Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy


Abstract
:1. Introduction
2. PDGF and PDGFR Family
3. PDGF/PDGFR and Vascular Development
4. PDGF are Differentially Expressed by Normal Tissues

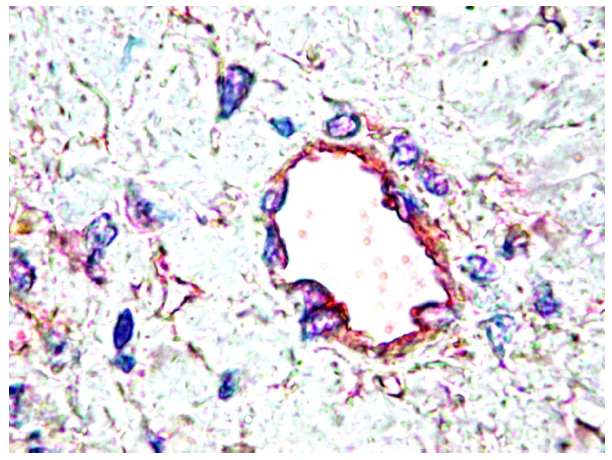{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | PDGF-A | PDGFRα | PDGF-B | PDGFRβ |
|---|---|---|---|---|
| Blood vessel | - | - | Endothelium | Perivascular cells |
| Heart | Muscle cells | Mesenchyme | Muscle cells Endothelium | Perivascular cells |
| Lung | Epithelium | Mesenchyme | - | - |
| Kidney | Early nephron epithelium | Mesenchyme | Glomerular endothelium | Mesenchyme |
| Pancreas | Epithelium | Mesenchyme | - | - |
| Gut | Epithelium | Mesenchyme | - | - |
| Skin | Epidermis Hair follicle epithelium | Dermis | - | - |
| Nervous | Neurons Astrocytes | Astrocytes Oligodendrocyte precursors | Postnatal neurons | Postnatal neurons |
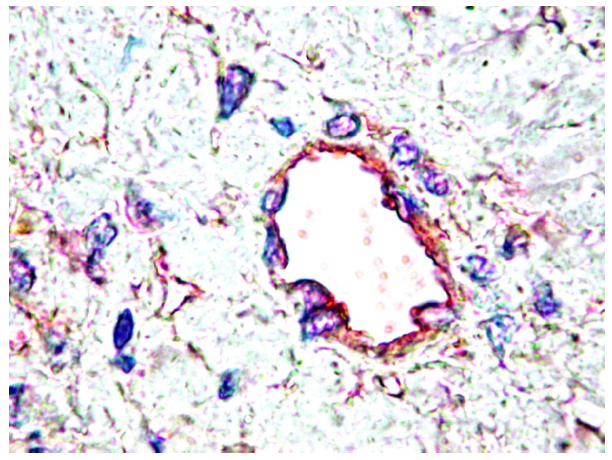
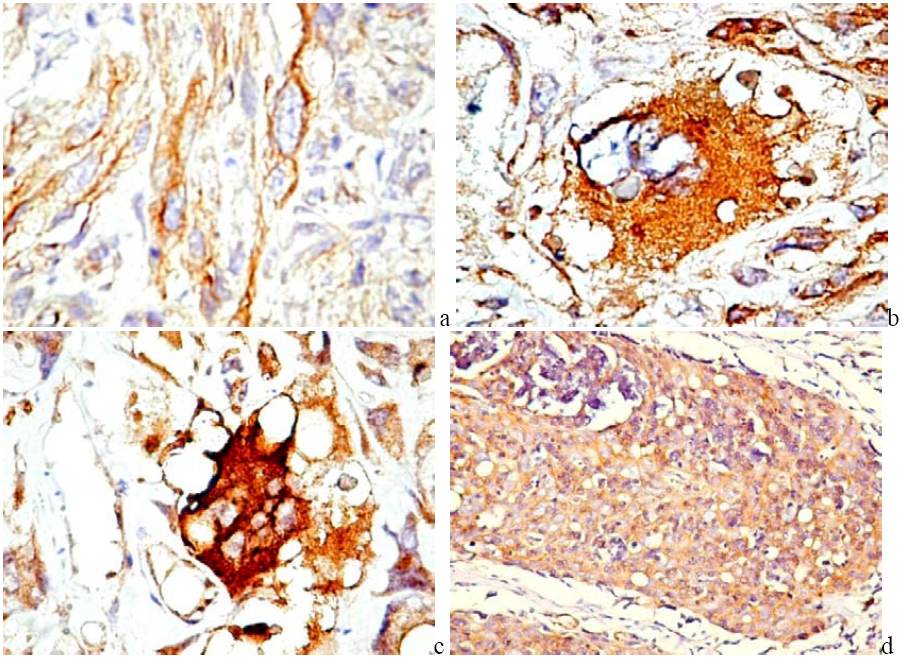
5. PDGF and PDGFR in Cancer
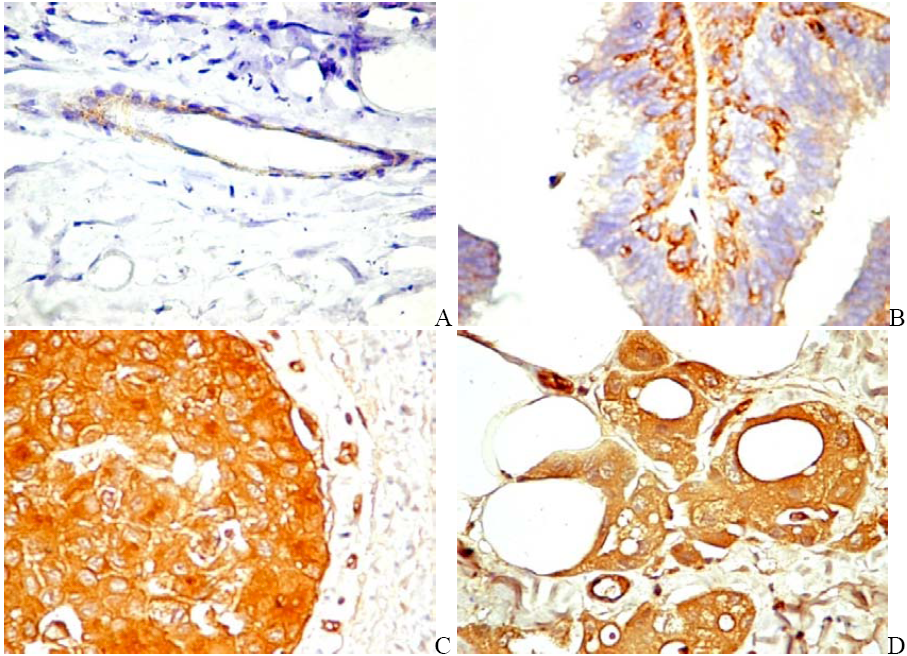
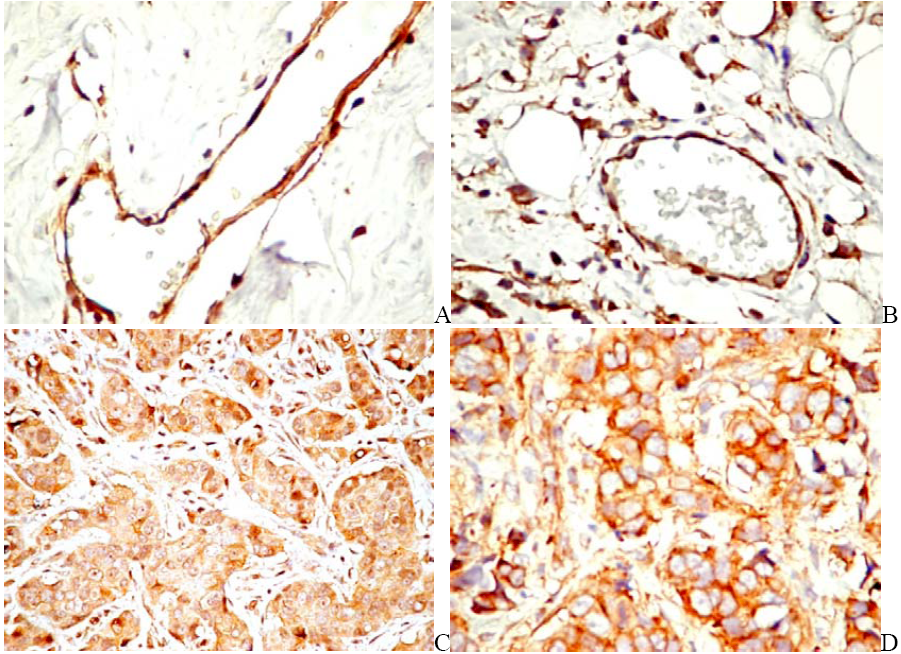
6. PDGF/PDGFR Are Expressed by Both Tumor and Stromal Cells
6.1. PDGF Expression in Tumor Cells
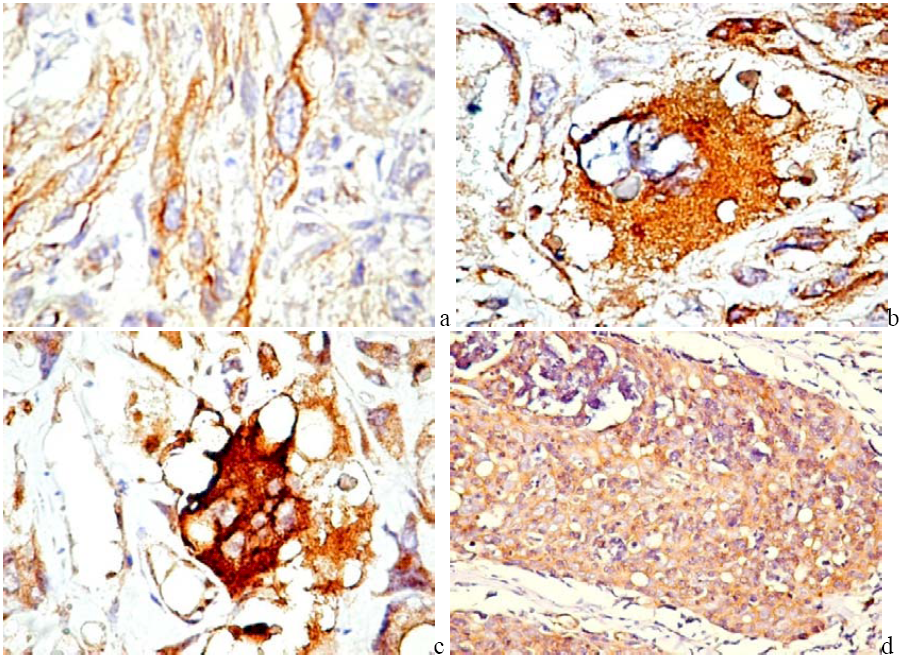
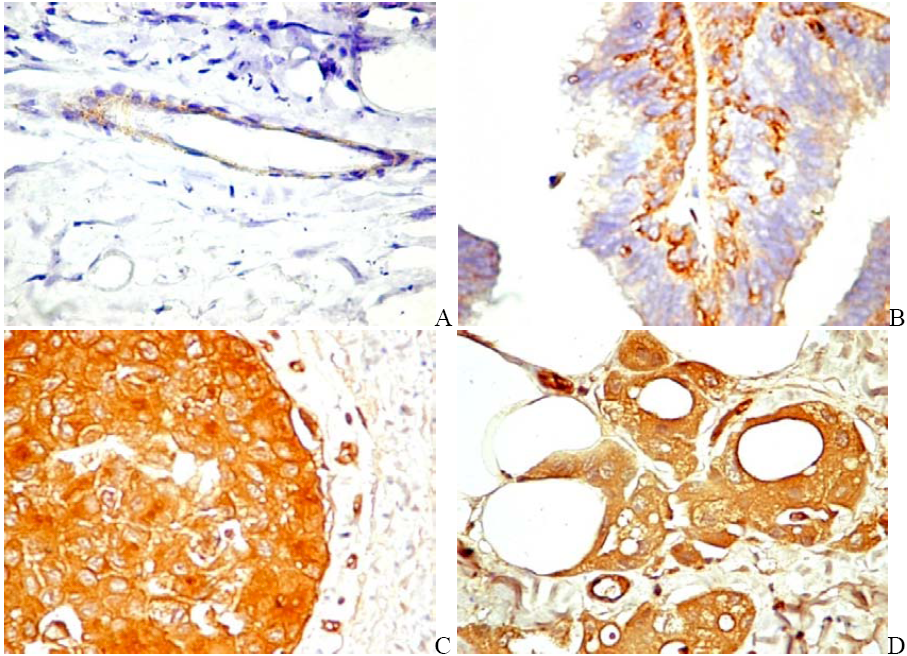
6.2. PDGFR in Experimental and Human Tumors

6.3. Targeting PDGF Signaling in the Tumor Stroma
7. PDGF and Tumor Experimental Models
8. PDGF/PDGFR Axis and Tumor Prognosis
| Tumor | PDGF-A | PDGF-B | PDGFRα | PDGFRβ | Correlation with | Ref |
|---|---|---|---|---|---|---|
| Nephroblastoma (n=62) | 50% | ND | 55% | ND | Progression | [93] |
| NSCLC (n=335) | 98% | 100% | 98% | 98% | LNM | [94] |
| Hodgkin lymphoma (n=65) | ND | ND | 95% | ND | ND | [95] |
| Non-Hodgkin lymphoma (n=50) | ND | ND | 48% | ND | No correlation | [96] |
| Recurrent ovarian cancer (n=44) | 88.4% | 69.8% | 90.9% | 88.6% | No response to imatinib therapy | [97] |
| Colorectal carcinoma (n=60) | ND | 60% | ND | ND | Vascular invasion | [98] |
| Osteosarcoma (n=54) | 80.4% | 75.4% | 79.6% | 86% | DFS for PDGF-A/PDGFRα | [99] |
| Cervical adenosquamous carcinoma (n=27) | ND | ND | 100% | ND | NS | [100] |
9. The Role of PDGF/PDGFR in Lymphangiogenesis
10. PDGF/PDGFR Axis and Therapy
11. Perspectives
Acknowledgements
References and Notes
- Folkman, J. Tumor angiogenesis: Therapeutic implications. New Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Ribatti, D. Endogenous inhibitors of angiogenesis: A historical review. Leuk. Res. 2009, 33, 638–644. [Google Scholar]
- Murukesh, N.; Five, C.; Jayson, G.C. Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br. J. Cancer 2010, 102, 8–18. [Google Scholar]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar]
- Ferrara, N. Molecular and biological properties of vascular endothelial growth factor. J. Mol. Med. 1999, 77, 527–543. [Google Scholar]
- Ferrara, N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am. J. Physiol. Cell. Physiol. 2001, 280, 1358–1366. [Google Scholar]
- Ferrara, N.; Kerbel, K.S. Angiogenesis as a therapeutic target. Nature Insight 2005, 438, 967–974. [Google Scholar] [CrossRef]
- Kohler, N.; Lipton, A. Platelets as a source of fibroblast growth-promoting activity. Exp. Cell. Res. 1974, 87, 297–301. [Google Scholar]
- Ross, R.; Glomset, J.; Kariya, B.; Harker, L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc. Natl. Acad. Sci. USA 1974, 71, 1207–1210. [Google Scholar] [CrossRef]
- Westermark, B.; Wasteson, A. A platelet factor stimulating human normal glial cells. Exp. Cell. Res. 1976, 98, 170–174. [Google Scholar]
- Heldin, C.H.; Westermark, B.; Wasteson, A. Platelet-derived growth factor: Purification and partial characterization. Proc. Natl. Acad. Sci. USA 1979, 76, 3722–3726. [Google Scholar]
