Brain "ouabain", a neurosteroid, mediates sympathetic hyperactivity in salt-sensitive hypertension. Clin. Exp. Hypertens. 1998, 20, 119–140. [Google Scholar] [CrossRef]
- van Huysse, J.W.; Leenen, F.H. Role of endogenous brain "ouabain" in the sympathoexcitatory and pressor effects of sodium. Clin. Exp. Hypertens. 1998, 20, 657–667. [Google Scholar] [CrossRef]
- Ono, A.; Kuwaki, T.; Kumada, M.; Fujita, T. Differential central modulation of the baroreflex by salt loading in normotensive and spontaneously hypertensive rats. Hypertension 1997, 29, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Leenen, F.H. Brain 'ouabain,' sodium, and arterial baroreflex in spontaneously hypertensive rats. Hypertension 1995, 25, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Chronic administration of aldosterone depresses baroreceptor reflex function in the dog. Hypertension 1994, 24, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.M.; Struthers, A.D. Endogenous angiotensin II and baroreceptor dysfunction: a comparative study of losartan and enalapril in man. Br. J. Clin. Pharmacol. 1998, 46, 583–588. [Google Scholar] [CrossRef]
- McMullan, S.; Goodchild, A.K.; Pilowsky, P.M. Circulating angiotensin II attenuates the sympathetic baroreflex by reducing the barosensitivity of medullary cardiovascular neurones in the rat. J. Physiol. 2007, 582, 711–722. [Google Scholar] [CrossRef]
- Ylitalo, A.; Airaksinen, K.E.; Hautanen, A.; Kupari, M.; Carson, M.; Virolainen, J.; Savolainen, M.; Kauma, H.; Kesäniemi, Y.A.; White, P.C.; Huikuri, H.V. Baroreflex sensitivity and variants of the renin angiotensin system genes. J. Am. Coll. Cardiol. 2000, 35, 194–200. [Google Scholar] [CrossRef]
- Coruzzi, P.; Parati, G.; Brambilla, L.; Brambilla, V.; Gualerzi, M.; Novarini, A.; Castiglioni, P.; di Rienzo, M. Effects of salt sensitivity on neural cardiovascular regulation in essential hypertension. Hypertension 2005, 46, 1321–1326. [Google Scholar] [CrossRef]
- Huang, B.S.; Leenen, F.H. Blockade of brain mineralocorticoid receptors or Na+ channels prevents sympathetic hyperactivity and improves cardiac function in rats post-MI. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2491–H2497. [Google Scholar] [CrossRef]
- Chan, J.Y.; Wang, L.L.; Wu, K.L.; Chan, S.H. Reduced functional expression and molecular synthesis of inducible nitric oxide synthase in rostral ventrolateral medulla of spontaneously hypertensive rats. Circulation 2001, 104, 1676–1681. [Google Scholar] [CrossRef]
- Dahl, L.K.; Heine, M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ. Res. 1975, 36, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, K.; Watanabe, T.X.; Shiono, K.; Sokabe, H. Influence on blood pressure of renal isografts between spontaneously hypertensive and normotensive rats, utilizing the F1 hybrids. Jpn. Heart J. 1978, 19, 886–894. [Google Scholar] [CrossRef] [PubMed]
- de Wardener, H.E.; Mac Gregor, G.A. The natriuretic hormone and essential hypertension. The Lancet 1982, 319, 1450–1454. [Google Scholar] [CrossRef]
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 2003, 348, 101–108. [Google Scholar] [CrossRef]
- Skov, K.; Nyengaard, J.R.; Korsgaard, N.; Mulvany, M.J. Number and size of renal glomeruli in spontaneously hypertensive rats. J. Hypertens. 1994, 12, 1373–1376. [Google Scholar] [CrossRef]
- Fogelgren, B.; Yang, S.; Sharp, I.C.; Huckstep, O.J.; Ma, W.; Somponpun, S.J.; Carlson, E.C.; Uyehara, C.F.T.; Lozanoff, S. Deficiency in Six2 during prenatal development is associated with reduced nephron number, chronic renal failure, and hypertension in Br/+ adult mice. Am. J. Physiol. Renal Physiol. 2009, 296, F1166–F1178. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef]
