Role of Glycogen Synthase Kinase-3β in APP Hyperphosphorylation Induced by NMDA Stimulation in Cortical Neurons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
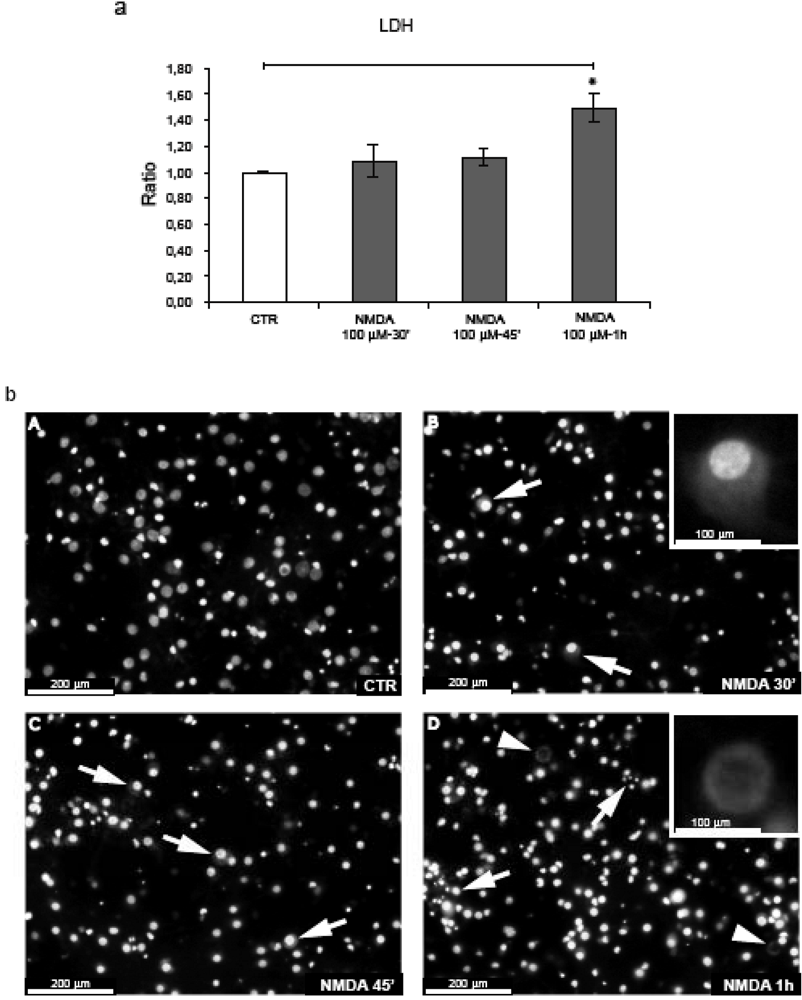
2.1. NMDA treatment of cortical neurons
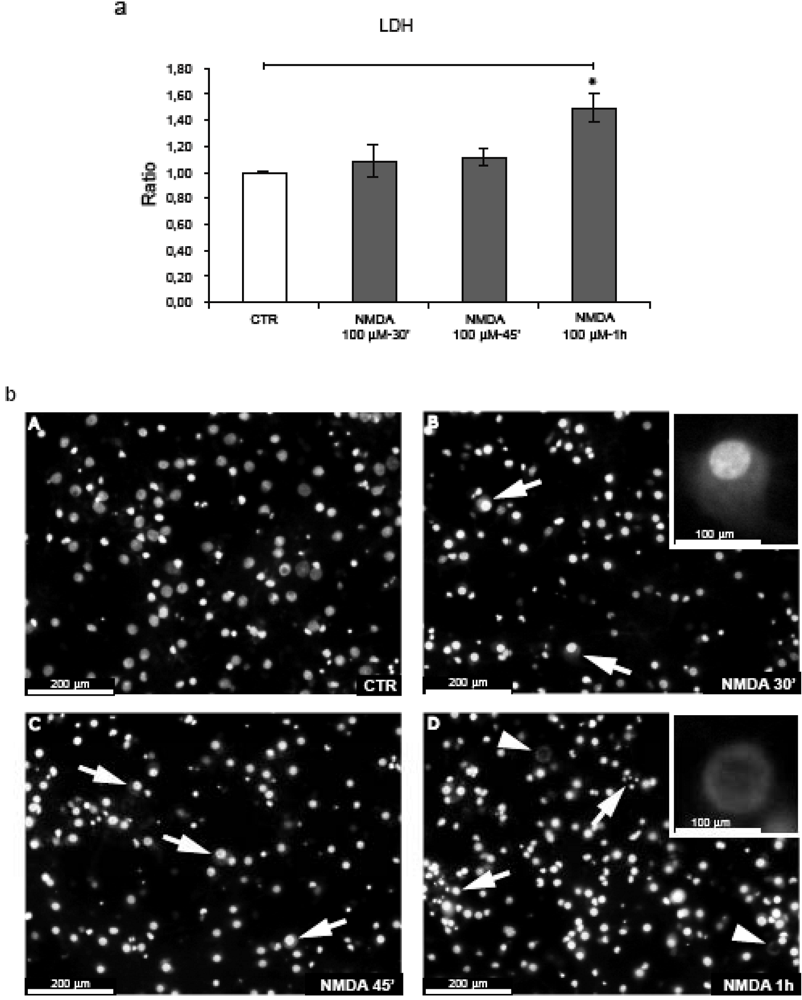
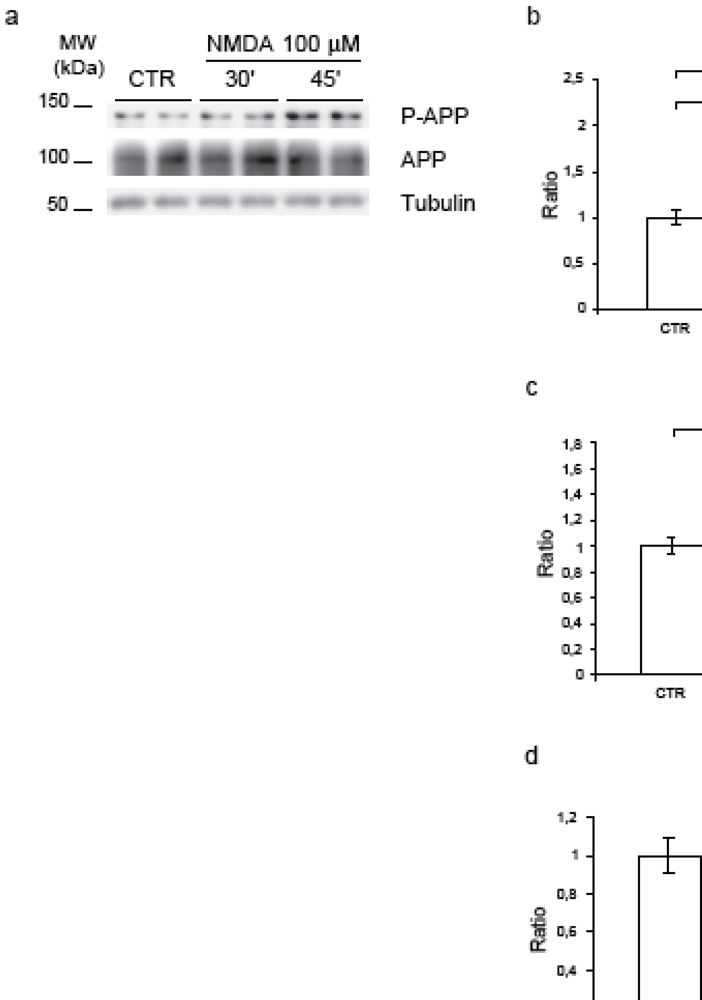