- Ek, B.; Heldin, C.H. Characterization of a tyrosine-specific kinase activity in human fibroblast membranes stimulated by platelet-derived growth factor. J. Biol. Chem. 1982, 257, 10486–10492. [Google Scholar]
- Li, X.; Pontén, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Bäckström, G.; Hellström, M.; Boström, H.; Li, H.; Soriano, P.; Betsholtz, C.; Heldin, C.H.; Alitalo, K.; Ostman, A.; Eriksson, U. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat. Cell. Biol. 2000, 2, 302–309. [Google Scholar]
- Bergsten, E.; Uutela, M.; Li, X.; Pietras, K.; Ostman, A.; Heldin, C.H.; Alitalo, K.; Eriksson, U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell. Biol. 2001, 3, 512–516. [Google Scholar]
- Frederiksson, L.; Li, H.; Eriksson, U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar]
- Heldin, C.H.; Eriksson, U.; Ostman, A. New members of the platelet-derived growth factor family of mitogens. Arch. Biochem. Biophys. 2002, 398, 284–290. [Google Scholar]
- Heldin, C.H.; Westermark, M. Mechanisms of action an in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [PubMed]
- Magnusson, P.U.; Looman, C.; Ahgren, A.; Wu, Y.; Claesson-Welsh, L.; Heuchel, R.L. Platelet-derived growth factor receptor-beta constitutive activity promotes angiogenesis in vivo and in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2142–2149. [Google Scholar] [PubMed]
- Betscholtz, C. Biology of platelet-derived growth factors in development. Birth Defects Res. 2003, 69, 272–285. [Google Scholar]
- Őstman, A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004, 15, 275–286. [Google Scholar]
- Stice, L.L.; Vaziri, C.; Faller, D.V. Regulation of platelet-derived growth factor signaling by activated p21Ras. Front. Biosci. 1999, 15, D72–D86. [Google Scholar]
- Khachigian, L.M.; Fries, J.W.U.; Benz, M.W.; Conthron, D.T.; Collins, T. Novel cis-acting elements in the human platelet-derived growth factor B-chain core promoter that mediate gene expression in cultured vascular endothelial cells. J. Biol. Chem. 1994, 269, 22647–22656. [Google Scholar]
- Liang, Y.; Robinson, D.F.; Dennig, J.; Suske, G.; Fabl, W.E. Transcriptional regulation of the SIS/PDGF-B gene in human osteosarcoma cells by the Sp family of transcription factors. J. Biol. Chem. 1996, 271, 11792–11797. [Google Scholar]
- Rafty, L.A.; Khachigian, L.M. Sp1 phosphorylation regulates inducible expression of platelet-derived growth factor B-chain gene via atypical protein kinase C-ξ. Nucleic Acids Res. 2001, 29, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Li, W.L.; Yamada, Y.; Ueno, M.; Nishikawa, S.; Nishikawa, S.I.; Takakura, N. Platelet-derived growth factor receptor alpha is essential for establishing a microenvironment that supports definitive erythropoiesis. J. Biochem. 2006, 140, 267–273. [Google Scholar]
- Board, R.; Jayson, G.C. Platelet-derived growth factor receptor (PDGFR): A target for anticancer therapeutics. Drug Resist. Update. 2005, 8, 75–83. [Google Scholar]
- Laschke, M.W.; Elitzsch, A.; Vollmer, B.; Vajkoczy, P.; Menger, M.D. Combined inhibition of vascular endothelial growth factor (VEGF), fibroblast growth factor and platelet-derived growth factor, but not inhibition of VEGF alone, effectively suppress angiogenesis and vessel maturation in endometriotic lesions. Hum. Reprod. 2006, 21, 262–268. [Google Scholar] [PubMed]
- von Tell, D.; Armulik, A.; Betsholtz, C. Pericyte and vascular stability. Exp. Cell. Res. 2006, 312, 623–629. [Google Scholar]
- Kano, M.R.; Morishita, Y.; Iwata, C.; Iwasaka, S.; Watabe, T.; Ouchi, Y.; Miyazono, K.; Miyazawa, K. VEGF-A and FGF-2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF-B-PDGFRbeta signaling. J. Cell. Sci. 2005, 118, 3759–3768. [Google Scholar]
