Inactivation of Anandamide Signaling: A Continuing Debate

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
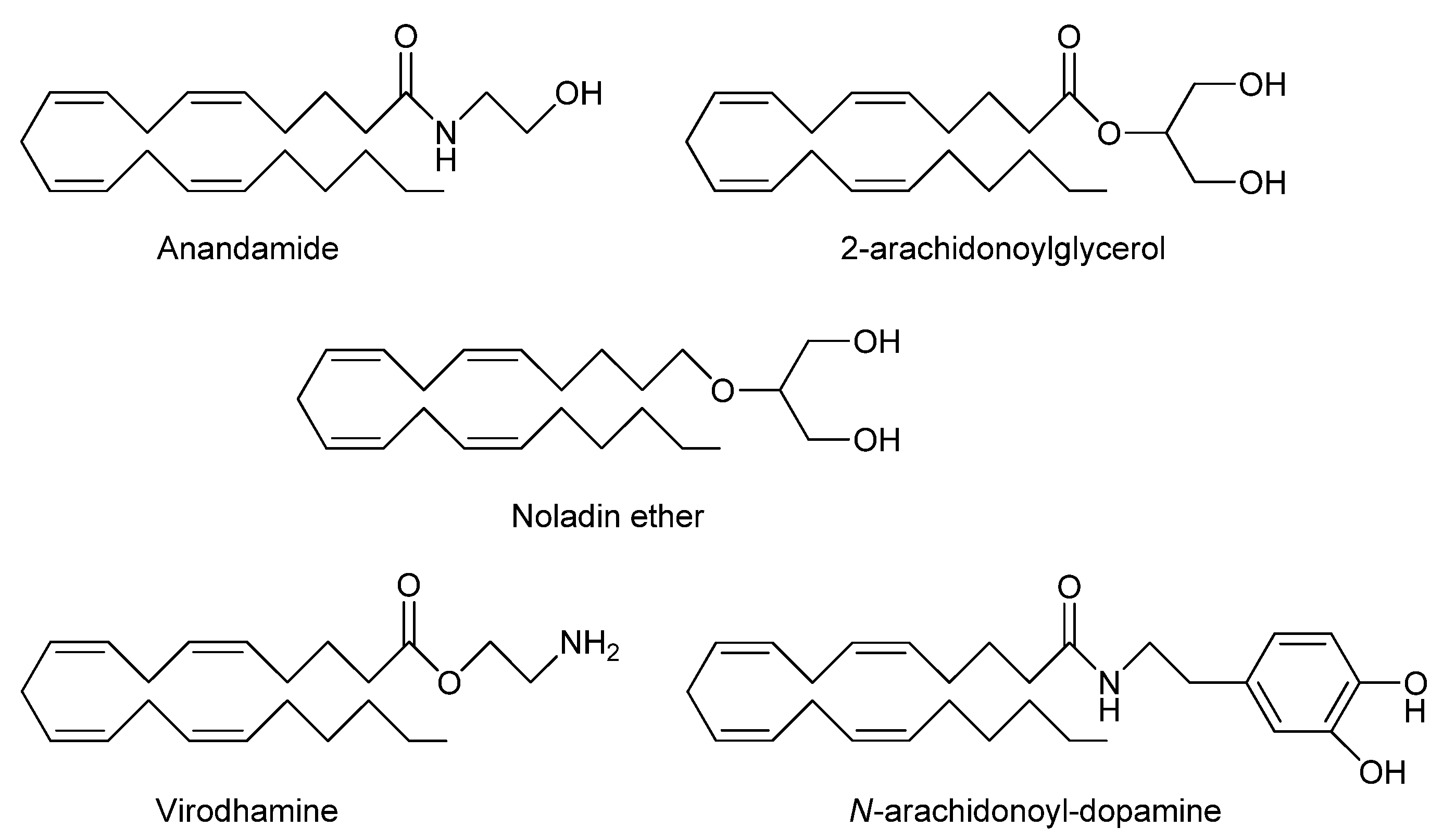
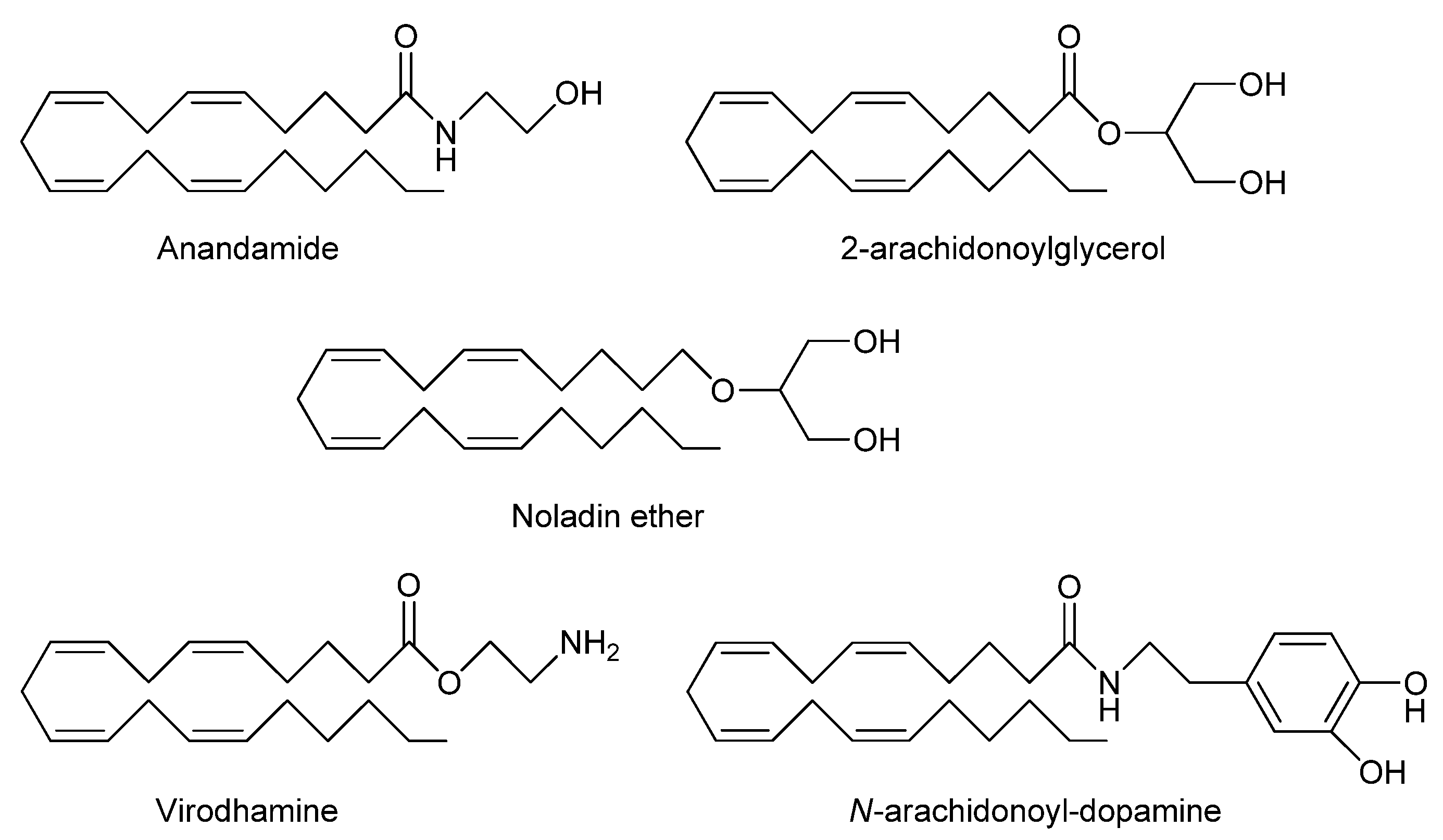
2. Endocannabinoid System

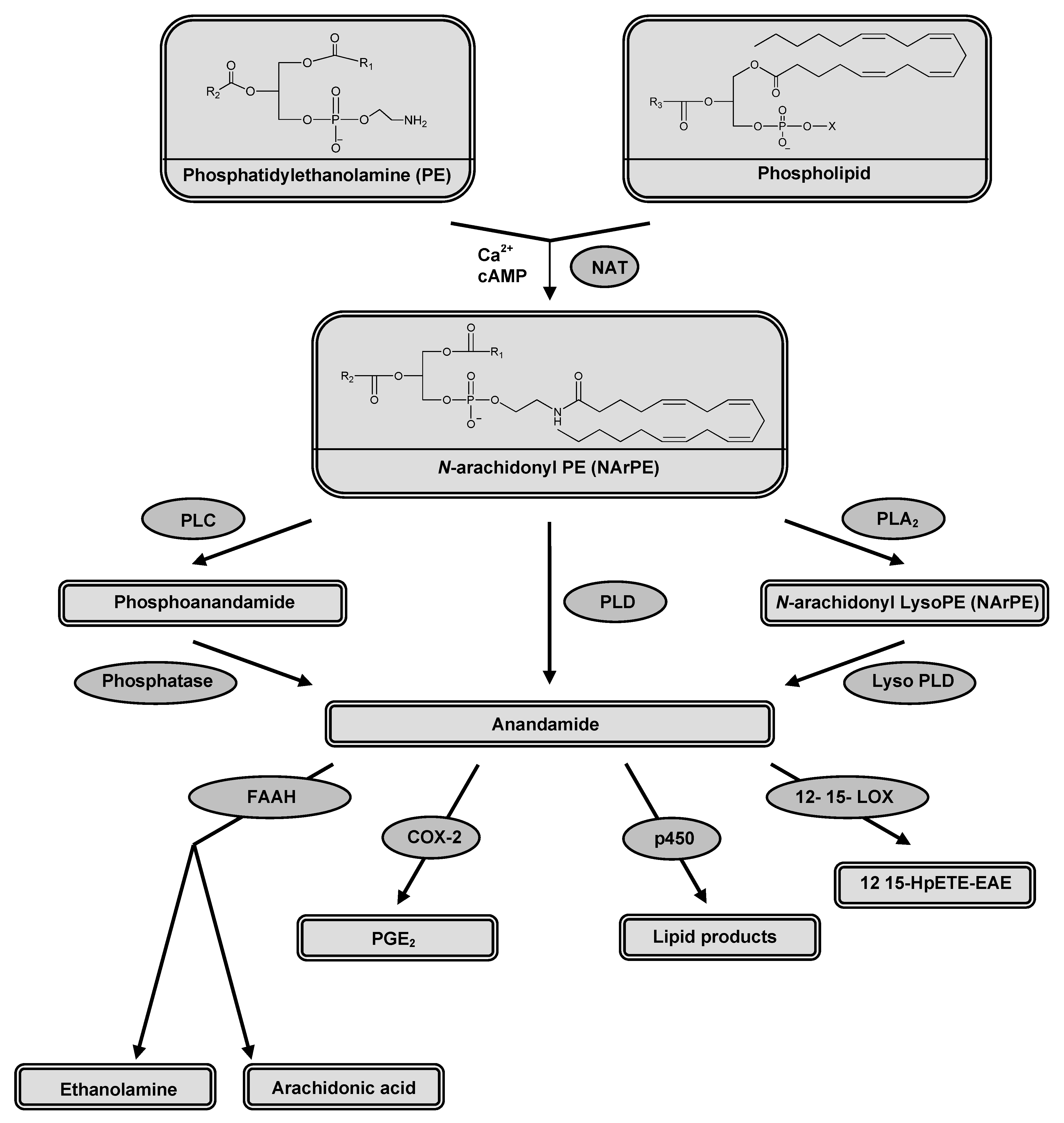
2.1. Synthesis and release
2.2. Functions and actions
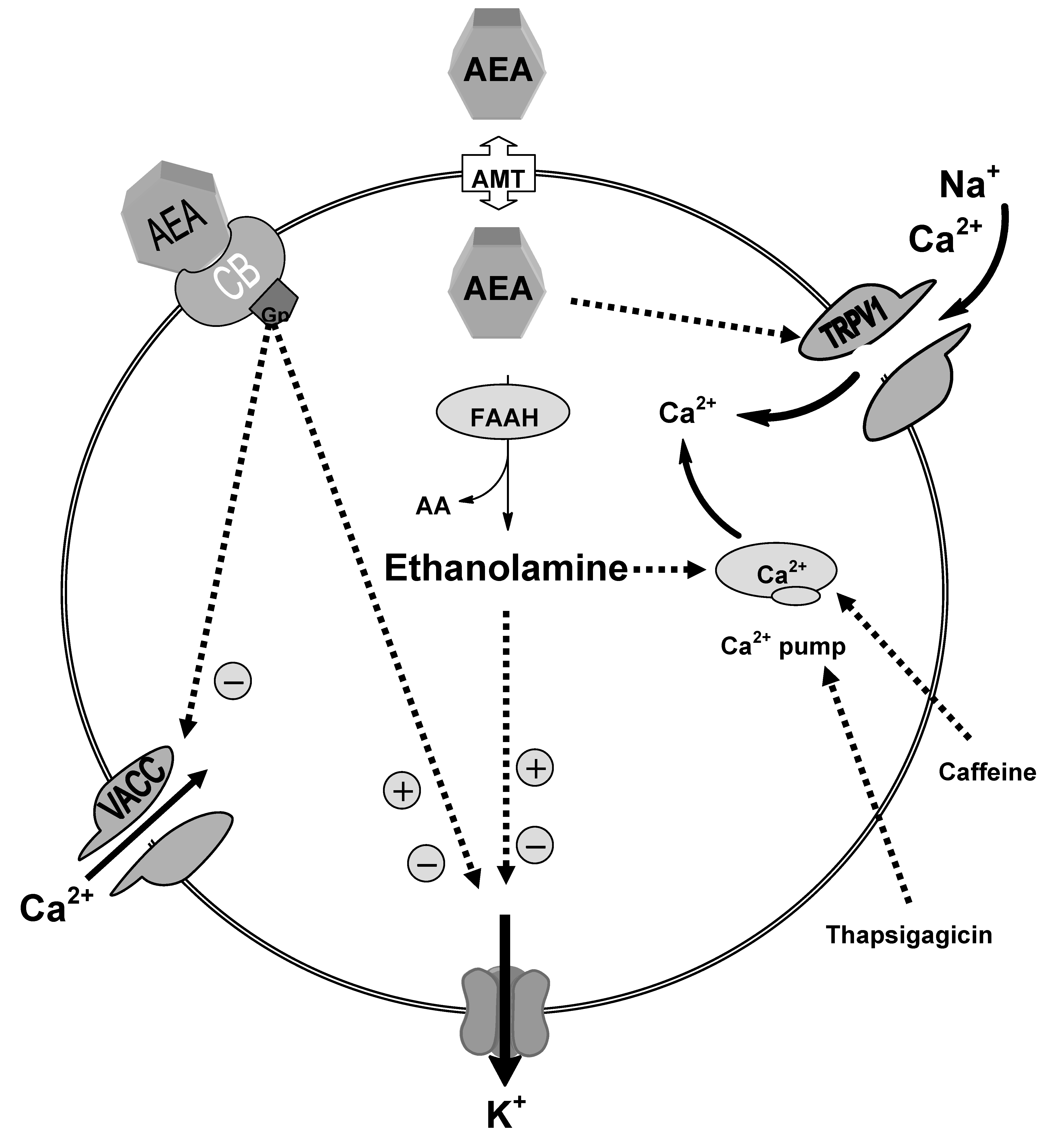
3. Reuptake
4. Hydrolysis

5. Oxidation
6. Concluding Remarks
Acknowledgments
References and notes
- Mechoulam, R.; Gaoni, Y. Recent advances in the chemistry of hashish. Fortschritte der Chemie Organischer Naturstoffe 1967, 25, 175–213. [Google Scholar]
- Wood, T.B.; Spivey, W.T.N.; Easterfield, T.H. III- Cannabinol. Part I. J. Chem. Soc. Trans. 1899, 75, 20–36. [Google Scholar]
- Mechoulam, R.; Carlini, E.A. Toward drugs derived from Cannabis. Naturwissenschaften 1978, 65, 174–179. [Google Scholar]
- Pertwee, R.G. Cannabinoids and multiple sclerosis. Pharmacol. Therap. 2002, 95, 165–174. [Google Scholar]
- Iversen, L.; Chapman, V. Cannabinoids: A real prospect for pain relief? Curr. Opin. Pharmacol. 2002, 2, 50–55. [Google Scholar]
- Bifulco, M. A new strategy to block tumor growth by inhibiting endocannabinoid inactivation. FASLB J. 2004, 18, 1606–1608. [Google Scholar]
- Horvath, T.L. Endocannabinoids and the regulation of body fat: The smoke is clearing. J. Clin. Invest. 2003, 112, 323–326. [Google Scholar]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar]
- Mechoulam, R.; Ben-Shabat, S.; Hanuš, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 3662–3665. [Google Scholar]
- Bisogno, T.; Melck, D.; Bobrov, M.Y.; Gretskaya, N.M.; Bezuglov, V.V.; De Petrocellis, L.; Di Marzo, V. N-acyl-dopamines: Novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem. J. 2000, 351, 817–824. [Google Scholar]
- Porter, A.C.; Sauer, J.M.; Knierman, M.D.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; Felder, C.C. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1020–1024. [Google Scholar]
- Thomas, A.; Hopfgartner, G.; Giroud, C.; Staub, C. Quantitative and qualitative profiling of endocannabinoids in human plasma using a triple quadrupole linear ion trap mass spectrometer with liquid chromatography. RCMS 2009, 23, 629–638. [Google Scholar]