- Zandi-Nejad, K.; Luyckx, V.A.; Brenner, B.M. Adult Hypertension and Kidney Disease. The Role of Fetal Programming. Hypertension 2006, 47, 502–508. [Google Scholar] [CrossRef]
- Marin, E.C.; Balbi, A.P.; Francescato, H.D.; Alves da Silva, C.G.; Costa, R.S.; Coimbra, T.M. Renal structure and function evaluation of rats from dams that received increased sodium intake during pregnancy and lactation submitted or not to 5/6 nephrectomy. Ren. Fail. 2008, 30, 547–555. [Google Scholar] [CrossRef]
- Johnson, R.J.; Schreiner, G.F. Hypothesis: the role of acquired tubulointerstitial disease in the pathogenesis of salt-dependent hypertension. Kidney Int. 1997, 52, 1169–1179. [Google Scholar] [CrossRef]
- Ray, P.E.; Suga, S.; Liu, X.H.; Huang, X.; Johnson, R.J. Chronic potassium depletion induces renal injury, salt sensitivity, and hypertension in young rats. Kidney Int. 2001, 59, 1850–1858. [Google Scholar] [CrossRef]
- Kurokawa, K. Role of the kidney in the genesis of hypertension: aberrant renal response to excess salt intake. Clin. Exp. Pharmacol. Physiol. 1995, 22, S417–S422. [Google Scholar] [PubMed]
- Persson, A.E.; Gutierrez, A.; Pittner, J.; Ring, A.; Ollerstam, A.; Brown, R.; Liu, R.; Thorup, C. Renal NO production and the development of hypertension. Acta Physiol. Scand. 2000, 168, 169–174. [Google Scholar] [CrossRef]
- Oparil, S. The sympathetic nervous system in clinical and experimental hypertension. Kidney Int. 1986, 30, 437–352. [Google Scholar] [CrossRef]
- Sato, Y.; Ando, K.; Ogata, E.; Fujita, T. High-potassium diet attenuates salt-induced acceleration of hypertension in SHR. Am. J. Physiol. 1991, 260, R21–R26. [Google Scholar] [PubMed]
- Wyss, J.M.; Oparil, S.; Sripairojthikoon, W. Neuronal control of the kidney: contribution to hypertension. Can. J. Physiol. Pharmacol. 1992, 70, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Pedrino, G.R.; Rosa, D.A.; Korim, W.S.; Cravo, S.L. Renal sympathoinhibition induced by hypernatremia: involvement of A1 noradrenergic neurons. Auton. Neurosci. 2008, 142, 55–63. [Google Scholar] [CrossRef]
- Esler, M.; Lambert, G.; Brunner-La Rocca, H.P.; Vaddadi, G.; Kaye, D. Sympathetic nerve activity and neurotransmitter release in humans: translation from pathophysiology into clinical practice. Acta Physiol. Scand. 2003, 177, 275–284. [Google Scholar] [CrossRef]
- Judy, W.V.; Farrell, S.K. Arterial baroreceptor reflex control of sympathetic nerve activity in the spontaneously hypertensive rat. Hypertension 1979, 1, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Caplea, A.; Seachrist, D.; Daneshvar, H.; Dunphy, G.; Ely, D. Noradrenergic content and turnover rate in kidney and heart shows gender and strain differences. J. Appl. Physiol. 2002, 92, 567–571. [Google Scholar] [PubMed]
- Guadagnini, D.; Gontijo, J.A. Altered renal sodium handling in spontaneously hypertensive rats (SHR) after hypertonic saline intracerebroventricular injection: role of renal nerves. Life Sci. 2006, 79, 1666–1673. [Google Scholar] [CrossRef]
- Huang, B.S.; Ahmad, M.; Deng, A.Y.; Leenen, F.H. Neuronal responsiveness to central Na+ in 2 congenic strains of Dahl salt-sensitive rats. Hypertension 2007, 49, 1315–1320. [Google Scholar] [CrossRef]
- Greenberg, S.; Osborn, J.L. Relationship between sodium balance and renal innervation during hypertension development in the spontaneously hypertensive rat. J. Hypertens. 1994, 12, 1359–1364. [Google Scholar] [CrossRef]
