Allosteric Modulation of αβδ GABAA Receptors

Abstract
:1. Introduction
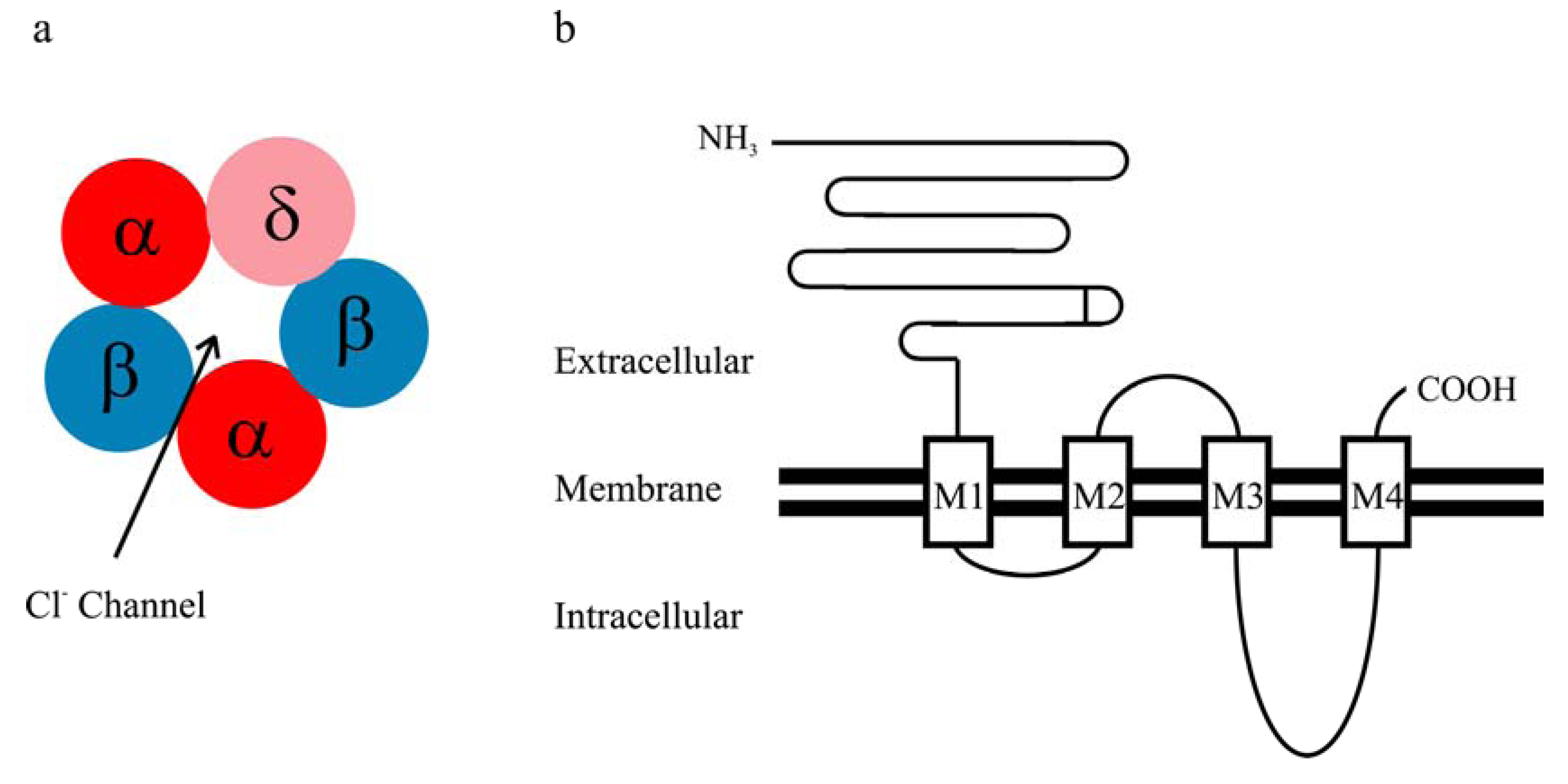
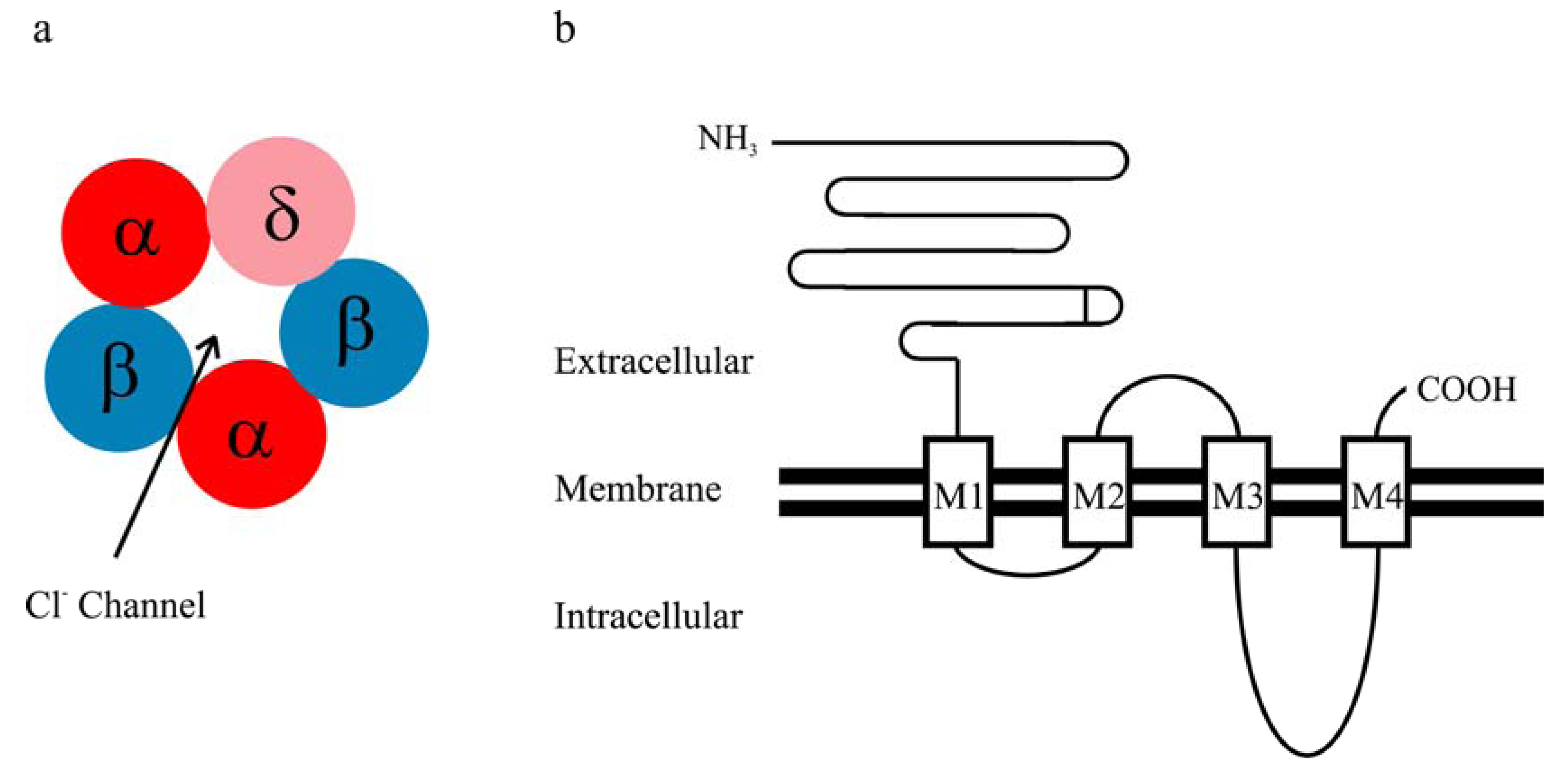
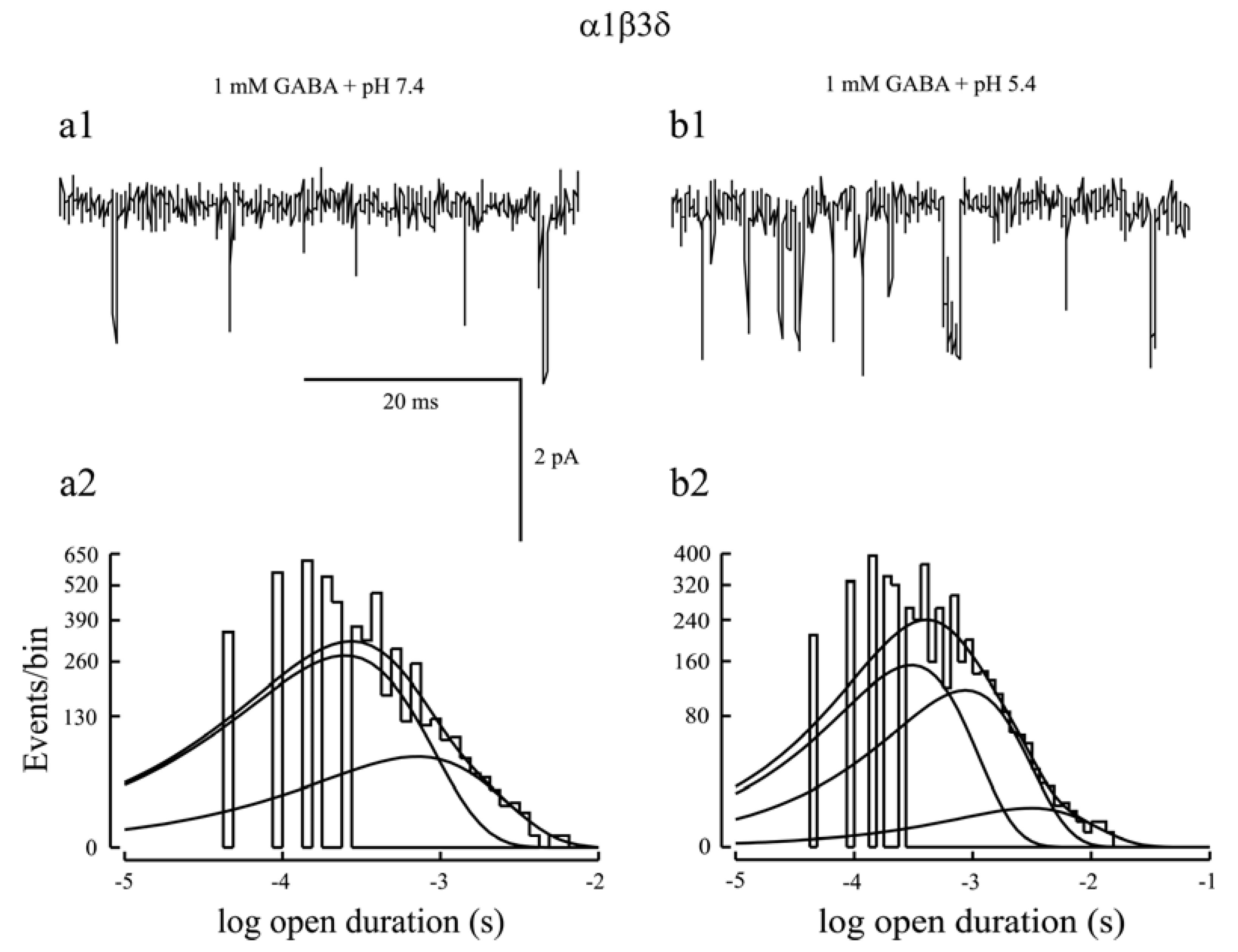
2. Kinetic Properties of αβδ GABAA Receptor Currents

{kind=link}
{kind=link}
{kind=link}
| α1β3δ | α4β3δ | α5β3δ | α6β3δ | |
|---|---|---|---|---|
| Desensitization | 24.8 ± 6.5% | 53.4 ± 2.1% | 36.2 ± 4.4% | 44.7 ± 3.9% |
| Deactivation | 125.3 ± 10.5 ms | 117.8 ± 13.5 ms | 345.7 ± 87.4 ms | 449.1 ± 80.9 ms |
3. Modulation of αβδ Receptors by Anesthetics
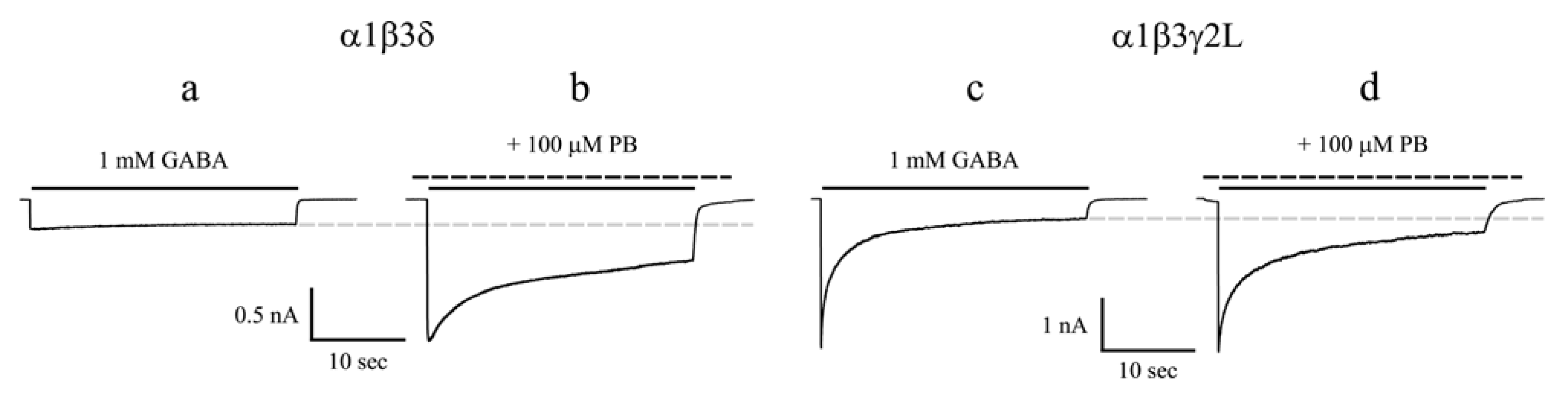
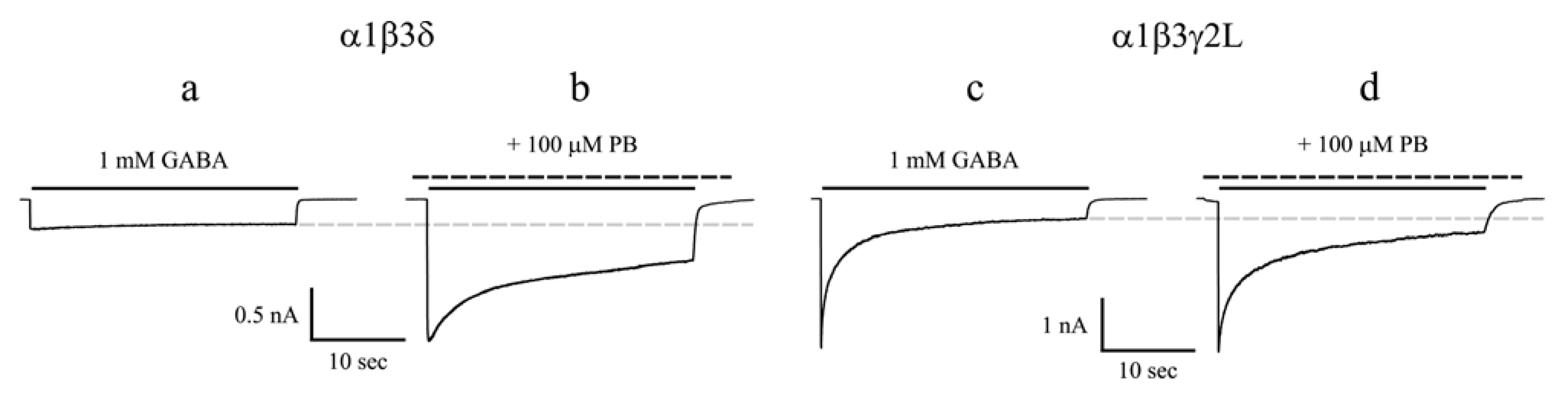
3.1. Barbiturates
3.2. Neurosteroids
3.3. Other Anesthetics
4. Modulation of αβδ Receptors by Ethanol
5. Mechanisms for Positive Modulation of αβδ Receptors
6. Conclusions
Acknowledgements
References
- Olsen, R.W.; Macdonald, R.L. GABAA receptor complex: structure and function. In Glutamate and GABA Receptors and Transporters: Structure, Function and Pharmacology; Egebjerg, J., Schousboe, A., Krogsgaard-Larsen, P., Eds.; Taylor and Francis: London, U.K., 2002; pp. 202–235. [Google Scholar]
- Beleboni, R.O.; Carolino, R.O.; Pizzo, A.B.; Castellan-Baldan, L.; Coutinho-Netto, J.; dos Santos, W.F.; Coimbra, N.C. Pharmacological and biochemical aspects of GABAergic neurotransmission: pathological and neuropsychobiological relationships. Cell. Mol. Neurobiol. 2004, 24, 707–728. [Google Scholar]
- Schousboe, A.; Waagepetersen, H.S. GABA: homeostatic and pharmacological aspects. Prog. Brain Res. 2007, 160, 9–19. [Google Scholar]
- Roberts, E. GABA: the road to neurotransmitter status. In Benzodiazepine/GABA Receptors and Chloride Channels: Structure and Functional Properties; Olsen, R.W., Venter, C.J., Eds.; Wiley: New York, USA, 1986; pp. 1–39. [Google Scholar]
- Otsuka, M. Establishment of GABA as an inhibitory neurotransmitter at Crustacean neuromuscular junction and in the mammalian central nervous system. In GABA: Receptors, Transporters and Metabolism; Tanaka, C., Bowery, N.G., Eds.; Birkhauser: Basel, Switzerland, 1996; pp. 1–6. [Google Scholar]
- Soudijn, W.; van Wijngaarden, I. The GABA transporter and its inhibitors. Curr. Med. Chem. 2000, 7, 1063–1079. [Google Scholar]
- Keros, S.; Hablitz, J.J. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J. Neurophysiol. 2005, 94, 2073–2085. [Google Scholar]
- Mody, I.; Pearce, R.A. Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci. 2004, 27, 569–575. [Google Scholar]
- Farrant, M.; Nusser, Z. Variation on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar]
- Belelli, D.; Harrison, N.L.; Maguire, J.; Macdonald, R.L.; Walker, M.C.; Cope, D.W. Extrasynaptic GABAA receptors: form, pharmacology, and function. J. Neurosci. 2009, 29, 12757–12763. [Google Scholar]
- Zheleznova, N.N.; Sedelnikova, A.; Weiss, D.S. Function and modulation of δ-containing GABAA receptors. Psychoneuroendocrinology 2009, 34S, S67–S73. [Google Scholar]
- Belelli, D.; Peden, D.R.; Rosahl, T.W.; Wafford, K.A.; Lambert, J.J. Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J. Neurosci. 2005, 25, 11513–11520. [Google Scholar]