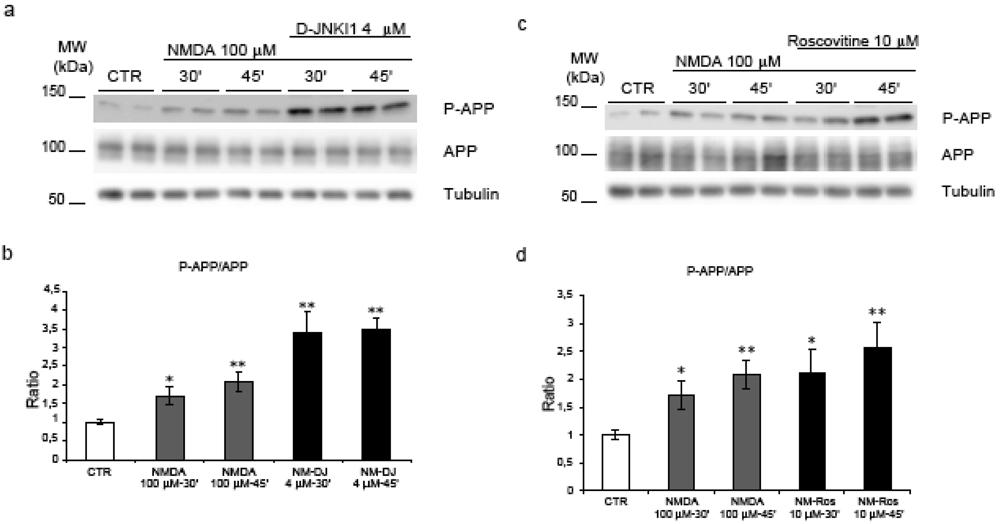
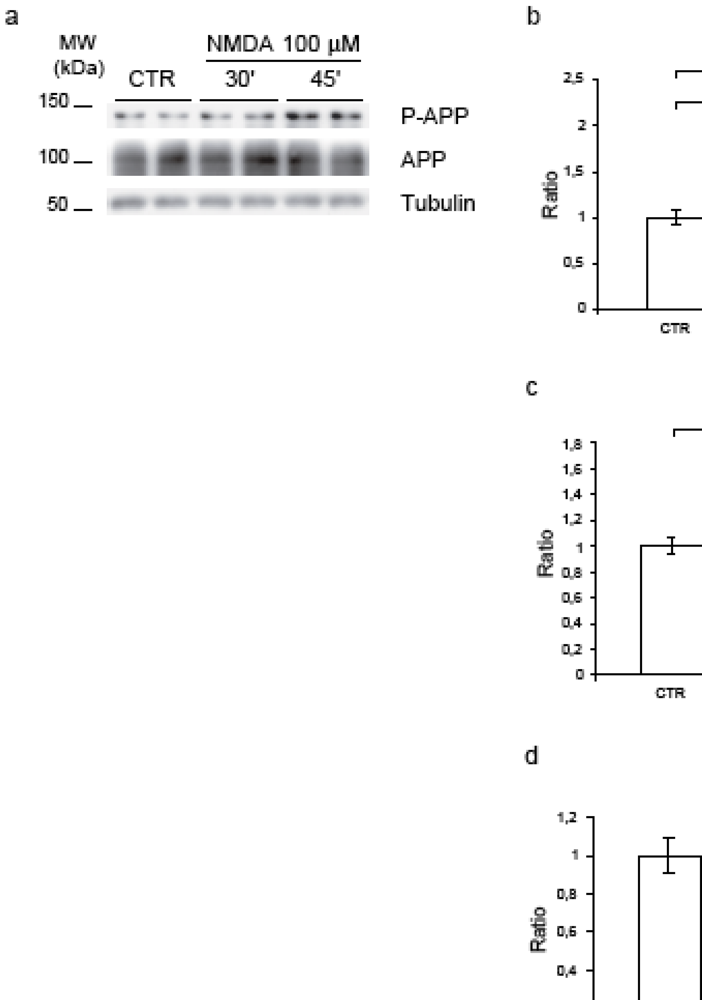
2.2. NMDA for 30’-45’ induces APP hyperphosphorylation at Thr668