- Grisk, O.; Rose, H.-J.; Lorenz, G.; Rettig, R. Sympathetic–renal interaction in chronic arterial pressure control. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R441–R450. [Google Scholar] [PubMed]
- Lucas-Teixeira, V.A.; Vieira-Coelho, M.A.; Serrão, P.; Pestana, M.; Soares-da-Silva, P. Salt intake and sensitivity of intestinal and renal Na+-K+ ATPase to inhibition by dopamine in spontaneous hypertensive and Wistar-Kyoto rats. Clin. Exp. Hypertens. 2000, 22, 455–469. [Google Scholar] [CrossRef]
- Zeng, C.; Luo, Y.; Asico, L.D.; Hopfer, U.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Perturbation of D1 dopamine and AT1 receptor interaction in spontaneously hypertensive rats. Hypertension 2003, 42, 787–792. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, Z.; Hopfer, U.; Asico, L.D.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Rat strain effects of AT1 receptor activation on D1 dopamine receptors in immortalized renal proximal tubule cells. Hypertension 2005, 46, 799–805. [Google Scholar] [CrossRef]
- Iimura, O.; Shimamoto, K. Salt and hypertension: water-sodium handling in essential hypertension. Ann. N. Y. Acad. Sci. 1993, 676, 105–121. [Google Scholar] [CrossRef]
- Katori, M.; Majima, M. The renal kallikrein-kinin system: its role as a safety valve for excess sodium intake, and its attenuation as a possible etiologic factor in salt-sensitive hypertension. Crit. Rev. Clin. Lab. Sci. 2003, 40, 43–115. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Fedorova, O.V. Cardenolide and bufadienolide ligands of the sodium pump. How they work together in NaCl sensitive hypertension. Front. Biosci. 2005, 10, 2250–2256. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Hamlyn, J.M. Role of a natriuretic factor in essential hypertension: an hypothesis. Ann. Intern. Med. 1983, 98, 785–792. [Google Scholar] [CrossRef] [PubMed]
- de Wardener, H.E.; Mac Gregor, G.A. The relation of the natriuretic hormone to essential hypertension. Postgrad. Med. J. 1983, 59, 74–77. [Google Scholar] [PubMed]
- Ferrari, P.; Ferrandi, M.; Valentini, G.; Manunta, P.; Bianchi, G. Targeting Ouabain- and Adducin-dependent mechanisms of hypertension and cardiovascular remodeling as a novel pharmacological approach. Med. Hypotheses 2007, 68, 1307–1314. [Google Scholar] [CrossRef]
- Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R553–R559. [Google Scholar] [PubMed]
- Dostanic, I.; Paul, R.J.; Lorenz, J.N.; Theriault, S.; van Huysse, J.W.; Lingrel, J.B. The alpha2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H477–H485. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, M.Y.; Cavalli, M.; Chen, L.; Berra-Romani, R.; Balke, C.W.; Bianchi, G.; Ferrari, P.; Hamlyn, J.M.; Iwamoto, T.; Lingrel, J.B.; Matteson, D.R.; Wier, W.G.; Blaustein, M.P. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J. Physiol. 2005, 569, 243–256. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Zhang, J.; Chen, L.; Song, H.; Raina, H.; Kinsey, S.P.; Izuka, M.; Iwamoto, T.; Kotlikoff, M.I.; Lingrel, J.B.; Philipson, K.D.; Wier, W.G.; Hamlyn, J.M. The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension 2009, 53, 291–298. [Google Scholar] [CrossRef]
- Cusi, D.; Barlassina, C.; Azzani, T.; Casari, G.; Citterio, L.; Devoto, M.; Glorioso, N.; Lanzani, C.; Manunta, P.; Righetti, M.; Rivera, R.; Stella, P.; Troffa, C.; Zagato, L.; Bianchi, G. Polymorphisms of alpha-adducin and salt sensitivity in patients with essential hypertension. Lancet 1997, 349(9062), 1353–1357. [Google Scholar] [CrossRef]