- Cope, D.W.; Hughes, S.W.; Crunelli, V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J. Neurosci. 2005, 25, 11553–11563. [Google Scholar]
- McKernan, R.M.; Whiting, P.J. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996, 19, 139–143. [Google Scholar]
- Pirker, S.; Schwarzer, C.; Wieselthaler, A.; Sieghart, W.; Sperk, G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 2000, 101, 815–850. [Google Scholar]
- Jones, A.; Korpi, E.R.; McKernan, R.M.; Pelz, R.; Nusser, Z.; Makela, R.; Mellor, J.R.; Pollard, S.; Bahn, S.; Stephenson, F.A.; Randall, A.D.; Sieghart, W.; Somogyi, P.; Smith, A.J.; Wisden, W. Ligand-gated ion channel subunit partnerships: GABAA receptor α6 subunit gene inactivation inhibits δ subunit expression. J. Neurosci. 1997, 17, 1350–1362. [Google Scholar]
- Poltl, A.; Hauer, B.; Fuchs, K.; Tretter, V.; Sieghart, W. Subunit composition and quantitative importance of GABAA receptor subtypes in the cerebellum of of mouse and rat. J. Neurochem. 2003, 87, 1444–1455. [Google Scholar]
- Sur, C.; Farrar, S.J.; Kerby, J.; Whiting, P.J.; Atack, J.R.; McKernan, R.M. Preferential coassembly of α4 and δ subunits of the γ-aminobutyric acidA receptor in rat thalamus. Mol. Pharmacol. 1999, 56, 110–115. [Google Scholar]
- Jia, F.; Pignataro, L.; Schofield, C.M.; Yue, M.; Harrison, N.L.; Goldstein, P.A. An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J. Neurophysiol. 2005, 94, 4491–4501. [Google Scholar]
- Chandra, D.; Jia, F.; Liang, J.; Peng, Z.; Suryanarayanan, A.; Werner, D.F.; Spigelman, I.; Houser, C.R.; Olsen, R.W.; Harrison, N.L.; Homanics, G.E. GABAA receptor α4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc. Natl. Acad. Sci. USA 2006, 103, 15230–15235. [Google Scholar]
- Drasbek, K.R.; Hoestgaard-Jensen, K.; Jensen, K. Modulation of extrasynaptic THIP conductances by GABAA-receptor modulators in mouse neocortex. J. Neurophysiol. 2007, 97, 2293–2300. [Google Scholar]
- Glykys, J.; Peng, Z.; Chandra, D.; Homanics, G.E.; Houser, C.R.; Mody, I. A new naturally occurring GABAA receptor subunit partnership with high sensitivity to ethanol. Nat. Neurosci. 2007, 10, 40–48. [Google Scholar]
- Saxena, N.C.; Macdonald, R.L. Assembly of GABAA receptor subunit: role of the δ subunit. J. Neurosci. 1994, 14, 7077–7086. [Google Scholar]
- Wohlfarth, K.M.; Bianchi, M.T.; Macdonald, R.L. Enhanced neurosteroid potentiation of ternary GABAA receptors containing the δ subunit. J. Neurosci. 2002, 22, 1541–1549. [Google Scholar]
- Feng, H.J.; Macdonald, R.L. Multiple actions of propofol on αβγ and αβδ GABAA receptors. Mol. Pharmacol. 2004, 66, 1517–1524. [Google Scholar]
- Feng, H.J.; Bianchi, M.T.; Macdonald, R.L. Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol. Pharmacol. 2004, 66, 988–1003. [Google Scholar]
- Brown, N.; Kerby, J.; Bonnert, T.P.; Whiting, P.J.; Wafford, K.A. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br. J. Pharmacol. 2002, 136, 965–974. [Google Scholar]
- Feng, H.J.; Kang, J.Q.; Song, L.; Dibbens, L.; Mulley, J.; Macdonald, R.L. δ subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of α4β2δ GABAA receptors. J. Neurosci. 2006, 26, 1499–1506. [Google Scholar]