- Stella, N.; Schweitzer, P.; Plomelli, D. A second endogenous' cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar]
- Beltramo, M.; Stella, N.; Calignano, A.; Lin, S.Y.; Makriyannis, A.; Piomelli, D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 1997, 277, 1094–1097. [Google Scholar]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar]
- Jonsson, K.-O.; Vandevoorde, S.; Lambert, D.M.; Tiger, G.; Fowler, C.J. Effects of homologues and analogues of palmitoylethanolamide upon the inactivation of the endocannabinoid anandamide. Br. J. Pharmacol. 2001, 133, 1263–1275. [Google Scholar]
- Fedorova, I.; Hashimoto, A.; Fecik, R.A.; Hedrick, M.P.; Hanuš, L.O.; Boger, D.L.; et al. Behavioral evidence for the interaction of oleamide with multiple neurotransmitter systems. J. Pharmacol. Exp. Ther. 2001, 299, 332–342. [Google Scholar]
- Walker, J.M.; Krey, J.F.; Chu, C.J.; Huang, S.M. Endocannabinoids and related fatty acid derivatives in pain modulation. Chem. Phys. Lipids 2002, 121, 159–172. [Google Scholar]
- Mechoulam, R.; Fride, E.; Di Marzo, V. Endocannabinoids. Eur. J. Pharmacol. 1998, 359, 1–18. [Google Scholar]
- Cadas, H.; Gaillet, S.; Beltramo, M.; Venance, L.; Piomelli, D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J. Neurosci. 1996, 16, 3934–3942. [Google Scholar]
- Okamoto, Y.; Tsuboi, K.; Ueda, N. Enzymatic Formation of Anandamide. Vitam. Horm. 2009, 81, 1–24. [Google Scholar]
- Simon, G.M.; Cravatt, B.F. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for α/β-hydrolase 4 in this pathway. J. Biol. Chem. 2006, 281, 26465–26472. [Google Scholar]
- Sun, Y.; Alexander, S.P.H.; Garle, M.J.; Gibson, C.L.; Hewitt, K.; Murphy, S.P.; Kendall, D.A.; Bennett, A.J. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br. J. Pharmacol. 2007, 152, 734–743. [Google Scholar]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A.; et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar]
- Tian, X.; Guo, J.; Yao, F.; Yang, D.P.; Makriyannis, A. The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. J. Biol. Chem. 2005, 280, 29788–29795. [Google Scholar]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nature Rev. Neurosci. 2003, 4, 873–884. [Google Scholar]