- Ferrandi, M.; Salardi, S.; Tripodi, G.; Barassi, P.; Rivera, R.; Manunta, P.; Goldshleger, R.; Ferrari, P.; Bianchi, G.; Karlish, S.J.D. Evidence for an interaction between adducin and Na+-K+-ATPase: relation to genetic hypertension. Am. J. Physiol. Heart Circ. Physiol. 1999, 277, H1338–H1349. [Google Scholar]
- Wang, J.G.; Staessen, J.A.; Messaggio, E.; Nawrot, T.; Fagard, R.; Hamlyn, J.M.; Bianchi, G.; Manunta, P. Salt, endogenous ouabain and blood pressure interactions in the general population. J. Hypertens. 2003, 21, 1475–1481. [Google Scholar] [CrossRef]
- Manunta, P.; Maillard, M.; Tantardini, C.; Simonini, M.; Lanzani, C.; Citterio, L.; Stella, P.; Casamassima, N.; Burnier, M.; Hamlyn, J.M.; Bianchi, G. Relationships among endogenous ouabain, alpha-adducin polymorphisms and renal sodium handling in primary hypertension. J. Hypertens. 2008, 26, 914–920. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Kolodkin, N.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Marinobufagenin, an endogenous alpha-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension 2001, 37, 462–466. [Google Scholar] [PubMed]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Lakatta, E.G.; Bagrov, A.Y. Endogenous ligand of alpha(1) sodium pump, marinobufagenin, is a novel mediator of sodium chloride--dependent hypertension. Circulation 2002, 105, 1122–1127. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I. Endogenous digitalis: pathophysiologic roles and therapeutic applications. Nat. Clin. Pract. Nephrol. 2008, 4, 378–392. [Google Scholar] [CrossRef]
- Anderson, D.E.; Fedorova, O.V.; Morrell, C.H.; Longo, D.L.; Kashkin, V.A.; Metzler, J.D.; Bagrov, A.Y.; Lakatta, E.G. Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1248–R1254. [Google Scholar] [PubMed]
- Fedorova, O.V.; Zhuravin, I.A.; Agalakova, N.I.; Yamova, L.A.; Talan, M.I.; Lakatta, E.G.; Bagrov, A.Y. Intrahippocampal microinjection of an exquisitely low dose of ouabain mimics NaCl loading and stimulates a bufadienolide Na/K-ATPase inhibitor. J. Hypertens. 2007, 25, 1834–1844. [Google Scholar] [CrossRef]
- Smallegange, C.; Kline, R.L.; Adams, M.A. Transplantation of enalapril-treated kidneys confers persistent lowering of arterial pressure in SHR. Hypertension 2003, 42, 932–936. [Google Scholar] [CrossRef]
- Hodge, G.; Ye, V.Z.; Duggan, K.A. Dysregulation of angiotensin II synthesis is associated with salt sensitivity in the spontaneous hypertensive rat. Acta Physiol. Scand. 2002, 174, 209–215. [Google Scholar] [CrossRef]
- Meng, Q.C.; Durand, J.; Chen, Y.F.; Oparil, S. Effects of dietary salt on angiotensin peptides in kidney. J. Am. Soc. Nephrol. 1995, 6, 1209–1215. [Google Scholar] [PubMed]
- Mizutani, S.; Ishii, M.; Hattori, A.; Nomura, S.; Numaguchi, Y.; Tsujimoto, M.; Kobayshi, H.; Murohara, T.; Wright, J. W. New insights into the importance of aminopeptidase A in hypertension. Heart Fail. Rev. 2008, 13, 273–284. [Google Scholar] [CrossRef]
- Pak, C.H. Plasma adrenaline and noradrenaline concentrations of the spontaneously hypertensive rat. Jpn. Heart J. 1981, 22, 987–995. [Google Scholar] [PubMed]
- Tsunoda, M.; Takezawa, K.; Santa, T.; Ina, Y.; Nagashima, K.; Ohmori, K.; Kobayashi, S.; Imai, K. New approach for measurement of sympathetic nervous abnormality in conscious, spontaneously hypertensive rats. Jpn. J. Pharmacol. 2000, 83, 39–45. [Google Scholar] [CrossRef]
- Lee, R.M.; Triggle, C.R.; Cheung, D.W.; Coughlin, M.D. Structural and functional consequence of neonatal sympathectomy on the blood vessels of spontaneously hypertensive rats. Hypertension 1987, 10, 328–338. [Google Scholar] [PubMed]