- Feng, H.J.; Botzolakis, E.J.; Macdonald, R.L. Context-dependent modulation of αβγ and αβδ GABAA receptors by penicillin: implication for phasic and tonic inhibition. Neuropharmacology 2009, 56, 161–173. [Google Scholar]
- Lagrange, A.H.; Botzolakis, E.J.; Macdonald, R.L. Enhanced macroscopic desensitization shapes the response of α4 subtype-containing GABAA receptors to synaptic and extrasynaptic GABA. J. Physiol. 2007, 578, 655–676. [Google Scholar]
- Dibbens, L.M.; Feng, H.J.; Richards, M.C.; Harkin, L.A.; Hodgson, B.L.; Scott, D.; Jenkins, M.; Petrou, S.; Sutherland, G.R.; Scheffer, I.E.; Berkovic, S.F.; Macdonald, R.L.; Mulley, J.C. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum. Mol. Genet. 2004, 13, 1315–1319. [Google Scholar]
- Bianchi, M.T.; Haas, K.F.; Macdonald, R.L. Structural determinants of fast desensitization and desensitization-deactivation coupling in GABAA receptors. J. Neurosci. 2001, 21, 1127–1136. [Google Scholar]
- Henschel, O.; Gipson, K.E.; Bordey, A. GABAA receptors, anesthetics and anticonvulsants in brain development. CNS Neurol. Disord. Drug Targets 2008, 7, 211–224. [Google Scholar]
- Nicoll, R.A.; Wojtowicz, J.M. The effects of pentobarbital and related compounds on frog motoneurons. Brain Res. 1980, 191, 225–237. [Google Scholar]
- Schulz, D.W.; Macdonald, R.L. Barbiturate enhancement of GABA-mediated inhibition and activation of chloride ion conductance: correlation with anticonvulsant and anesthetic actions. Brain Res. 1981, 209, 177–188. [Google Scholar]
- Krampfl, K.; Wolfes, H.; Dengler, R.; Bufler, J. Kinetic analysis of the agonistic and blocking properties of pentobarbital on recombinant rat α1β2γ2S GABAA receptor channels. Eur. J. Pharmacol. 2002, 435, 1–8. [Google Scholar]
- Akaike, N.; Maruyama, T.; Tokutomi, N. Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurones. J. Physiol. 1987, 394, 85–98. [Google Scholar]
- Rho, J.M.; Donevan, S.D.; Rogawski, M.A. Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons. J. Physiol. 1996, 497, 509–522. [Google Scholar]
- Akk, G.; Steinbach, J.H. Activation and block of recombinant GABAA receptors by pentobarbitone: a single-channel study. Br. J. Pharmacol. 2000, 130, 249–258. [Google Scholar]
- Lambert, J.J.; Belelli, D.; Hill-Venning, C.; Peters, J.A. Neurosteroids and GABAA receptor function. Trends Pharmacol. Sci. 1995, 16, 295–303. [Google Scholar]
- Rupprecht, R.; Holsboer, F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999, 22, 410–416. [Google Scholar]
- Lin, L.H.; Wang, L.H. Region-specific changes in GABAA receptor δ subunit mRNA level by tolerance to and withdrawal from pentobarbital. Neurosci. Lett. 1996, 202, 149–152. [Google Scholar]
- Adkins, C.E.; Pillai, G.V.; Kerby, J.; Bonnert, T.P.; Haldon, C.; McKeman, R.M.; Gonzalez, J.E.; Oades, K.; Whiting, P.J.; Simpson, P.B. α4β3δ GABAA receptors characterized by fluorescence resonance energy transfer-derived measurements of membrane potential. J. Biol. Chem. 2001, 276, 38934–38939. [Google Scholar]
- Feng, H.J.; Macdonald, R.L. Barbiturates require the N terminus and first transmembrane domain of the δ subunit for enhancement of α1β3δ GABAA receptor currents. J. Biol. Chem. 2010, 285, 23614–23621. [Google Scholar]