- Guzmán, M.; Sánchez, C.; Galve-Roperh, I. Cannabinoids and cell fate. Pharmacol. Therap. 2002, 95, 175–184. [Google Scholar]
- Wenger, T.; Moldrich, G. The role of endocannabinoids in the hypothalamic regulation of visceral function. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 301–307. [Google Scholar]
- Randall, M.D.; Harris, D.; Kendall, D.A.; Ralevic, V. Cardiovascular effects of cannabinoids. Pharmacol. Therap. 2002, 95, 191–202. [Google Scholar]
- Schmid, K.; Niederhoffer, N.; Szabo, B. Analysis of the respiratory effects of cannabinoids in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 368, 301–308. [Google Scholar]
- Di Carlo, G.; Izzo, A.A. Cannabinoids for gastrointestinal diseases: Potential therapeutic applications. Expert Opin. Investig. Drugs 2003, 12, 39–49. [Google Scholar]
- Klein, T.W.; Newton, C.; Larsen, K.; Lu, L.; Perkins, I.; Nong, L.; Friedman, H. The cannabinoid system and immune modulation. J. Leukoc. Biol. 2003, 74, 486–496. [Google Scholar]
- Howlett, A.C. Cannabinoid receptor signaling. In Handbook of Experimental Pharmacology; Pertwee, P.G., Ed.; Springer-Verlag: Heidelburg, 2005; Volume 168, pp. 53–79. [Google Scholar]
- Bosier, B.; Muccioli, G.G.; Hermans, E.; Lambert, D.M. Functionally selective cannabinoid receptor signalling: Therapeutic implications and opportunities. Biochem. Pharmacol. 2010, 80, 1–12. [Google Scholar]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar]
- Sánchez, M.G.; Ruiz-Llorente, L.; Sánchez, A.M.; Díaz-Laviada, I. Activation of phosphoinositide 3-kinase/PKB pathway by CB1 and CB2 cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell. Signal. 2003, 15, 851–859. [Google Scholar]
- Miller, A.M.; Stella, N. CB2 receptor-mediated migration of immune cells: It can go either way. Br. J. Pharmacol. 2008, 153, 299–308. [Google Scholar]
- Evans, R.M.; Scott, R.H.; Ross, R.A. Multiple actions of anandamide on neonatal rat cultured sensory neurones. Br. J. Pharmacol. 2004, 141, 1223–1233. [Google Scholar]