- Sofola, O.A.; Knill, A.; Hainsworth, R.; Drinkhill, M. Change in endothelial function in mesenteric arteries of Sprague-Dawley rats fed a high salt diet. J. Physiol. 2002, 543, 255–260. [Google Scholar] [CrossRef]
- Rossoni, L.V.; Cunha, V.; Franca, A.; Vassallo, D.V. The influence of nanomolar ouabain on vascular pressor responses is modulated by the endothelium. J. Cardiovasc. Pharmacol. 1999, 34, 887–892. [Google Scholar] [CrossRef]
- Padilha, A.S.; Rossoni, L.V.; Xavier, F.E.; Vassallo, D.V. Ouabain at nanomolar concentration promotes synthesis and release of angiotensin II from the endothelium of the tail vascular bed of spontaneously hypertensive rats. J. Cardiovasc. Pharmacol. 2004, 44, 372–380. [Google Scholar] [CrossRef]
- Schwarz, P.; Diem, R.; Dun, N.J.; Förstermann, U. Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circ. Res. 1995, 77, 841–848. [Google Scholar] [PubMed]
- Vo, P.A.; Reid, J.J.; Rand, M.J. Attenuation of vasoconstriction by endogenous nitric oxide in rat caudal artery. Br. J. Pharmacol. 1992, 107, 1121–1128. [Google Scholar] [PubMed]
- Costa, F.; Christensen, N.J.; Farley, G.; Biaggioni, I. NO modulates norepinephrine release in human skeletal muscle: implications for neural preconditioning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1494–R1498. [Google Scholar] [PubMed]
- Patil, R.D.; di Carlo, S.E.; Collins, H.L. Acute exercise enhances nitric oxide modulation of vascular response to phenylephrine. Am. J. Physiol. 1993, 265, H1184–H1188. [Google Scholar] [PubMed]
- Toda, N.; Kitamura, Y.; Okamura, T. Neural mechanism of hypertension by nitric oxide synthase inhibitor in dogs. Hypertension 1993, 21, 3–8. [Google Scholar] [PubMed]
- Ni, Z.; Oveisi, F.; Vaziri, N.D. Nitric Oxide Synthase Isotype Expression in Salt-Sensitive and Salt-Resistant Dahl Rats. Hypertension 1999, 34, 552–557. [Google Scholar] [PubMed]
- Fujiwara, N.; Osanai, T.; Kamada, T.; Katoh, T.; Takahashi, K.; Okumura, K. Study on the Relationship Between Plasma Nitrite and Nitrate Level and Salt Sensitivity in Human Hypertension. Modulation of Nitric Oxide Synthesis by Salt Intake. Circulation 2000, 101, 856–861. [Google Scholar] [PubMed]
- Iaccarino, G.; Ciccarelli, M.; Sorriento, D.; Cipolletta, E.; Cerullo, V.; Iovino, G.L.; Paudice, A.; Elia, A.; Santulli, G.; Campanile, A.; Arcucci, O.; Pastore, L.; Salvatore, F.; Condorelli, G.; Trimarco, B. AKT participates in endothelial dysfunction in hypertension. Circulation 2004, 109, 2587–2593. [Google Scholar] [CrossRef]
- Hare, J.M.; Loh, E.; Creager, M.A.; Colucci, W.S. Nitric oxide inhibits the positive inotropic response to beta-adrenergic stimulation in humans with left ventricular dysfunction. Circulation 1995, 92, 2198–2203. [Google Scholar] [PubMed]
- Iwamoto, T.; Kita, S.; Zhang, J.; Blaustein, M.P.; Arai, Y.; Yoshida, S.; Wakimoto, K.; Komuro, I.; Katsuragi, T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat. Med. 2004, 10, 1193–1199. [Google Scholar] [CrossRef]
- Kramer, H.J.; Glanzer, K.; Sorger, M. The role of endogenous inhibition of Na-K-ATPase in human hypertension--sodium pump activity as a determinant of peripheral vascular resistance. Clin. Exp. Hypertens. A 1985, 7, 769–782. [Google Scholar] [CrossRef]
- Laredo, J.; Shah, J.R.; Lu, Z.; Hamilton, B.P.; Hamlyn, J.M. Angiotensin II Stimulates Secretion of Endogenous Ouabain From Bovine Adrenocortical Cells via Angiotensin Type 2 Receptors. Hypertension 1997, 29, 401–407. [Google Scholar] [PubMed]