- Akk, G.; Bracamontes, J.; Steinbach, J.H. Activation of GABAA receptors containing the α4 subunit by GABA and pentobarbital. J. Physiol. 2004, 556, 387–399. [Google Scholar]
- Akk, G.; Covey, D.F.; Evers, A.S.; Steinbach, J.H.; Zorumski, C.F.; Mennerick, S. Mechanisms of neurosteroid interactions with GABAA receptors. Pharmacol. Ther. 2007, 116, 35–57. [Google Scholar]
- Mitchell, E.A.; Herd, M.B.; Gunn, B.G.; Lambert, J.J.; Belelli, D. Neurosteroid modulation of GABAA receptors: molecular determinants and significance in health and disease. Neurochem. Int. 2008, 52, 588–595. [Google Scholar]
- Stell, B.M.; Brickley, S.G.; Tang, C.Y.; Farrant, M.; Mody, I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 14439–14444. [Google Scholar]
- Maguire, J.L.; Stell, B.M.; Rafizadeh, M.; Mody, I. Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 2005, 8, 797–804. [Google Scholar]
- Sanna, E.; Mostallino, M.C.; Murru, L.; Carta, M.; Talani, G.; Zucca, S.; Mura, M.L.; Maciocco, E.; Biggio, G. Changes in expression and function of extrasynaptic GABAA receptors in the rat hippocampus during pregnancy and after delivery. J. Neurosci. 2009, 29, 1755–1765. [Google Scholar]
- Maguire, J.; Mody, I. Steroid hormone fluctuations and GABAAR plasticity. Psychoneuroendocrinology 2009, 34S, S84–S90. [Google Scholar]
- Smith, S.S.; Aoki, C.; Shen, H. Puberty, steroids and GABAA receptor plasticity. Psychoneuroendocrinology 2009, 34S, S91–S103. [Google Scholar]
- Shen, H.; Sabaliauskas, N.; Sherpa, A.; Fenton, A.A.; Stelzer, A.; Aoki, C.; Smith, S.S. A critical role for α4βd GABAA receptors in shaping learning deficits at puberty in mice. Science 2010, 327, 1515–1518. [Google Scholar]
- Kaur, K.H.; Baur, R.; Sigel, E. Unanticipated structural and functional properties of δ-subunit-containing GABAA receptors. J. Biol. Chem. 2009, 284, 7889–7896. [Google Scholar]
- Meera, P.; Olsen, R.W.; Otis, T.S.; Wallner, M. Etomidate, propofol and the neurosteroid THDOC increase the GABA efficacy of recombinant α4β3δ and α4β3 GABAA receptors expressed in HEK cells. Neuropharmacology 2009, 56, 155–160. [Google Scholar]
- Fancsik, A.; Linn, D.M.; Tasker, J.G. Neurosteroid modulation of GABA IPSCs is phosphorylation dependent. J. Neurosci. 2000, 20, 3067–3075. [Google Scholar]
- Leidenheimer, N.J.; Chapell, R. Effects of PKC activation and receptor desensitization on neurosteroid modulation of GABAA receptors. Brain Res. Mol. Brain Res. 1997, 52, 173–181. [Google Scholar]
- Tang, X.; Hernandez, C.C.; Macdonald, R.L. Modulation of spontaneous and GABA-evoked tonic α4β3δ and α4β3γ2L GABAA receptor currents by protein kinase A. J. Neurophysiol. 2010, 103, 1007–1019. [Google Scholar]
- Lees, G.; Edwards, M.D. Modulation of recombinant human γ-aminobutyric acidA receptors by isoflurane: influence of the δ subunit. Anesthesiology 1998, 88, 206–217. [Google Scholar]
- Yamashita, M.; Marszalec, W.; Yeh, J.Z.; Narahashi, T. Effects of ethanol on tonic GABA currents in cerebellar granule cells and mammalian cells recombinantly expressing GABAA receptors. J. Pharmacol. Exp. Ther. 2006, 319, 431–438. [Google Scholar]
- Caraiscos, V.B.; Newell, J.G.; You-Ten, K.E.; Elliott, E.M.; Rosahl, T.W.; Wafford, K.A.; MacDonald, J.F.; Orser, B.A. Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentration of the volatile anesthetic isoflurane. J. Neurosci. 2004, 24, 8454–8458. [Google Scholar]
