Implications of Inter-Individual Differences in Clopidogrel Metabolism, with Focus on Pharmacogenetics

Abstract
:1. Introduction
2. CYP2C19 and Clopidogrel Pharmacokinetics and Pharmacodynamics
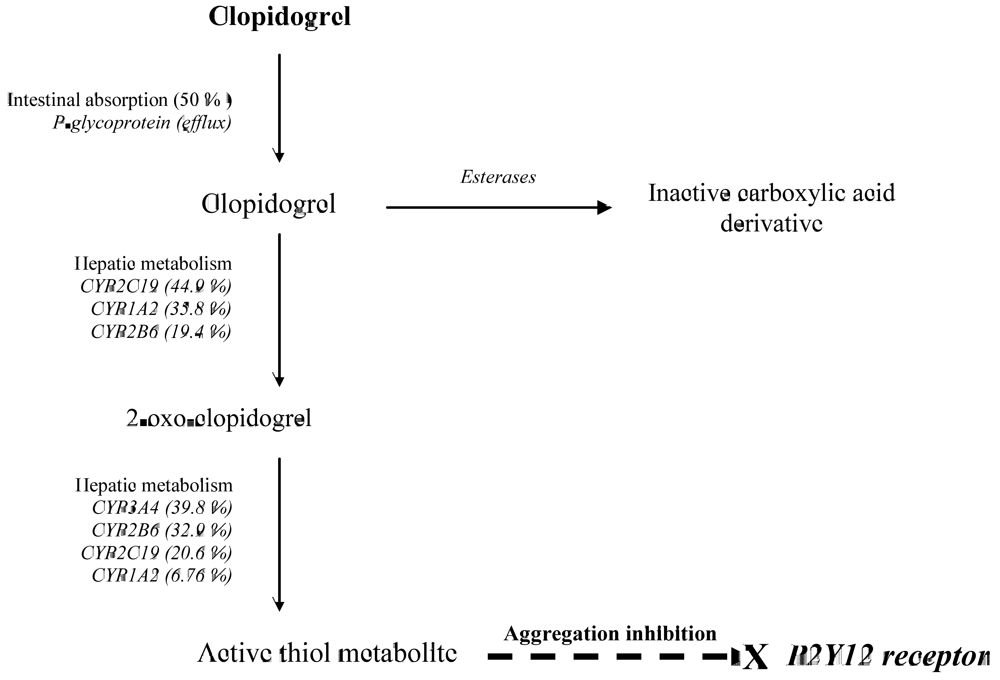

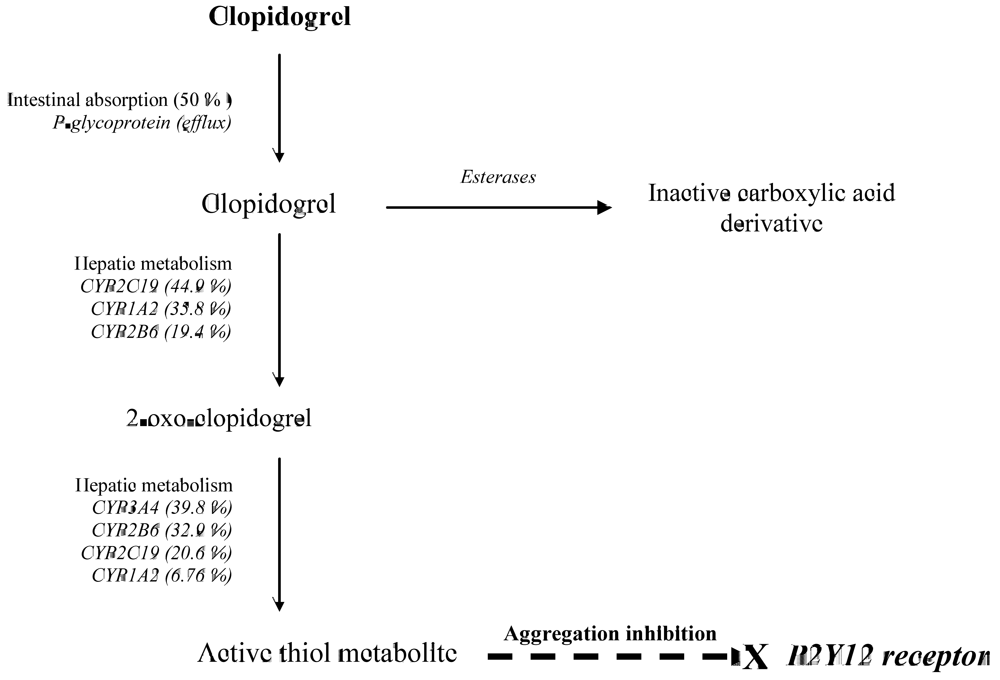{kind=link}
| Study population | Treatment dose (mg) | Reference |
|---|---|---|
| Homozygous b | ||
| HV Dutch mostly Caucasian (n =74) | 300 | [13] |
| (induced by 20 µM ADP) | 1.9–fold ↓ | 10.3–fold ↓ |
| HV Mixed (n =162) | [14] | |
| (induced by 20 µM ADP) | ||
| HV Japanese (n =47) | 300 | [15] |
| (4 h after dosing) | 1.2– fold ↑ | 1.4–fold ↑ |
| HV Korean (n =24) | 300 | [16] |
| (induced by 5 µM ADP) | 1.1–fold ↓ | 2.2–fold ↓ |
3. CYP2C19 Genotype and Long-Term Outcome of Clopidogrel Treatment
4. Other CYP and ABCB1 Genotypes in Relation to Clopidogrel Response
5. Drug Interactions
6. Clinical Implications
Conflict of Interest
References
- Gurbel, P.A.; Bliden, K.P.; Hiatt, B.L.; O'Connor, C.M. Clopidogrel for coronary stenting: response variability, drug resistance, and the effect of pretreatment platelet reactivity. Circulation 2003, 107, 2908–2913. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Fernandez-Ortiz, A.; Bernardo, E.; Ramirez, C.; Cavallari, U.; Trabetti, E.; Sabate, M.; Hernandez, R.; Moreno, R.; Escaned, J.; Alfonso, F.; Banuelos, C.; Costa, M.A.; Bass, T.A.; Pignatti, P.F.; Macaya, C. Contribution of gene sequence variations of the hepatic cytochrome P450 3A4 enzyme to variability in individual responsiveness to clopidogrel. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1895–900. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L. P2Y(12) inhibitors: Differences in properties and mechanisms of action and potential consequences for clinical use. Eur. Heart J. 2009, 30, 1964–77. [Google Scholar] [CrossRef] [PubMed]
- Plavix (Sanofi Aventis) Label Information. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label-/-2009/020839s044lbl.pdf/ 11 December 2009.
- Kazui, M.; Nishiya, Y.; Ishizuka, T.; Hagihara, K.; Farid, N.A.; Okazaki, O.; Ikeda, T.; Kurihara, A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2010, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.; Zhao, X.; Shin, J.G.; Flockhart, D.A. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 2002, 41, 913–958. [Google Scholar] [CrossRef] [PubMed]
- Human cytochrome P450 allele nomenclature. Available online: http://www.cypalleles.ki.se/cyp2c19.htm 23 March 2010.