- Evans, R.M.; Wease, K.N.; MacDonald, C.J.; Khairy, H.A.; Ross, R.A.; Scott, R.H. Modulation of sensory neuron potassium conductances by anandamide indicates roles for metabolites. Br. J. Pharmacol. 2008, 154, 480–492. [Google Scholar]
- Poling, J.S.; Rogawski, M.A.; Salem, N.; Vicini, S. Anandamide, an endogenous cannabinoid, inhibits Shaker-related voltage-gated K+ channel. Neuropharmacol. 1996, 35, 983–991. [Google Scholar]
- Van den Bossche, I.; Vanheel, B. Influence of cannabinoids on the delayed rectifier in freshly dissociated smooth muscle cells of the rat aorta. Br. J. Pharmacol. 2000, 131, 85–93. [Google Scholar]
- Hillard, C.J.; Edgemond, W.S.; Jarrahian, A.; Campbell, W.B. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997, 69, 631–638. [Google Scholar]
- Maccarrone, M.; Bari, M.; Lorenzon, T.; Bisogno, T.; Di Marzo, V.; Finazzi-Agrò, A. Anandamide uptake by human endothelial cells and its regulation by nitric oxide. J. Biol. Chem. 2000, 275, 13484–13492. [Google Scholar]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar]
- Bisogno, T.; Maurelli, S.; Melck, D.; De Petrocellis, L.; Di Marzo, V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J. Biol. Chem. 1997, 272, 3315–3323. [Google Scholar]
- Moore, S.A.; Nomikos, G.G.; Dickason-Chesterfield, A.K.; Schober, D.A.; Schaus, J.M.; Ying, B.-P.; et al. Identification of a high-affinity binding site involved in the transport of endocannabinoids. Proc. Natl. Acad. Sci. USA 2005, 102, 17852–17857. [Google Scholar]
- Day, T.A.; Rakhshan, F.; Deutsch, D.G.; Barker, E.L. Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol. Pharmacol. 2001, 59, 1369–1375. [Google Scholar]
- Deutsch, D.G.; Glaser, S.T.; Howell, J.M.; Kunz, J.S.; Puffenbarger, R.A.; Hillard, C.J.; Abumrad, N. The Cellular uptake of anandamide is coupled to its breakdown by fatty-acid amide hydrolase. J. Biol. Chem. 2001, 276, 6967–6973. [Google Scholar]
- Glaser, S.T.; Abumrad, N.A.; Fatade, F.; Kaczocha, M.; Studholme, K.M.; Deutsch, D.G. Evidence against the presence of an anandamide transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 4269–4274. [Google Scholar]
- Alexander, J.P.; Cravatt, B.F. The Putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J. Am. Chem. Soc. 2006, 128, 9699–9704. [Google Scholar]