- Hamlyn, J.M.; Hamilton, B.P.; Manunta, P. Endogenous ouabain, sodium balance and blood pressure: a review and a hypothesis. J. Hypertens. 1996, 14, 151–167. [Google Scholar] [CrossRef]
- Hu, W.Y.; Fukuda, N.; Kanmatsuse, K. Growth characteristics, angiotensin II generation, and microarray-determined gene expression in vascular smooth muscle cells from young spontaneously hypertensive rats. J. Hypertens. 2002, 20, 1323–1333. [Google Scholar] [CrossRef]
- Puddu, P.; Puddu, G.M.; Zaca, F.; Muscari, A. Endothelial dysfunction in hypertension. Acta Cardiol. 2000, 55, 221–232. [Google Scholar] [CrossRef]
- Moreau, P.; d'Uscio, L.V.; Shaw, S.; Takase, H.; Barton, M.; Lüscher, T.F. Angiotensin II increases tissue endothelin and induces vascular hypertrophy - Reversal by ETA-receptor antagonist. Circulation 1997, 96, 1593–1597. [Google Scholar] [PubMed]
- Ortiz, M.C.; Manriquez, M.C.; Romero, J.C.; Juncos, L.A. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension 2001, 38, 655–659. [Google Scholar] [CrossRef]
- Zerrouk, A.; Auguet, M.; Chabrier, P.E. Augmented endothelium-dependent contraction to angiotensin II in the SHR aorta: Role of an inducible cyclooxygenase metabolite. J. Cardiovasc. Pharmacol. 1998, 31, 525–533. [Google Scholar] [CrossRef]
- Sasser, J.M.; Pollock, J.S.; Pollock, D.M. Renal endothelin in chronic angiotensin II hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R243–R248. [Google Scholar] [PubMed]
- Goto, K.; Fujii, K.; Onaka, U.; Abe, I.; Fujishima, M. Renin-Angiotensin System Blockade Improves Endothelial Dysfunction in Hypertension. Hypertension 2000, 36, 575–580. [Google Scholar] [PubMed]
- Cai, H.; Li, Z.; Dikalov, S.; Holland, S.M.; Hwang, J.; Jo, H.; Dudley Jr., S.C.; Harrison, D.G. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J. Biol. Chem. 2002, 277, 48311–48317. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef]
- Mollnau, H.; Wendt, M.; Szöcs, K.; Lassègue, B.; Schulz, E.; Oelze, M.; Li, H.; Bodenschatz, M.; August, M.; Kleschyov, A.L.; Tsilimingas, N.; Walter, U.; Förstermann, U.; Meinertz, T.; Griendling, K.; Münzel, T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 2002, 90, E58–E65. [Google Scholar] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J. Hypertens. 2001, 19, 1245–1254. [Google Scholar] [CrossRef]
- Rueckschloss, U.; Quinn, M.T.; Holtz, J.; Morawietz, H. Dose-dependent regulation of NAD(P)H oxidase expression by angiotensin II in human endothelial cells: protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1845–1851. [Google Scholar] [CrossRef]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [PubMed]
- Touyz, R.M.; Tabet, F.; Schiffrin, E.L. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin. Exp. Pharmacol. Physiol. 2003, 30, 860–866. [Google Scholar] [CrossRef]
- Matsui, H.; Ando, K.; Kawarazaki, H.; Nagae, A.; Fujita, M.; Shimosawa, T.; Nagase, M.; Fujita, T. Salt excess causes left ventricular diastolic dysfunction in rats with metabolic disorder. Hypertension 2008, 52, 287–294. [Google Scholar] [CrossRef]
- Matsui, H.; Barry, W.H.; Livsey, C.; Spitzer, K.W. Angiotensin II stimulates sodium-hydrogen exchange in adult rabbit ventricular myocytes. Cardiovasc. Res. 1995, 29, 215–221. [Google Scholar] [PubMed]
- Aviv, A. Cytosolic Ca2+, Na+/H+ antiport, protein kinase C trio in essential hypertension. Am. J. Hypertens. 1994, 7, 205–212. [Google Scholar] [PubMed]
- Schiffrin, E.L.; Park, J.B.; Intengan, H.D.; Touyz, R.M. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation 2000, 101, 1653–1659. [Google Scholar] [PubMed]