- Lo, I.C.; Lin, T.M.; Chou, L.H.; Liu, S.L.; Wu, L.W.; Shi, G.Y.; Wu, H.L.; Jiang, M.J. Ets-1 mediates platelet-derived growth factor-BB-induced thrombomodulin expression in human vascular smooth muscle cells. Cardiovascular Res. 2009, 91, 771–779. [Google Scholar]
- Sun, B.; Chen, B.; Zhao, Y.; Sun, W.; Chen, K.; Zhang, J.; Wei, Z.; Xiao, Z.; Dai, J. Crosslinking heparin to collagen scaffolds for the delivery of human platelet-derived growth factor. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2009, 91B, 366–372. [Google Scholar] [CrossRef]
- Cao, R.; Bråkenhielm, E.; Li, X.; Pietras, K.; Widenfalk, J.; Ostman, A.; Eriksson, U.; Cao, Y. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-alphaalpha and -alphabeta receptors. FASEB J. 2002, 16, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Betscholtz, C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004, 15, 215–228. [Google Scholar]
- Andrae, J.; Gallini, R.; Betscholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Develop. 2008, 22, 1276–1312. [Google Scholar]
- Lange, S.; Heger, J.; Euler, G.; Wartenberg, M.; Piper, H.M.; Sauer, H. Platelet-derived growth factor BB stimulates vasculogenesis of embryonic cell-derives endothelial cells by calcium-mediated generation of reactive oxygen species. Cardiovascular Res. 2009, 81, 159–168. [Google Scholar]
- Oikawa, T.; Onozawa, C.; Sakaguchi, M.; Morita, I.; Murota, S. Three isoforms of platelet-derived growth factors all have the capability to induce angiogenesis in vivo. Biol. Pharm. Bull. 1994, 17, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Uutela, M.; Laurén, J.; Bergsten, E.; Li, X.; Horelli-Kuitunen, N.; Eriksson, U.; Alitalo, K. Chromosomal location, exon structure, and vascular expression patterns of the human PDGFC and PDGFD genes. Circulation 2001, 103, 2242–2247. [Google Scholar] [PubMed]
- Shih, A.H.; Holland, E.C. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett. 2006, 232, 139–147. [Google Scholar]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar]
- Levéen, P.; Pekny, M.; Gebre-Medhin, S.; Swolin, B.; Larsson, E.; Betsholtz, C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994, 8, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar]
- French, W.J.; Creemers, E.E.; Tallquist, M.D. Platelet-derived growth factor receptors direct vascular development independent of vascular smooth muscle cell function. Mol. Cell. Biol. 2008, 28, 5646–5657. [Google Scholar]
- Lindahl, P.; Boström, H.; Karlsson, L.; Hellström, M.; Kalén, M.; Betsholtz, C. Role of platelet-derived growth factors in angiogenesis and alveogenesis. Curr. Top. Pathol. 1999, 93, 27–33. [Google Scholar]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Battegay, E.J.; Rupp, J.; Iruela-Arispe, L.; Sage, E.H.; Pech, M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J. Cell. Biol. 1994, 125, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Paye, J.M.; Phng, L.K.; Lanahan, A.A.; Gerhard, H.; Simons, M. Synectin-dependent regulation of arterial maturation. Dev. Dyn. 2009, 238, 604–610. [Google Scholar]
- Rolny, C.; Nilsson, I.; Magnusson, P.; Armulik, A.; Jakobsson, L.; Wentzel, P.; Lindblom, P.; Norlin, J.; Betsholtz, C.; Heuchel, R.; Welsh, M.; Claesson-Welsh, L. Platelet-derived growth factor receptor-β promotes early endothelial cell differentiation. Blood 2006, 108, 1877–1886. [Google Scholar]
- Dong, J.; Grunstein, J.; Tejada, M.; Peale, F.; Frantz, G.; Liang, W.C.; Bai, W.; Yu, L.; Kowalski, J.; Liang, X.; Fuh, G.; Gerber, H.P.; Ferrara, N. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004, 23, 2800–2810. [Google Scholar]