- Jia, F.; Yue, M.; Chandra, D.; Homanics, G.E.; Goldstein, P.A.; Harrison, N.L. Isoflurane is a potent modulator of extrasynaptic GABAA receptors in the thalamus. J. Pharmacol. Exp. Ther. 2008, 324, 1127–1135. [Google Scholar]
- Wang, X. Propofol and isoflurane enhancement of tonic γ-aminobutyric acid type A current in cardiac vagal neurons in the nucleus ambiguus. Anesth. Analg. 2009, 108, 142–148. [Google Scholar]
- Bai, D.; Zhu, G.; Pennefather, P.; Jackson, M.F.; MacDonald, J.F.; Orser, B.A. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by γ-aminobutyric acidA receptors in hippocampal neurons. Mol. Pharmacol. 2001, 59, 814–824. [Google Scholar]
- Bieda, M.C.; MacIver, M.B. Major role for tonic GABAA conductances in anesthetic suppression of intrinsic neuronal excitability. J. Neurophysiol. 2004, 92, 1658–1667. [Google Scholar]
- Takahashi, A.; Mashimo, T.; Uchida, I. GABAergic tonic inhibition of substantia gelatinosa neurons in mouse spinal cord. Neuroreport 2006, 17, 1331–1335. [Google Scholar]
- McDougall, S.J.; Bailey, T.W.; Mendelowitz, D.; Andresen, M.C. Propofol enhances both tonic and phasic inhibitory currents in second-order neurons of the solitary tract nucleus (NTS). Neuropharmacology 2008, 54, 552–563. [Google Scholar]
- Dong, X.P.; Xu, T.L. The actions of propofol on γ-aminobutyric acid-A and glycine receptors in acutely dissociated spinal dorsal horn neurons of the rat. Anesth. Analg. 2002, 95, 907–914. [Google Scholar]
- Faingold, C.L.; N’Gouemo, P.; Riaz, A. Ethanol and neurotransmitter interactions-from molecular to integrative effects. Prog. Neurobiol. 1998, 55, 509–535. [Google Scholar]
- Feng, H.J.; Faingold, C.L. The effects of chronic ethanol administration on amygdala neuronal firing and ethanol withdrawal seizures. Neuropharmacology 2008, 55, 648–653. [Google Scholar]
- Silberman, Y.; Bajo, M.; Chappell, A.M.; Christian, D.T.; Cruz, M.; Diaz, M.R.; Kash, T.; Lack, A.K.; Messing, R.O.; Siggins, G.R.; Winder, D.; Roberto, M.; McCool, B.A.; Weiner, J.L. Neurobiological mechanisms contributing to alcohol-stress-anxiety interactions. Alcohol 2009, 43, 509–519. [Google Scholar]
- Mihalek, R.M.; Bowers, B.J.; Wehner, J.M.; Kralic, J.E.; VanDoren, M.J.; Morrow, A.L.; Homanics, G.E. GABAA-receptor δ subunit knockout mice have multiple defects in behavioral responses to ethanol. Alcohol Clin. Exp. Res. 2001, 25, 1708–1718. [Google Scholar]
- Sundstrom-Poromaa, I.; Smith, D.H.; Gong, Q.H.; Sabado, T.N.; Li, X.; Light, A.; Wiedmann, M.; Williams, K.; Smith, S.S. Hormonally regulated α4β2δ GABAA receptors are a target for alcohol. Nat. Neurosci. 2002, 5, 721–722. [Google Scholar]
- Wallner, M.; Hanchar, H.J.; Olsen, R.W. Ethanol enhances α4β3δ and α6β3δγ-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc. Natl. Acad. Sci. USA 2003, 100, 15218–15223. [Google Scholar]
- Santhakumar, V.; Wallner, M.; Otis, T.S. Ethanol acts directly on extrasynaptic subtypes of GABAA receptors to increase tonic inhibition. Alcohol 2007, 41, 211–221. [Google Scholar]