- Sim, S.C.; Risinger, C.; Dahl, M.L.; Aklillu, E.; Christensen, M.; Bertilsson, L.; Ingelman-Sundberg, M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin. Pharmacol. Ther. 2006, 79, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kurzawski, M.; Gawronska-Szklarz, B.; Wrzesniewska, J.; Siuda, A.; Starzynska, T.; Drozdzik, M. Effect of CYP2C19*17 gene variant on Helicobacter pylori eradication in peptic ulcer patients. Eur. J. Clin. Pharmacol. 2006, 62, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Rudberg, I.; Mohebi, B.; Hermann, M.; Refsum, H.; Molden, E. Impact of the ultrarapid CYP2C19*17 allele on serum concentration of escitalopram in psychiatric patients. Clin. Pharmacol. Ther. 2008, 83, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Ragia, G.; Arvanitidis, K.I.; Tavridou, A.; Manolopoulos, V.G. Need for reassessment of reported CYP2C19 allele frequencies in various populations in view of CYP2C19*17 discovery: The case of Greece. Pharmacogenomics 2009, 10, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Uno, T.; Yamazaki, H.; Tateishi, T. Limited frequency of the CYP2C19*17 allele and its minor role in a Japanese population. Br. J. Clin. Pharmacol. 2008, 65, 437–439. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.T.; Close, S.L.; Iturria, S.J.; Payne, C.D.; Farid, N.A.; Ernest, C.S.; Lachno, D. R.; Salazar, D.; Winters, K. J. Common polymorphisms of CYP2C19 and CYP2C9 affect the pharmacokinetic and pharmacodynamic response to clopidogrel but not prasugrel. J. Thromb. Haemost. 2007, 5, 2429–2436. [Google Scholar] [CrossRef]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; Sabatine, M.S. Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Umemura, K.; Furuta, T.; Kondo, K. The common gene variants of CYP2C19 affect pharmacokinetics and pharmacodynamics in an active metabolite of clopidogrel in healthy subjects. J. Thromb. Haemost. 2008, 6, 1439–1441. [Google Scholar] [CrossRef]
- Kim, K.A.; Park, P.W.; Hong, S.J.; Park, J.Y. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: A possible mechanism for clopidogrel resistance. Clin. Pharmacol. Ther. 2008, 84, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Hulot, J.S.; Bura, A.; Villard, E.; Azizi, M.; Remones, V.; Goyenvalle, C.; Aiach, M.; Lechat, P.; Gaussem, P. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 2006, 108, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Hulot, J.S.; De Moerloose, P.; Gaussem, P. Influence of CYP2C19 and CYP3A4 gene polymorphisms on clopidogrel responsiveness in healthy subjects. J. Thromb. Haemost. 2007, 5, 2153–2155. [Google Scholar] [CrossRef]
- Geisler, T.; Schaeffeler, E.; Dippon, J.; Winter, S.; Buse, V.; Bischofs, C.; Zuern, C.; Moerike, K.; Gawaz, M.; Schwab, M. CYP2C19 and nongenetic factors predict poor responsiveness to clopidogrel loading dose after coronary stent implantation. Pharmacogenomics 2008, 9, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Gori, A.M.; Marcucci, R.; Saracini, C.; Sestini, I.; Paniccia, R.; Valente, S.; Antoniucci, D.; Abbate, R.; Gensini, G.F. Cytochrome P450 2C19 loss-of-function polymorphism, but not CYP3A4 IVS10 + 12G/A and P2Y12 T744C polymorphisms, is associated with response variability to dual antiplatelet treatment in high-risk vascular patients. Pharmacogenet. Genomics 2007, 17, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Frere, C.; Cuisset, T.; Morange, P.E.; Quilici, J.; Camoin-Jau, L.; Saut, N.; Faille, D.; Lambert, M.; Juhan-Vague, I.; Bonnet, J.L.; Alessi, M.C. Effect of cytochrome p450 polymorphisms on platelet reactivity after treatment with clopidogrel in acute coronary syndrome. Am. J. Cardiol. 2008, 101, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Gladding, P.; Webster, M.; Zeng, I.; Farrell, H.; Stewart, J.; Ruygrok, P.; Ormiston, J.; El-Jack, S.; Armstrong, G.; Kay, P.; Scott, D.; Gunes, A.; Dahl, M.L. The pharmacogenetics and pharmacodynamics of clopidogrel response: An analysis from the PRINC (Plavix Response in Coronary Intervention) trial. JACC Cardiovasc. Interv. 2008, 1, 620–627. [Google Scholar] [CrossRef]