- Guo, P.; Hu, B.; Gu, W.; Xu, L.; Wang, D.; Huang, H.J.S.; Cavenee, W.K.; Cheng, S.Y. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am. J. Pathol. 2003, 162, 1083–1093. [Google Scholar]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar]
- Di Tomaso, E.; London, N.; Fuja, D.; Logie, J.; Tyrrell, J.A.; Kamoun, W.; Munn, L.L.; Jain, R.K. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS ONE 2009, 4, e5123. [Google Scholar]
- Li, C.; Shintani, S.; Terakado, N.; Klosek, S.K.; Ishikawa, T.; Nakashiro, K.; Hamakawa, H. Microvessel density and expression of vascular endothelial growth factor, basic fibroblast growth factor, and platelet-derived growth factor in oral squamous cell carcinoma. Int. J. Oral Maxilofac. Surg. 2005, 34, 559–563. [Google Scholar] [CrossRef]
- Fujimoto, K.; Hosotani, R.; Wada, M.; Lee, J.-U.; Koshiba, T.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Doi, R.; Imamura, R. Expression of two angiogenic factors, vascular endothelial growth factor and platelet-derived endothelial cell growth factor in human pancreatic cancer, and its relationship to angiogenesis. Eur. J. Cancer. 1998, 34, 1439–1447. [Google Scholar] [PubMed]
- Saeki, T.; Tanada, M.; Takashima, S.; Saeki, H.; Takiyama, W.; Nishimoto, N.; Moriwaki, S. Correlation between expression of platelet-derived endothelial cell growth factor (thymidine phosphorylase) and microvessel density in early stage human colon carcinomas. Jpn. J. Clin. Oncol. 1997, 27, 227–230. [Google Scholar]
- Takahashi, Y.; Bucana, C.D.; Liu, W.; Yoneda, J.; Kitadai, Y.; Cleary, K.R.; Ellis, L.M. Platelet-derived endothelial cell growth factor in human colon cancer angiogenesis: Role of infiltrating cells. J. Natl. Cancer Inst. 1996, 88, 1146–1151. [Google Scholar]
- Abramsson, A.; Lindblom, P.; Betsholtz, C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Invest. 2003, 112, 1142–1151. [Google Scholar]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar]
- Shih, A.H.; Dai, C.; Hu, X.; Rosenblum, M.K.; Koutcher, J.A.; Holland, E.C. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004, 64, 4783–4789. [Google Scholar]
- Uehara, H.; Kim, S.J.; Karashima, T.; Shepherd, D.L.; Fan, D.; Tsan, R.; Killion, J.J.; Logothetis, C.; Mathew, P.; Fidler, I.J. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. J. Natl. Cancer Inst. 2003, 95, 558–570. [Google Scholar]
- Seki, N.; Kodama, J.; Hongo, A.; Miyagi, M.; Yoshinouchi, M.; Kudo, T. Vascular endothelial growth factor and platelet-derived endothelial cell growth factor expression are implicated in the angiogenesis of endometrial cancer. Eur. J. Cancer 2000, 36, 68–73. [Google Scholar]
- De Marchis, F.; Ribatti, D.; Giampietri, C.; Lentini, A.; Faraone, D.; Scoccianti, M.; Capogrossi, M.C.; Facchiano, A. Platelet-derived growth factor inhibits basic fibroblast growth factor angiogenic properties in vitro and in vivo through its alpha receptor. Blood 2002, 99, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Werth, C.; Stuhlmann, D.; Cat, B.; Steinbrenner, H.; Alili, L.; Sies, H.; Brenneisen, P. Stromal resistance of fibroblasts against oxidative damage: Involvement of tumor cell-secreted platelet-derived growth factor (PDGF) and phosphoinositide 3-kinase (PI3K) activation. Carcinogenesis 2008, 29, 404–410. [Google Scholar]
- Tejada, M.L.; Yu, L.; Dong, J.; Jung, K.; Meng, G.; Peale, F.V.; Frantz, G.D.; Hall, L.; Liang, X.; Gerber, H.P.; Ferrara, N. Tumor-driven paracrine platelet-derived growth factor receptor alpha signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin. Cancer Res. 2006, 12, 2676–2688. [Google Scholar]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 2003, 111, 1287–1295. [Google Scholar]