- Hanchar, H.J.; Chutsrinopkun, P.; Meera, P.; Supavilai, P.; Sieghart, W.; Wallner, M.; Olsen, R.W. Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro 15-4513 to α4/6β3δ GABAA receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 8546–8551. [Google Scholar]
- Wallner, M.; Hanchar, H.J.; Olsen, R.W. Low-dose alcohol actions on α4β3δ GABAA receptors are reversed by the behavioral alcohol antagonist Ro 15-4513. Proc. Natl. Acad. Sci. USA 2006, 103, 8540–8545. [Google Scholar]
- Kash, T.L.; Jenkins, A.; Kelley, J.C.; Trudell, J.R.; Harrison, N.L. Coupling of agonist binding to channel gating in the GABAA receptor. Nature 2003, 421, 272–275. [Google Scholar]
- Crawford, D.K.; Trudell, J.R.; Bertaccini, E.J.; Li, K.; Davies, D.L.; Alkana, R.L. Evidence that ethanol acts on a target in loop 2 of the extracellular domain of α1 glycine receptors. J. Neurochem. 2007, 102, 2097–2109. [Google Scholar]
- Perkins, D.I.; Trudell, J.R.; Crawford, D.K.; Alkana, R.L.; Davies, D.L. Targets for ethanol action and antagonism in loop 2 of the extracellular domain of glycine receptors. J. Neurochem. 2008, 106, 1337–1349. [Google Scholar]
- Perkins, D.I.; Trudell, J.R.; Crawford, D.K.; Asatryan, L.; Alkana, R.L.; Davies, D.L. Loop 2 structure in glycine and GABAA receptors plays a key role in determining ethanol sensitivity. J. Biol. Chem. 2009, 284, 27304–27314. [Google Scholar]
- Wei, W.; Faria, L.C.; Mody, I. Low ethanol concentrations selectively augment the tonic inhibition mediated by δ subunit-containing GABAA receptors in hippocampal neurons. J. Neurosci. 2004, 24, 8379–8382. [Google Scholar]
- Liang, J.; Zhang, N.; Cagetti, E.; Houser, C.R.; Olsen, R.W.; Spigelman, I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. J. Neurosci. 2006, 26, 1749–1758. [Google Scholar]
- Fleming, R.L.; Wilson, W.A.; Swartzwelder, H.S. Magnitude and ethanol sensitivity of tonic GABAA receptor-mediated inhibition in dentate gyrus changes from adolescence to adulthood. J. Neurophysiol. 2007, 97, 3806–3811. [Google Scholar]
- Jia, F.; Chandra, D.; Homanics, G.E.; Harrison, N.L. Ethanol modulates synaptic and extrasynaptic GABAA receptors in the thalamus. J. Pharmacol. Exp. Ther. 2008, 326, 475–482. [Google Scholar]
- Glykys, J.; Peng, Z.; Chandra, D.; Homanics, G.E.; Houser, C.R.; Mody, I. A new naturally occurring GABAA receptor subunit partnership with high sensitivity to ethanol. Nat. Neurosci. 2007, 10, 40–48. [Google Scholar]
- Saba, L.; Porcella, A.; Congeddu, E.; Colombo, G.; Peis, M.; Pistis, M.; Gessa, G.L.; Pani, L. The R100Q mutation of the GABAA α6 receptor subunit may contribute to voluntary aversion to ethanol in the sNP rat line. Brain Res. Mol. Brain Res. 2001, 87, 263–270. [Google Scholar]
- Carr, L.G.; Spence, J.P.; Peter Eriksson, C.J.; Lumeng, L.; Li, T.K. AA and ANA rats exhibit the R100Q mutation in the GABAA receptor α6 subunit. Alcohol 2003, 31, 93–97. [Google Scholar]
- Santhakumar, V.; Hanchar, H.J.; Wallner, M.; Olsen, R.W.; Otis, T.S. Contributions of the GABAA receptor α6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the α6 gene. J. Neurosci. 2006, 26, 3357–3364. [Google Scholar]