- Ortar, G.; Cascio, M.G.; Moriello, A.S.; Camalli, M.; Morera, E.; Nalli, M.; Di Marzo, V. Carbamoyl tetrazoles as inhibitors of endocannabinoid inactivation: A critical revisitation. Eur. J. Med. Chem. 2008, 43, 62–72. [Google Scholar]
- Thors, L.; Fowler, C.J. Is there a temperature-dependent uptake of anandamide into cells? Br. J. Pharmacol. 2006, 149, 73–81. [Google Scholar]
- Hillard, C.J.; Jarrahian, A. The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem. Phys. Lipids 2000, 108, 123–134. [Google Scholar]
- McFarland, M.J.; Porter, A.C.; Rakhshan, F.R.; Rawat, D.S.; Gibbs, R.A.; Barker, E.L. A role for caveolae/lipid rafts in the uptake and recycling of the endogenous cannabinoid anandamide. J. Biol. Chem. 2004, 279, 41991–41997. [Google Scholar]
- Stremmel, W.; Pohl, J.; Ring, A.; Herrmann, T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids 2001, 36, 981–989. [Google Scholar]
- Hillard, C.J.; Jarrahian, A. Cellular accumulation of anandamide: consensus and controversy. Br. J. Pharmacol. 2003, 140, 802–808. [Google Scholar]
- Jarrahian, A.; Manna, S.; Edgemond, W.S.; Campbell, W.B.; Hillard, C.J. Structure-activity relationships among N-arachidonylethanolamine (anandamide) head group analogues for the anandamide transporter. J. Neurochem. 2000, 74, 2597–2606. [Google Scholar]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar]
- De Petrocellis, L.; Davis, J.B.; Di Marzo, V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001, 506, 253–256. [Google Scholar]
- Van Der Stelt, M.; Trevisani, M.; Vellani, V.; De Petrocellis, L.; Moriello, A.S.; Campi, B.; McNaughton, P.; Geppetti, P.; Di Marzo, V. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J. 2005, 24, 3026–3037. [Google Scholar]
- De Petrocellis, L.; Bisogno, T.; Maccarrone, M.; Davis, J.B.; Finazzi-Agrò, A; Di Marzo, V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J. Biol. Chem. 2001, 276, 12856–12863. [Google Scholar]
- Numazaki, M.; Tominaga, T.; Toyooka, H.; Tominaga, M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cε and identification of two target serine residues. J. Biol. Chem. 2002, 277, 13375–13378. [Google Scholar]