- Carvalho, I.; Milanezi, F.; Martins, A.; Reis, R.M.; Schmitt, F. Overexpression of platelet-derived growth factor receptor alpha in breast cancer is associated with tumour progression. Breast Cancer Res. 2005, 7, R788–R795. [Google Scholar]
- Plate, K.H.; Breier, G.; Farrell, C.L.; Risau, W. Platelet-derived growth factor receptor-beta is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab. Invest. 1992, 67, 529–534. [Google Scholar] [PubMed]
- Schmitt, J.; Matei, D. Platelet-derived growth factor pathway inhibitors in ovarian cancer. Clin. Ovarian Cancer 2008, 1, 120–126. [Google Scholar]
- Vrekoussis, T.; Stathopoulos, E.N.; Kafousi, M.; Navrozoglou, I.; Zoras, O. Expression of endothelial PDGF receptors alpha and beta in breast cancer: Up-regulation of endothelial PDGF receptor beta. Oncol. Rep. 2007, 17, 1115–1119. [Google Scholar]
- Lev, D.C.; Kim, S.J.; Onn, A.; Stone, V.; Nam, D.H.; Yazici, S.; Fidler, I.J.; Price, J.E. Inhibition of platelet-derived growth factor receptor signaling restricts the growth of human breast cancer in the bone of nude mice. Clin. Cancer Res. 2005, 11, 306–314. [Google Scholar]
- Chott, A.; Sun, Z.; Morganstern, D.; Pan, J.; Li, T.; Susani, M.; Mosberger, I.; Upton, M.P.; Bubley, G.J.; Balk, S.P. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor. Am. J. Pathol. 1999, 155, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, N.; Yonemitsu, Y.; Shikada, Y.; Onimaru, M.; Tanii, M.; Okano, S.; Kaneko, K.; Hasegawa, M.; Hashizume, M.; Maehara, Y.; Sueishi, K. Essential role of PDGFRalpha-p70S6K signaling in mesenchymal cells during therapeutic and tumor angiogenesis in vivo: Role of PDGFRalpha during angiogenesis. Circ. Res. 2004, 94, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Bran, B.; Bran, G.; Hörmann, K.; Riedel, F. The platelet-derived growth factor receptor as a target for vascular endothelial growth factor-mediated anti-angiogenetic therapy in head and neck cancer. Int. J. Oncol. 2009, 34, 255–261. [Google Scholar]
- Furuhashi, M.; Sjöblom, T.; Abramsson, A.; Ellingsen, J.; Micke, P.; Li, H.; Bergsten-Folestad, E.; Eriksson, U.; Heuchel, R.; Betsholtz, C.; Heldin, C.H.; Ostman, A. Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Res. 2004, 64, 2725–2733. [Google Scholar]
- Suzuki, S.; Heldin, C.H.; Heuchel, R.L. Platelet-derived growth factor receptor-beta, carrying the activating mutation D849N, accelerates the establishment of B16 melanoma. BMC Cancer 2007, 7, 224. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.S.; Johnson, M.M.; Bauer, R.L.; Hudkins, K.L.; Gilbertson, D.G.; Riehle, K.J.; Yeh, M.M.; Alpers, C.E.; Fausto, N. Targeting stromal cells for the treatment of platelet-derived growth factor C-induced hepatocellular carcinogenesis. Differentiation 2007, 75, 843–852. [Google Scholar]
- Nissen, L.J.; Cao, R.; Hedlund, E.M.; Wang, Z.; Zhao, X.; Wetterskog, D.; Funa, K.; Bråkenhielm, E.; Cao, Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J. Clin. Invest. 2007, 117, 2766–2777. [Google Scholar]
- Lederle, W.; Stark, H.J.; Skobe, M.; Fusenig, N.E.; Mueller, M.M. Platelet-derived growth factor-BB controls epithelial tumor phenotype by differential growth factor regulation in stromal cells. Am. J. Pathol. 2006, 169, 1767–1783. [Google Scholar]
- Beppu, K.; Jaboine, J.; Merchant, M.S.; Mackall, C.L.; Thiele, C.J. Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J. Natl. Cancer Inst. 2004, 96, 46–55. [Google Scholar]
- Bäckman, U.; Christofferson, R. The selective class III/V receptor tyrosine kinase inhibitor SU11657 inhibits tumor growth and angiogenesis in experimental neuroblastomas grown in mice. Pediatr. Res. 2005, 57, 690–695. [Google Scholar]