- Hanchar, H.J.; Dodson, P.D.; Olsen, R.W.; Otis, T.S.; Wallner, M. Alcohol-induced motor impairment caused by increased extrasynaptic GABAA receptor activity. Nat. Neurosci. 2005, 8, 339–345. [Google Scholar]
- Borghese, C.M.; Harris, R.A. Studies of ethanol actions on recombinant δ-containingγ-aminobutyric acid type A receptors yield contradictory results. Alcohol 2007, 41, 155–162. [Google Scholar]
- Korpi, E.R.; Debus, F.; Linden, A.M.; Malecot, C.; Leppa, E.; Vekovischeva, O.; Rabe, H.; Bohme, I.; Aller, M.I.; Wisden, W.; Luddens, H. Does ethanol act preferentially via selected brain GABAA receptor subtypes? The current evidence is ambiguous. Alcohol 2007, 41, 163–176. [Google Scholar]
- Lovinger, D.M.; Homanics, G.E. Tonic for what ails us? High-affinity GABAA receptors and alcohol. Alcohol 2007, 41, 139–143. [Google Scholar]
- Borghese, C.M.; Storustovu, S.; Ebert, B.; Herd, M.B.; Belelli, D.; Lambert, J.J.; Marshall, G.; Wafford, K.A.; Harris, R.A. The δ subunit of γ-aminobutyric acid type A receptors does not confer sensitivity to low concentrations of ethanol. J. Pharmacol. Exp. Ther. 2006, 316, 1360–1368. [Google Scholar]
- Mehta, A.K.; Marutha Ravindran, C.R.; Ticku, M.K. Low concentrations of ethanol do not affect radioligand binding to the δ-subunit-containing GABAA receptors in the brain. Brain Res. 2007, 1165, 15–20. [Google Scholar]
- Baur, R.; Kaur, K.H.; Sigel, E. Structure of α6β3δ GABAA receptors and their lack of ethanol sensitivity. J. Neurochem. 2009, 111, 1172–1181. [Google Scholar]
- Carta, M.; Mameli, M.; Valenzuela, C.F. Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J. Neurosci. 2004, 24, 3746–3751. [Google Scholar]
- Casagrande, S.; Cupello, A.; Pellistri, F.; Robello, M. Only high concentrations of ethanol affect GABAA receptors of rat cerebellum granule cells in culture. Neurosci. Lett. 2007, 414, 273–276. [Google Scholar]
- Botta, P.; Mameli, M.; Floyd, K.L.; Radcliffe, R.A.; Valenzuela, C.F. Ethanol sensitivity of GABAergic currents in cerebellar granule neurons is not increased by a single amino acid change (R100Q) in the α6 GABAA receptor subunit. J. Pharmacol. Exp. Ther. 2007, 323, 684–691. [Google Scholar]
- Choi, D.S.; Wei, W.; Deitchman, J.K.; Kharazia, V.N.; Lesscher, H.M.B.; McMahon, T.; Wang, D.; Qi, Z.H.; Sieghart, W.; Zhang, C.; Shokat, K.M.; Mody, I.; Messing, R.O. Protein kinase Cδ regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. J. Neurosci. 2008, 28, 11890–11899. [Google Scholar]
- Krishek, B.J.; Amato, A.; Connolly, C.N.; Moss, S.J.; Smart, T.G. Proton sensitivity of the GABAA receptor is associated with the receptor subunit composition. J. Physiol. 1996, 492, 431–443. [Google Scholar]
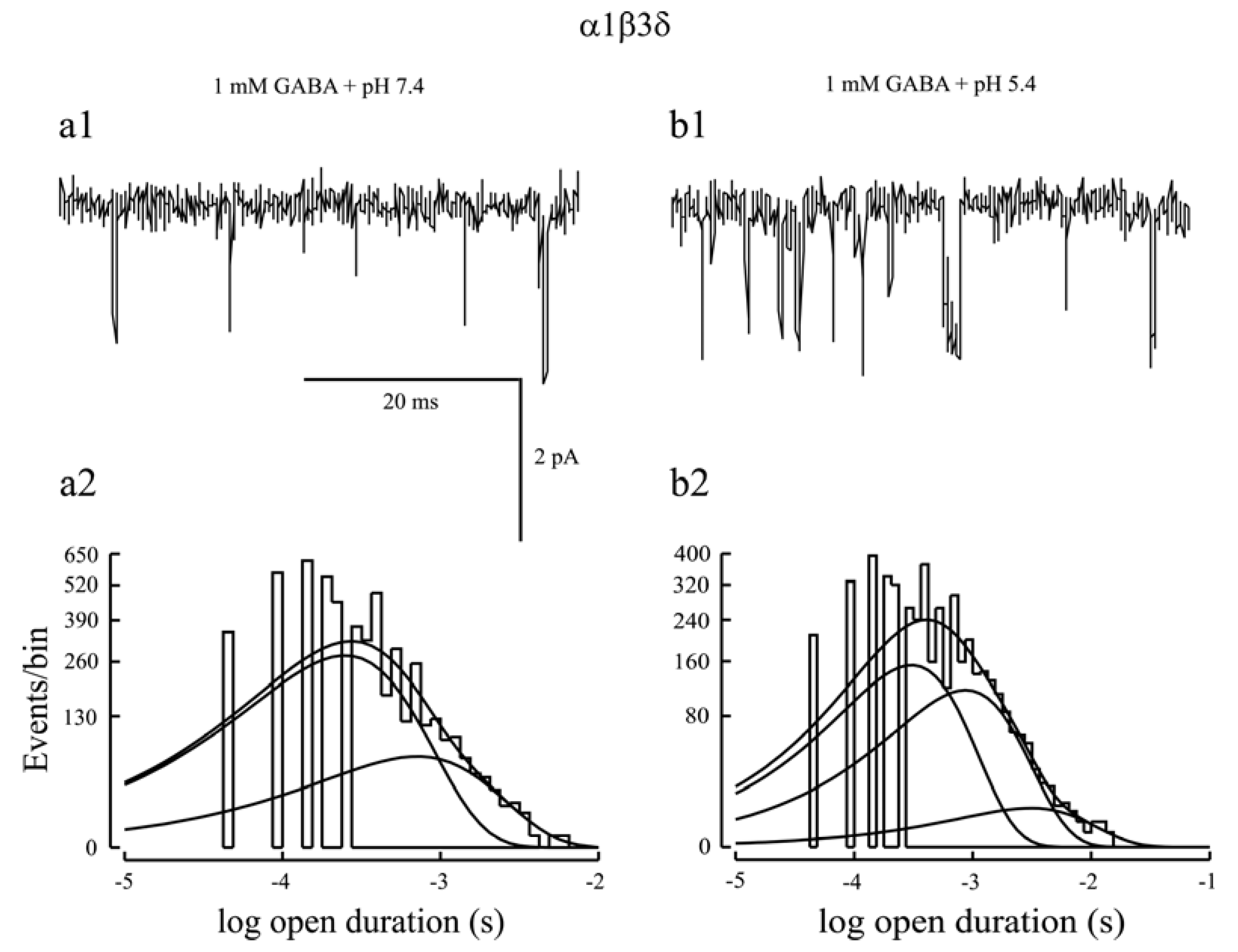
- Feng, H.J.; Macdonald, R.L. Proton modulation of α1β3δ GABAA receptor channel gating and desensitization. J. Neurophysiol. 2004, 92, 1577–1585. [Google Scholar]
- Drasbek, K.R.; Jensen, K. THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABAA receptor-mediated conductance in mouse neocortex. Cereb. Cortex 2006, 16, 1134–1141. [Google Scholar]
- Lewis, R.W.; Mabry, J.; Polisar, J.G.; Eagen, K.P.; Ganem, B.; Hess, G.P. Dihydropyrimidinone positively modulation of δ-subunit-containing γ-aminobutyric acid type A receptors, including an epilepsy-linked mutant variant. Biochemistry 2010, 49, 4841–4851. [Google Scholar]