- Malcher-Lopes, R.; Di, S.; Marcheselli, V.S.; Weng, F.J.; Stuart, C.T.; Bazan, N.G.; Tasker, J.G. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J. Neurosci. 2006, 26, 6643–6650. [Google Scholar]
- Premkumar, L.S.; Ahern, G.P. Induction of vanilloid receptor channel activity by protein kinase C. Nature 2000, 408, 985–990. [Google Scholar]
- Bhave, G.; Hu, H.J.; Glauner, K.S.; Zhu, W.; Wang, H.; Brasier, D.J.; Oxford, G.S.; Gereau, R.W. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc. Natl. Acad. Sci. USA 2003, 100, 12480–12485. [Google Scholar]
- Chuang, H.H.; Prescott, E.D.; Kong, H.; Shields, S.; Jordt, S.E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar]
- Evans, R.M.; Scott, R.H.; Ross, R.A. Chronic exposure of sensory neurones to increased levels of nerve growth factor modulates CB1/TRPV1 receptor crosstalk. Br. J. Pharmacol. 2007, 152, 404–413. [Google Scholar]
- Ahluwalia, J.; Urban, L.; Capogna, M.; Bevan, S.; Nagy, I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience 2000, 100, 685–688. [Google Scholar]
- Kulkarni-Narla, A.; Brown, D.R. Opioid, cannabinoid and vanilloid receptor localization on porcine cultured myenteric neurons. Neurosci. Lett. 2001, 308, 153–156. [Google Scholar]
- Cristino, L.; de Petrocellis, L.; Pryce, G.; Baker, D.; Guglielmotti, V.; Di Marzo, V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 2006, 139, 1405–1415. [Google Scholar]
- De Petrocellis, L.; Di Marzo, V. Role of endocannabinoids and endovanilloids in Ca2+ signaling. Cell Calcium 2009, 45, 611–624. [Google Scholar]
- Di Marzo, V.; Cristino, L. Why endocannabinoids are not all alike. Nat. Neurosci. 2008, 11, 124–126. [Google Scholar]
- Deutsch, D.G.; Chin, S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993, 46, 791–796. [Google Scholar]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar]
- Desarnaud, F.; Cadas, H.; Piomelli, D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J. Biol. Chem. 1995, 270, 6030–6035. [Google Scholar]
- Katayama, K.; Ueda, N.; Kurahashi, Y.; Suzuki, H.; Yamamoto, S.; Kato, I. Distribution of anandamide amidohydrolase in rat tissues with special reference to small intestine. Biochim. Biophys. Acta 1997, 1347, 212–218. [Google Scholar]