- Roberts, W.G.; Whalen, P.M.; Soderstrom, E.; Moraski, G.; Lyssikatos, J.P.; Wang, H.F.; Cooper, B.; Baker, D.A.; Savage, D.; Dalvie, D.; Atherton, J.A.; Ralston, S.; Szewc, R.; Kath, J.C.; Lin, J.; Soderstrom, C.; Tkalcevic, G.; Cohen, B.D.; Pollack, V.; Barth, W.; Hungerford, W.; Ung, E. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005, 65, 957–966. [Google Scholar]
- Shen, J.; Vil, M.D.; Zhang, H.; Tonra, J.R.; Rong, L.L.; Damoci, C.; Prewett, M.; Deevi, D.S.; Kearney, J.; Surguladze, D.; Jimenez, X.; Iacolina, M.; Bassi, R.; Zhou, K.; Balderes, P.; Mangalampalli, V.R.M.; Loizos, N.; Ludwig, D.L.; Zhu, Z. An antibody directed against PDGF receptor β enhances the antitumor and anti-angiogenic activities of an anti-VEGF receptor 2 antibody. Biochem. Biophys. Res. Commun. 2007, 357, 1142–1147. [Google Scholar]
- Henriksen, R.; Funa, K.; Wilander, E.; Bäckström, T.; Ridderheim, M.; Oberg, K. Expression and prognostic significance of platelet-derived growth factor and its receptors in epithelial ovarian neoplasms. Cancer Res. 1993, 53, 4550–4554. [Google Scholar]
- Antoniades, H.N.; Galanopoulos, T.; Neville-Golden, J.; O'Hara, C.J. Malignant epithelial cells in primary human lung carcinomas coexpress in vivo platelet-derived growth factor (PDGF) and PDGF receptor mRNAs and their protein products. Proc. Natl. Acad. Sci. USA 1992, 89, 3942–3946. [Google Scholar]
- Chung, C.K.; Antoniades, H.N. Expression of c-sis/platelet-derived growth factor B, insulin-like growth factor I, and transforming growth factor alpha messenger RNAs and their respective receptor messenger RNAs in primary human gastric carcinomas: In vivo studies with in situ hybridization and immunocytochemistry. Cancer Res. 1992, 52, 3453–3459. [Google Scholar] [PubMed]
- Seymour, L.; Dajee, D.; Bezwoda, W.R. Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res. Treat. 1993, 26, 247–252. [Google Scholar]
- Krasagakis, K.; Garbe, C.; Orfanos, C.E. Cytokines in human melanoma cells: Synthesis, autocrine stimulation and regulatory functions - an overview. Melanoma Res. 1993, 3, 425–433. [Google Scholar]
- Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Busund, L.-T.; Bremnes, R. Prognostic impact of platelet-derived growth factors in non-small cell lung cancer tumor and stromal cells. J. Thoracic. Oncol. 2008, 3, 963–970. [Google Scholar]
- Faraone, D.; Aguzzi, M.S.; Toietta, G.; Facchiano, A.M.; Facchiano, F.; Magenta, A.; Martelli, F.; Truffa, S.; Cesareo, E.; Ribatti, D.; Capogrossi, M.C.; Facchiano, A. Platelet-derived growth factor-receptor α strongly inhibits melanoma growth in vitro and in vivo. Neoplasia 2009, 11, 732–742. [Google Scholar] [PubMed]
- Wyman, K.; Atkins, M.B.; Prieto, V.; Eton, O.; McDermott, D.F.; Hubbard, F.; Byrnes, C.; Sanders, K.; Sosman, J.A. Multicenter Phase II trial of high-dose imatinib mesylate in metastatic melanoma: Significant toxicity with no clinical efficacy. Cancer 2006, 106, 2005–2011. [Google Scholar]
- Ghanem, M.; Nijman, R.; Safan, M.; van der Kwast, T.; Vansteenbrugge, G. Expression and prognostic value of platelet-derived growth factor AA and its receptor α in nephroblastoma. BJU Intern. 2010. [Google Scholar]
- Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Busund, L.-T.; Bremnes, R.M. Co-expression of PDGF-B and VEGFR-3 strongly correlates with lymph node metastasis and poor survival in non-small-cell lung cancer. Ann. Oncol. 2010, 21, 223–231. [Google Scholar]