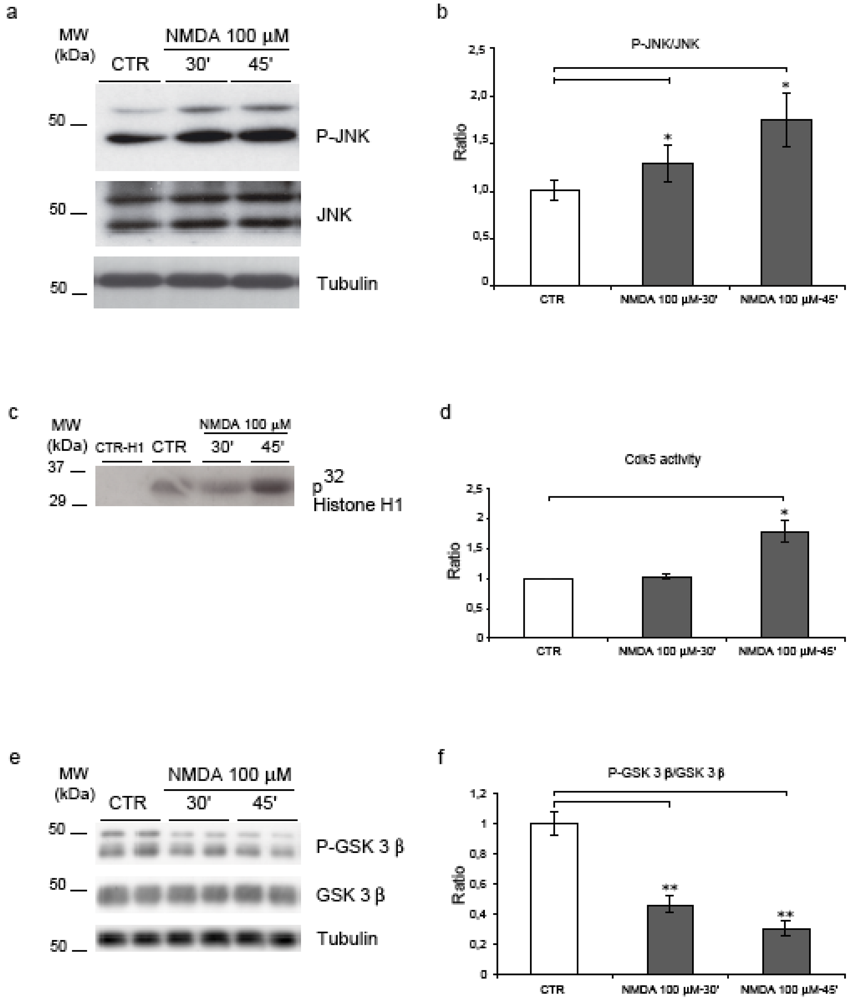
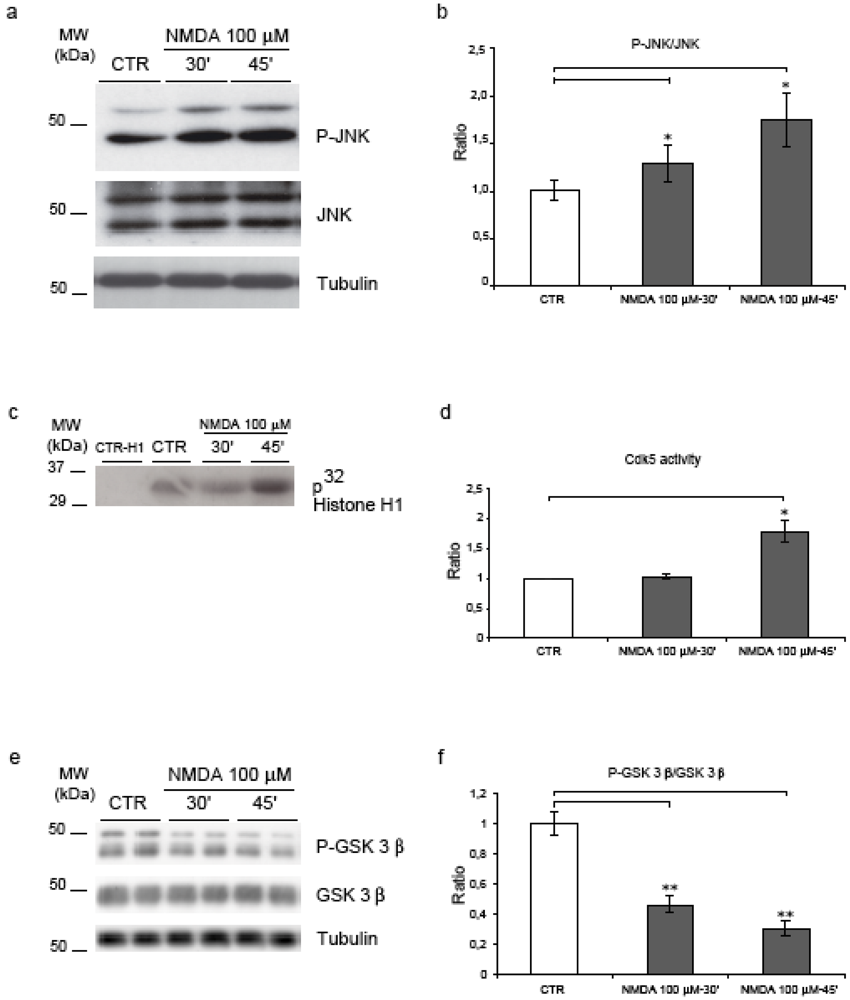
2.3. NMDA stimulation induces JNK, Cdk5 and GSK-3β
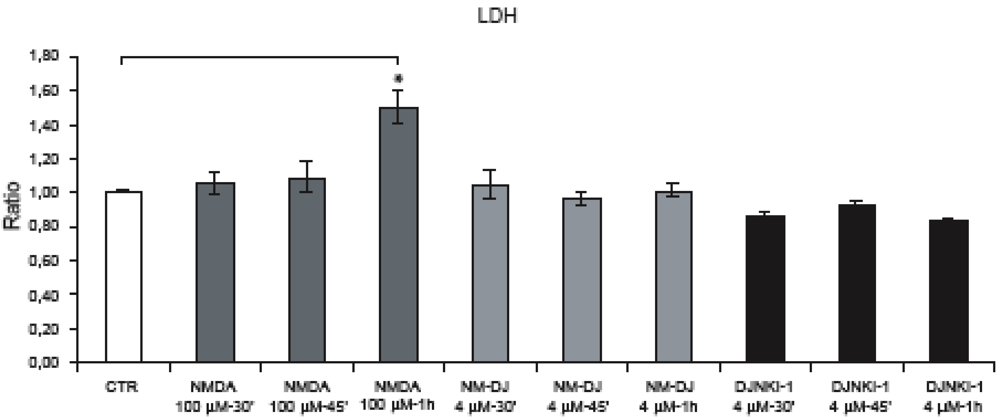