- Thompson, S.A.; Wingrove, P.B.; Connelly, L.; Whiting, P.J.; Wafford, K.A. Tracazolate reveals a novel type of allosteric interaction with recombinant γ-aminobutyric acidA receptors. Mol. Pharmacol. 2002, 61, 861–869. [Google Scholar]
- Zheleznova, N.; Sedelnikova, A.; Weiss, D.S. α1β2δ, a silent GABAA receptor: recruitment by tracazolate and neurosteroids. Br. J. Pharmacol. 2008, 153, 1062–1071. [Google Scholar]
- Hoestgaard-Jensen, K.; Dalby, N.O.; Wolinsky, T.D.; Murphey, C.; Jones, K.A.; Rottlander, M.; Frederiksen, K.; Watson, W.P.; Jensen, K.; Ebert, B. Pharmacological characterization of a novel positive modulator at α4β3δ-containing extrasynaptic GABAA receptors. Neuropharmacology 2010, 58, 702–711. [Google Scholar]
- Mihalek, R.M.; Banerjee, P.K.; Korpi, E.R.; Quinlan, J.J.; Firestone, L.L.; Mi, Z.P.; Lagenaur, C.; Tretter, V.; Sieghart, W.; Anagnostaras, S.G.; Sage, J.R.; Fanselow, M.S.; Guidotti, A.; Spigelman, I.; Li, Z.; DeLorey, T.M.; Olsen, R.W.; Homanics, G.E. Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor δ subunit knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12905–12910. [Google Scholar]
- Fisher, J.L.; Macdonald, R.L. Single channel properties of recombinant GABAA receptors containing γ2 or δ subtypes expressed with α1 and β3 subtypes in mouse L929 cells. J. Physiol. 1997, 505, 283–297. [Google Scholar]
- Haas, K.F.; Macdonald, R.L. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J. Physiol. 1999, 514, 27–45. [Google Scholar]
- Steinbach, J.H.; Akk, G. Modulation of GABAA receptor channel gating by pentobarbital. J. Physiol. 2001, 537, 715–733. [Google Scholar]
- Bianchi, M.T.; Macdonald, R.L. Neurosteroids shift partial agonist activation of GABAA receptor channels from low- to high-efficacy gating patterns. J. Neurosci. 2003, 23, 10934–10943. [Google Scholar]
- Qi, J.S.; Yao, J.; Fang, C.; Luscher, B.; Chen, G. Downregulation of tonic GABA currents following epileptogenic stimulation of rat hippocampal cultures. J. Physiol. 2006, 577, 579–590. [Google Scholar]
- Cope, D.W.; Di Giovanni, G.; Fyson, S.J.; Orban, G.; Errington, A.C.; Lorincz, M.L.; Gould, T.M.; Carter, D.A.; Crunelli, V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 2009, 15, 1392–1398. [Google Scholar]
- Fritsch, B.; Qashu, F.; Figueiredo, T.H.; Aroniadou-Anderjaska, V.; Rogawski, M.A.; Braga, M.F. Pathological alterations in GABAergic interneurons and reduced tonic inhibition in the basolateral amygdale during epileptogenesis. Neuroscience 2009, 163, 415–429. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, H.-J. Allosteric Modulation of αβδ GABAA Receptors. Pharmaceuticals 2010, 3, 3461-3477. https://doi.org/10.3390/ph3113461
Feng H-J. Allosteric Modulation of αβδ GABAA Receptors. Pharmaceuticals. 2010; 3(11):3461-3477. https://doi.org/10.3390/ph3113461
Chicago/Turabian StyleFeng, Hua-Jun. 2010. "Allosteric Modulation of αβδ GABAA Receptors" Pharmaceuticals 3, no. 11: 3461-3477. https://doi.org/10.3390/ph3113461
APA StyleFeng, H.-J. (2010). Allosteric Modulation of αβδ GABAA Receptors. Pharmaceuticals, 3(11), 3461-3477. https://doi.org/10.3390/ph3113461