- Egertova, M.; Giang, D.K.; Cravatt, B.F.; Elphick, M.R. A new perspective on cannabinoid signalling: Complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc. Biol. Sci. 1998, 265, 2081–2085. [Google Scholar]
- Tsou, K.; Nogueron, M.I.; Muthian, S.; Sañudo-Peña, M.C.; Hillard, C.J.; Deutsch, D.G.; Walker, J.M. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci. Lett. 1998, 254, 137–140. [Google Scholar]
- Kaczocha, M.; Glaser, S.T.; Deutsch, D.G. Identification of intracellular carriers for the endocannabinoid anandamide. PNAS 2009, 106, 6375–6380. [Google Scholar]
- Koutek, B.; Prestwich, G.D.; Howlett, A.C.; Chin, S.A.; Salehani, D.; Akhavan, N.; Deutsch, D.G. Inhibitors of arachidonoyl ethanolamide hydrolysis. J. Biol. Chem. 1994, 269, 22937–22940. [Google Scholar]
- Ueda, N.; Kurahashi, Y.; Yamamoto, S.; Tokunaga, T. Partial purification and characterization of the porcine brain enzyme hydrolyzing and synthesizing anandamide. J. Biol. Chem. 1995, 270, 23823–23827. [Google Scholar]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valino, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar]
- Schlosburg, J.E.; Kinsey, S.G.; Lichtman, A.H. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009, 11, 39–44. [Google Scholar]
- Ahn, K.; Johnson, D.S.; Fitzgerald, L.R.; Liimatta, M.; Arendse, A.; Stevenson, T.; Lund, E.T.; Nugent, R.A.; Nomanbhoy, T.K.; Alexander, J.P.; Cravatt, B.F. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 2007, 46, 13019–13030. [Google Scholar]
- Clapper, J.R.; Mangieri, R.A.; Piomelli, D. The endocannabinoid system as a target for the treatment of cannabis dependence. Neuropharmacology 2009, 56, 235–243. [Google Scholar]
- Long, J.Z.; Nomura, D.K.; Vann, R.E.; Walentiny, D.M.; Booker, L.; Jin, X.; et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. PNAS 2009, 106, 20270–20275. [Google Scholar]
- Kaczocha, M.; Glaser, S.T.; Chae, J.; Brown, D.A.; Deutsch, D.G. Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J. Biol. Chem. 2010, 285, 2796–2806. [Google Scholar]
- Tsuboi, K.; Sun, Y.; Okamoto, Y.; Araki, N.; Tonai, T.; Ueda, N. J. Biol. Chem. 2005, 280, 11082–11092.