- Pessam, F.H.; Alexandrakis, M.G.; Kafousi, M.; Darivianaki, K.; Tsirakis, G.; Roussou, P.A.; Stathoupoulos, E.N.; Siafakas, N.M. Histological expression of angiogenic factors: VEGF, PDGFRalpha, and HIF-1alpha in Hodgkin lymphoma. Pathol. Res. Pract. 2009, 205, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-P.; Chang, K.-C.; Su, W.-C.; Chen, T.-Y. The expression and prognostic significance of platelet-derived growth factor receptor alpha in mature T- and natural killer-cell lymphomas. Ann. Hematol. 2008, 87, 985–990. [Google Scholar]
- Schilder, R.J.; Sill, M.W.; Lee, R.B.; Shaw, T.J.; Sentermann, M.K.; Klein-Szanto, A.J.; Miner, Z.; Vanderhyden, B.C. Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: A gynecologic oncology group study. J. Clin. Oncol. 2008, 26, 3418–3425. [Google Scholar]
- Nakamura, Y.; Tanaka, F.; Yoshikawa, Y.; Mimori, K.; Inoue, H.; Yanaga, K.; Mori, M. PDGF-BB is novel prognostic factor in colorectal cancer. Ann. Surg. Oncol. 2008, 15, 2129–2136. [Google Scholar]
- Kubo, T.; Piperdi, S.; Rosenblum, J.; Antonescu, C.R.; Chen, W.; Kim, H.S.; Huvos, A.G.; Sowers, R.; Meyers, P.A.; Healey, J.H.; Gorlick, R. Platelet-derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer 2008, 112, 2119–2129. [Google Scholar]
- Longatto-Filho, A.; Pinheiro, C.; Martinho, O.; Moreira, M.A.R.; Ribeiro, L.F.J.; Queiroz, G.S.; Schmitt, F.C.; Baltazar, F.; Reis, R.M. Molecular characterization of EGFR, PDGFRA amd VEGFR2 in cervical adenosquamous carcinoma. BMC Cancer 2009, 9, 212. [Google Scholar]
- Wang, Z.; Kong, D.; Li, Y.; Sarkar, F.H. PDGF-D signaling: A novel target in cancer therapy. Curr. Drug Targets 2009, 10, 38–41. [Google Scholar]
- Coltrera, M.D.; Wang, J.; Porter, P.L.; Gown, A.M. Expression of platelet-derived growth factor B-chain and the platelet-derived growth factor receptor beta subunit in human breast tissue and breast carcinoma. Cancer Res. 1995, 55, 2703–2708. [Google Scholar]
- Cao, R.; Bjorndahl, M.A.; Religa, P.; Clasper, S.; Garvin, S.; Galter, D.; Neister, B.; Ikomi, F.; Tritsaris, K.; Dissing, S.; Ohhashi, T.; Jackson, D.G.; Cao, Y. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell 2004, 6, 333–345. [Google Scholar]
- Cao, R.; Bråkenhielm, E.; Pawliuk, R.; Wariaro, D.; Post, M.J.; Wahlberg, E.; Leboulch, P.; Cao, Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat. Med. 2003, 9, 604–613. [Google Scholar]
- Cristofanilli, M.; Morandi, P.; Krishnamurthy, S.; Reuben, J.M.; Lee, B.-N.; Francis, D.; Booser, D.J.; Green, M.C.; Arun, G.K.; Pusztai, L.; Lopez, A.; Islam, R.; Valero, V.; Hortobagyi, G.N. Imatinib mesylate (GleevecR) in advanced breast cancer-expressing c-kit or PDGFR-β: Clinical activity and biological correlations. Ann. Oncol. 2008, 19, 1713–1719. [Google Scholar]
- Kim, S.J.; Uehara, H.; Yazici, S.; Busby, J.E.; Nakamura, T.; He, J.; Maya, M.; Logothetis, C.; Mathew, P.; Wang, X.; Do, K.A.; Fan, D.; Fidler, I.J. Targeting platelet-derived growth factor receptor on endothelial cells of multidrug-resistant prostate cancer. J. Natl. Cancer Inst. 2006, 98, 783–793. [Google Scholar]
- Mathew, P.; Thall, P.F.; Jones, D.; Perez, C.; Bucana, C.; Troncoso, P.; Kim, S.J.; Fidler, I.J.; Logothetis, C. Platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel: A modular phase I trial in androgen-independent prostate cancer. J. Clin. Oncol. 2004, 22, 3323–3329. [Google Scholar]
- Servidei, T.; Riccardi, A.; Sanguinetti, M.; Dominici, C.; Riccardi, R. Increased sensitivity to the platelet-derived growth factor (PDGF) receptor inhibitor STI571 in chemoresistant glioma cells is associated with enhanced PDGF-BB-mediated signaling and STI571-induced Akt inactivation. J. Cell. Physiol. 2006, 208, 220–228. [Google Scholar]