- Lee, J.M.; Park, S.; Shin, D.J.; Choi, D.; Shim, C.Y.; Ko, Y.G.; Kim, J.S.; Shin, E.S.; Chang, C.W.; Lee, J.E.; Jang, Y. Relation of genetic polymorphisms in the cytochrome P450 gene with clopidogrel resistance after drug-eluting stent implantation in Koreans. Am. J. Cardiol. 2009, 104, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Jinnai, T.; Horiuchi, H.; Makiyama, T.; Tazaki, J.; Tada, T.; Akao, M.; Ono, K.; Hoshino, K.; Naruse, Y.; Takahashi, K.; Watanabe, H.; Kita, T.; Kimura, T. Impact of CYP2C19 polymorphisms on the antiplatelet effect of clopidogrel in an actual clinical setting in Japan. Circ. J. 2009, 73, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Varenhorst, C.; James, S.; Erlinge, D.; Brandt, J.T.; Braun, O.O.; Man, M.; Siegbahn, A.; Walker, J.; Wallentin, L.; Winters, K.J.; Close, S.L. Genetic variation of CYP2C19 affects both pharmacokinetic and pharmacodynamic responses to clopidogrel but not prasugrel in aspirin-treated patients with coronary artery disease. Eur. Heart J. 2009, 30, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Frere, C.; Cuisset, T.; Gaborit, B.; Alessi, M.C.; Hulot, J.S. The CYP2C19*17 allele is associated with better platelet response to clopidogrel in patients admitted for non-ST acute coronary syndrome. J. Thromb. Haemost. 2009, 7, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Shuldiner, A.R.; O'Connell, J.R.; Bliden, K.P.; Gandhi, A.; Ryan, K.; Horenstein, R.B.; Damcott, C.M.; Pakyz, R.; Tantry, U.S.; Gibson, Q.; Pollin, T.I.; Post, W.; Parsa, A.; Mitchell, B.D.; Faraday, N.; Herzog, W.; Gurbel, P.A. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009, 302, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Trenk, D.; Hochholzer, W.; Fromm, M.F.; Chialda, L.E.; Pahl, A.; Valina, C.M.; Stratz, C.; Schmiebusch, P.; Bestehorn, H.P.; Buttner, H.J.; Neumann, F.J. Cytochrome P450 2C19 681G>A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J. Am. Coll. Cardiol. 2008, 51, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Verstuyft, C.; Mary-Krause, M.; Quteineh, L.; Drouet, E.; Meneveau, N.; Steg, P.G.; Ferrieres, J.; Danchin, N.; Becquemont, L. Genetic determinants of response to clopidogrel and cardiovascular events. N. Engl. J. Med. 2009, 360, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Giusti, B.; Gori, A.M.; Marcucci, R.; Saracini, C.; Sestini, I.; Paniccia, R.; Buonamici, P.; Antoniucci, D.; Abbate, R.; Gensini, G.F. Relation of cytochrome P450 2C19 loss-of-function polymorphism to occurrence of drug-eluting coronary stent thrombosis. Am. J. Cardiol. 2009, 103, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Stegherr, J.; Latz, W.; Koch, W.; Mehilli, J.; Dorrler, K.; Morath, T.; Schomig, A.; Kastrati, A.; von Beckerath, N. Cytochrome P450 2C19 loss-of-function polymorphism and stent thrombosis following percutaneous coronary intervention. Eur. Heart. J. 2009, 30, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Hulot, J.S.; Pena, A.; Villard, E.; Esteve, J.B.; Silvain, J.; Payot, L.; Brugier, D.; Cayla, G.; Beygui, F.; Bensimon, G.; Funck-Brentano, C.; Montalescot, G. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: A cohort study. Lancet 2009, 373, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Turpeinen, M.; Klein, K.; Schwab, M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem. 2008, 392, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; McLeod, H.L. Pharmacogenomics--drug disposition, drug targets, and side effects. N. Engl. J. Med. 2003, 348, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.W.; Koo, B.K.; Zhang, S.Y.; Park, K.W.; Cho, J.Y.; Jang, I.J.; Lee, D.S.; Sohn, D.W.; Lee, M.M.; Kim, H.S. Increased risk of atherothrombotic events associated with cytochrome P450 3A5 polymorphism in patients taking clopidogrel. CMAJ 2006, 174, 1715–1722. [Google Scholar] [PubMed]