- de Resende, M.M.; Mill, J.G. Effect of high salt intake on local renin-angiotensin system and ventricular dysfunction following myocardial infarction in rats. Clin. Exp. Pharmacol. Physiol. 2007, 34, 274–279. [Google Scholar] [PubMed]
- Liang, B.; Leenen, F.H. Prevention of salt-induced hypertension and fibrosis by AT1–receptor blockers in Dahl S rats. J. Cardiovasc. Pharmacol. 2008, 51, 457–466. [Google Scholar] [CrossRef]
- Liang, B.; Leenen, F.H. Prevention of salt induced hypertension and fibrosis by angiotensin converting enzyme inhibitors in Dahl S rats. Br. J. Pharmacol. 2007, 152, 903–914. [Google Scholar] [CrossRef]
- Linz, W.; Schölkens, B.A.; Ganten, D. Converting Enzyme Inhibition Specifically Prevents the Development and Induces Regression of Cardiac Hypertrophy in Rats. Clin. Exp. Hypertens. 1989, 11, 1325–1350. [Google Scholar] [CrossRef]
- Frohlich, E.D. Left ventricular hypertrophy: dissociation of structural and functional effects by therapy. Adv. Exp. Med. Biol. 1991, 308, 175–190. [Google Scholar] [PubMed]
- Biagini, G.; Zoli, M.; Torri, C.; Boschi, S.; Vantaggiato, G.; Ballestri, M.; Baraldi, A.; Agnati, L. F. Protective effects of delapril, indapamide and their combination chronically administered to stroke-prone spontaneously hypertensive rats fed a high-sodium diet. Clin. Sci. (Lond.) 1997, 93, 401–411. [Google Scholar] [PubMed]
- Migdalof, B.H.; Antonaccio, M.J.; McKinstry, D.N.; Singhvi, S.M.; Lan, S.J.; Egli, P.; Kripalani, K.J. Captopril: pharmacology, metabolism and disposition. Drug Metab. Rev. 1984, 15, 841–869. [Google Scholar] [CrossRef]
- Inada, Y.; Tanabe, M.; Shibouta, Y.; Kawazoe, K.; Nishikawa, K.; Kikuchi, S. Antihypertensive action of a non-sulfhydryl angiotensin converting enzyme inhibitor (CV-3317) in various hypertensive models. Jpn. J. Pharmacol. 1986, 42, 1–8. [Google Scholar] [CrossRef]
- Dunn, F.G.; Oigman, W.; Ventura, H.O.; Messerli, F.H.; Kobrin, I.; Frohlich, E.D. Enalapril improves systemic and renal hemodynamics and allows regression of left ventricular mass in essential hypertension. Am. J. Cardiol. 1984, 53, 105–108. [Google Scholar] [CrossRef]
- di Nicolantonio, R. Failure of captopril to lower blood pressure in spontaneously hypertensive rats offered water and saline to drink. Clin. Exp. Pharmacol. Physiol. 1983, 10, 269–272. [Google Scholar] [CrossRef]
- Fliser, D.; Nowack, R.; Wolf, G.; Ritz, E. Differential effects of ACE inhibitors and vasodilators on renal function curve in patients with primary hypertension. Blood Press. 1993, 2, 296–300. [Google Scholar] [CrossRef]
- Bouaziz, H.; Joulin, Y.; Safar, M.; Benetos, A. Effects of bradykinin B2 receptor antagonism on the hypotensive effects of ACE inhibition. Br. J. Pharmacol. 1994, 113, 717–722. [Google Scholar] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rassler, B. The Renin-Angiotensin System in the Development of Salt-Sensitive Hypertension in Animal Models and Humans. Pharmaceuticals 2010, 3, 940-960. https://doi.org/10.3390/ph3040940
Rassler B. The Renin-Angiotensin System in the Development of Salt-Sensitive Hypertension in Animal Models and Humans. Pharmaceuticals. 2010; 3(4):940-960. https://doi.org/10.3390/ph3040940
Chicago/Turabian StyleRassler, Beate. 2010. "The Renin-Angiotensin System in the Development of Salt-Sensitive Hypertension in Animal Models and Humans" Pharmaceuticals 3, no. 4: 940-960. https://doi.org/10.3390/ph3040940
APA StyleRassler, B. (2010). The Renin-Angiotensin System in the Development of Salt-Sensitive Hypertension in Animal Models and Humans. Pharmaceuticals, 3(4), 940-960. https://doi.org/10.3390/ph3040940