- Pietras, K.; Hanahan, D. A multitargeted, metronomic, and maximum-tolerated dose "chemo-switch" regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J. Clin. Oncol. 2005, 23, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Tam, B.Y.; Sennino, B.; Gray, J.T.; Yuan, J.; Jocson, A.; Nayak, N.R.; Mulligan, R.C.; McDonald, D.M.; Kuo, C.J. Soluble receptor-mediated selective inhibition of VEGFR and PDGFRbeta signaling during physiologic and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 10185–10190. [Google Scholar]
- Timke, C.; Zieher, H.; Roth, A.; Hauser, K.; Lipson, K.E.; Weber, K.J.; Debus, J.; Abdollahi, A.; Huber, P.E. Combination of vascular endothelial growth factor receptor/platelet-derived growth factor receptor inhibition markedly improves radiation tumor therapy. Clin. Cancer Res. 2008, 14, 2210–2219. [Google Scholar]
- Kontovinis, L.F.; Papazisis, K.T.; Touplikioti, P.; Andreadis, C.; Mouratidou, D.; Kortsaris, A.H. Sunitinib treatment for patients with clear-cell metastatic renal cell carcinoma: Clinical outcome and plasma angiogenesis markers. BMC Cancer 2009, 9, 82. [Google Scholar]
- Coluccia, A.M.L.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammaco, D.; Tassone, P.; Ribatti, D.; Gambacorti-Passerini, C.; Vacca, A. Validation of PDGFR2 and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 112, 1345–1356. [Google Scholar]
- Sennino, B.; Kuhnert, F.; Tabruyn, S.P.; Mancuso, M.R.; Hu-Lowe, D.D.; Kuo, C.J.; McDonald, D.M. Cellular source and amount of vascular endothelial growth factor and platelet-derived growth factor in tumors determine response to angiogenesis inhibitors. Cancer Res. 2009, 69, 4527–4536. [Google Scholar]
- Kim, K.B.; Eton, O.; Davis, D.W.; Frazier, M.L.; McConkey, D.J.; Diwan, A.H.; Papadopoulos, N.E.; Bedikian, A.T.; Camacho, L.H.; Ross, M.I.; Cormier, J.N.; Gershenwald, J.E.; Lee, J.E.; Mansfield, P.F.; Billings, L.A.; Ng, C.S.; Charnsangavej, C.; Bar-Eli, M.; Johnson, M.M.; Murgo, A.J.; Prieto, V.G. Phase II trial of imatinib mesylate in patients with metastatic melanoma. Br. J. Cancer 2008, 99, 734–740. [Google Scholar]
- Dispenzieri, A.; Gertz, M.A.; Lacy, M.Q.; Geyer, S.M.; Greipp, P.R.; Rajkumar, S.V.; Kimlinger, T.; Lust, J.A.; Fonseca, R.; Allred, J.; Witzig, T.E. A phase II trial of imatinib in patients with refractory/relapsed myeloma. Leuk. Lymphoma 2006, 47, 39–42. [Google Scholar]
- Eckel, F.; von Delius, S.; Mayr, M.; Dobritz, M.; Fend, F.; Hosius, C.; Schleyer, E.; Schulte-Frohlinde, E.; Schmid, R.M.; Lersch, C. Pharmacokinetic and clinical phase II trial of imatinib in patients with impaired liver function and advanced hepatocellular carcinoma. Oncology 2005, 69, 363–371. [Google Scholar]
- Sebti, S.M.; Hamilton, A.D. Design of growth factor antagonists with antiangiogenic and antitumor properties. Oncogene 2000, 19, 6566–6573. [Google Scholar]
- Lamy, S.; Beaulieu, E.; Labbé, D.; Bédard, V.; Moghrabi, A.; Barrette, S.; Gingras, D.; Béliveau, R. Delphinidin, a dietary anthocyanidin, inhibits platelet-derived growth factor ligand/receptor (PDGF/PDGFR) signaling. Carcinogenesis 2008, 29, 1033–10341. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Alitalo, K. Double target for tumor mass destruction. J. Clin. Invest. 2003, 111, 1277–1280. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572-599. https://doi.org/10.3390/ph3030572
Raica M, Cimpean AM. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals. 2010; 3(3):572-599. https://doi.org/10.3390/ph3030572
Chicago/Turabian StyleRaica, Marius, and Anca Maria Cimpean. 2010. "Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy" Pharmaceuticals 3, no. 3: 572-599. https://doi.org/10.3390/ph3030572
APA StyleRaica, M., & Cimpean, A. M. (2010). Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals, 3(3), 572-599. https://doi.org/10.3390/ph3030572