- Dally, H.; Bartsch, H.; Jager, B.; Edler, L.; Schmezer, P.; Spiegelhalder, B.; Dienemann, H.; Drings, P.; Kayser, K.; Schulz, V.; Risch, A. Genotype relationships in the CYP3A locus in Caucasians. Cancer Lett. 2004, 207, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Taubert, D.; von Beckerath, N.; Grimberg, G.; Lazar, A.; Jung, N.; Goeser, T.; Kastrati, A.; Schomig, A.; Schomig, E. Impact of P-glycoprotein on clopidogrel absorption. Clin. Pharmacol. Ther. 2006, 80, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.C.; Waskell, L.A.; Watkins, P.B.; Neer, C.J.; Horowitz, K.; Hopp, A.S.; Tait, A.R.; Carville, D.G.; Guyer, K.E.; Bates, E.R. Atorvastatin reduces the ability of clopidogrel to inhibit platelet aggregation: A new drug-drug interaction. Circulation 2003, 107, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Gilard, M.; Arnaud, B.; Cornily, J.C.; Le Gal, G.; Lacut, K.; Le Calvez, G.; Mansourati, J.; Mottier, D.; Abgrall, J.F.; Boschat, J. Influence of omeprazole on the antiplatelet action of clopidogrel associated with aspirin: The randomized, double-blind OCLA (Omeprazole CLopidogrel Aspirin) study. J. Am. Coll. Cardiol. 2008, 51, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.A.; Small, D.S.; Payne, C.D.; Jakubowski, J.A.; Brandt, J.T.; Li, Y.G.; Ernest, C.S.; Salazar, D.E.; Konkoy, C.S.; Winters, K.J. Effect of atorvastatin on the pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel in healthy subjects. Pharmacotherapy 2008, 28, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Pezalla, E.; Day, D.; Pulliadath, I. Initial assessment of clinical impact of a drug interaction between clopidogrel and proton pump inhibitors. J. Am. Coll. Cardiol. 2008, 52, 1038–1039. [Google Scholar] [CrossRef] [PubMed]
- Juurlink, D.N.; Gomes, T.; Ko, D.T.; Szmitko, P.E.; Austin, P.C.; Tu, J.V.; Henry, D.A.; Kopp, A.; Mamdani, M.M. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009, 180, 713–718. [Google Scholar] [PubMed]
- Ho, P.M.; Maddox, T.M.; Wang, L.; Fihn, S.D.; Jesse, R.L.; Peterson, E.D.; Rumsfeld, J.S. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009, 301, 937–944. [Google Scholar] [CrossRef] [PubMed]
- von Beckerath, N.; Taubert, D.; Pogatsa-Murray, G.; Schomig, E.; Kastrati, A.; Schomig, A. Absorption, metabolization, and antiplatelet effects of 300-, 600-, and 900-mg loading doses of clopidogrel: Results of the ISAR-CHOICE (Intracoronary Stenting and Antithrombotic Regimen: Choose Between 3 High Oral Doses for Immediate Clopidogrel Effect) Trial. Circulation 2005, 112, 2946–2950. [Google Scholar] [PubMed]
- Pena, A.; Collet, J.P.; Hulot, J.S.; Silvain, J.; Barthelemy, O.; Beygui, F.; Funck-Brentano, C.; Montalescot, G. Can we override clopidogrel resistance? Circulation 2009, 119, 2854–2857. [Google Scholar] [CrossRef] [PubMed]
- Hjemdahl, P. Should we monitor platelet function during antiplatelet therapy? Heart 2008, 94, 685–687. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dahl, M.-L.; Gunes, A. Implications of Inter-Individual Differences in Clopidogrel Metabolism, with Focus on Pharmacogenetics. Pharmaceuticals 2010, 3, 782-794. https://doi.org/10.3390/ph3040782
Dahl M-L, Gunes A. Implications of Inter-Individual Differences in Clopidogrel Metabolism, with Focus on Pharmacogenetics. Pharmaceuticals. 2010; 3(4):782-794. https://doi.org/10.3390/ph3040782
Chicago/Turabian StyleDahl, Marja-Liisa, and Arzu Gunes. 2010. "Implications of Inter-Individual Differences in Clopidogrel Metabolism, with Focus on Pharmacogenetics" Pharmaceuticals 3, no. 4: 782-794. https://doi.org/10.3390/ph3040782
APA StyleDahl, M.-L., & Gunes, A. (2010). Implications of Inter-Individual Differences in Clopidogrel Metabolism, with Focus on Pharmacogenetics. Pharmaceuticals, 3(4), 782-794. https://doi.org/10.3390/ph3040782