- Sun, Y.; Tsuboi, K.; Zhao, L.; Okamoto, Y.; Lambert, D.; Ueda, N. Biochim. Biophys. Acta 2005, 1736, 211–220.
- Khairy, H.; Adjei, G.; Allen-Redpath, K.; Scott, R.H. Actions of ethanolamine on cultured sensory neurones from neonatal rats. Neurosci. Lett. 2010, 468, 326–329. [Google Scholar]
- Alexander, S.P.H.; Kendall, D.A. The complications of promiscuity: Endocannabinoid action and metabolism. Br. J. Pharmacol. 2007, 152, 602–623. [Google Scholar]
- Harizi, H.; Corcuff, J.B.; Gualde, N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 2008, 14, 461–469. [Google Scholar]
- Jiang, X.; Zhang, Y.H.; Clark, J.D.; Tempel, B.L.; Nicol, G.D. Prostaglandin E2 inhibits the potassium current in sensory neurons from hyperalgesic Kv1.1 knockout mice. Neurosci. 2003, 119, 65–72. [Google Scholar]
- Chen, J.K.; Chen, J.; Imig, J.D.; Wei, S.; Hachey, D.L.; Guthi, J.S.; Falck, J.R.; Capdevila, J.H.; Harris, R.C. Identification of novel endogenous cytochrome P450 arachidonate metabolites with high affinity for cannabinoid receptors. J. Biol. Chem. 2008, 283, 24514–24524. [Google Scholar]
- Yu, M.; Ives, D.; Ramesha, C.S. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 1997, 272, 21181–21186. [Google Scholar]
- Woodward, D.F.; Carling, R.W.C.; Cornell, C.L.; Fliri, H.G.; Martos, J.L.; Pettit, S.N.; et al. The pharmacology and therapeutic relevance of endocannabinoid derived cyclo-oxygenase (COX)-2 products. Pharmacol. Therap. 2008, 120, 71–80. [Google Scholar]
- Weber, A.; Ni, J.; Ling, K.H.; Acheampong, A.; Tang-Liu, D.D.; Burk, R.; Cravatt, B.F.; Woodward, D. Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J. Lipid Res. 2004, 45, 757–763. [Google Scholar]
- Ueda, N.; Yamamoto, K.; Yamamoto, S.; Tokunaga, T.; Shirakawa, E.; Shinkai, H.; et al. Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim. Biophys. Acta 1995, 1254, 127–134. [Google Scholar]
- Hampson, A.J.; Hill, W.A.G.; Zan-Phillips, M.; Makriyannis, A.; Leung, E.; Eglen, R.M.; Bornheim, L.M. Anandamide hydroxylation by brain lipoxygenase: metabolite structures and potencies at the cannabinoid receptor. Biochim. Biophys. Acta 1995, 1259, 173–179. [Google Scholar]
- Mulder, A.M.; Cravatt, B.F. Endocannabinoid metabolism in the absence of fatty acid amide hydrolase (FAAH): Discovery of phosphorylcholine derivatives of N-acyl ethanolamines. Biochem. 2006, 45, 11267–11277. [Google Scholar]
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khairy, H.; Houssen, W.E. Inactivation of Anandamide Signaling: A Continuing Debate. Pharmaceuticals 2010, 3, 3355-3370. https://doi.org/10.3390/ph3113355
Khairy H, Houssen WE. Inactivation of Anandamide Signaling: A Continuing Debate. Pharmaceuticals. 2010; 3(11):3355-3370. https://doi.org/10.3390/ph3113355
Chicago/Turabian StyleKhairy, Hesham, and Wael E. Houssen. 2010. "Inactivation of Anandamide Signaling: A Continuing Debate" Pharmaceuticals 3, no. 11: 3355-3370. https://doi.org/10.3390/ph3113355
APA StyleKhairy, H., & Houssen, W. E. (2010). Inactivation of Anandamide Signaling: A Continuing Debate. Pharmaceuticals, 3(11), 3355-3370. https://doi.org/10.3390/ph3113355