2.4. Neither JNK nor Cdk5 are responsible for NMDA induced APP hyperphosphorylation
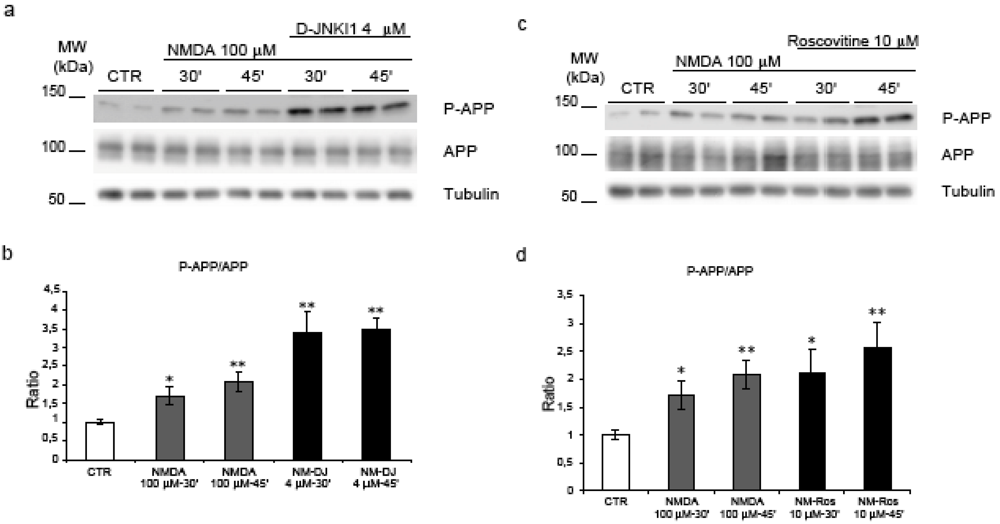
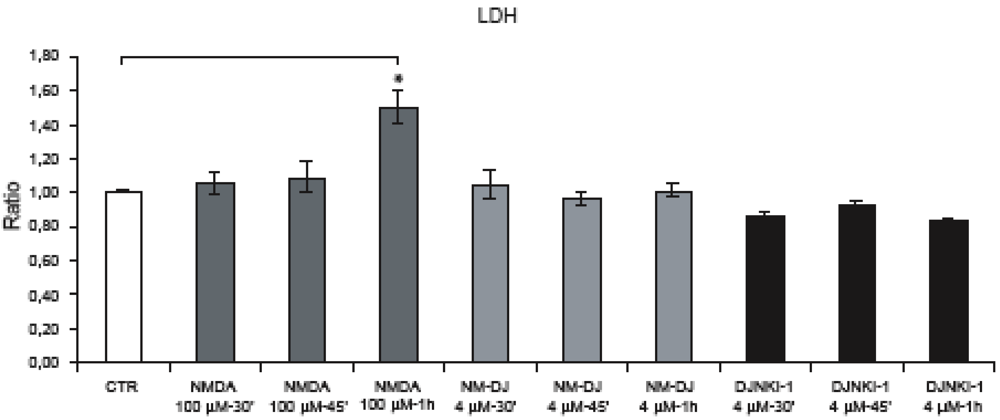
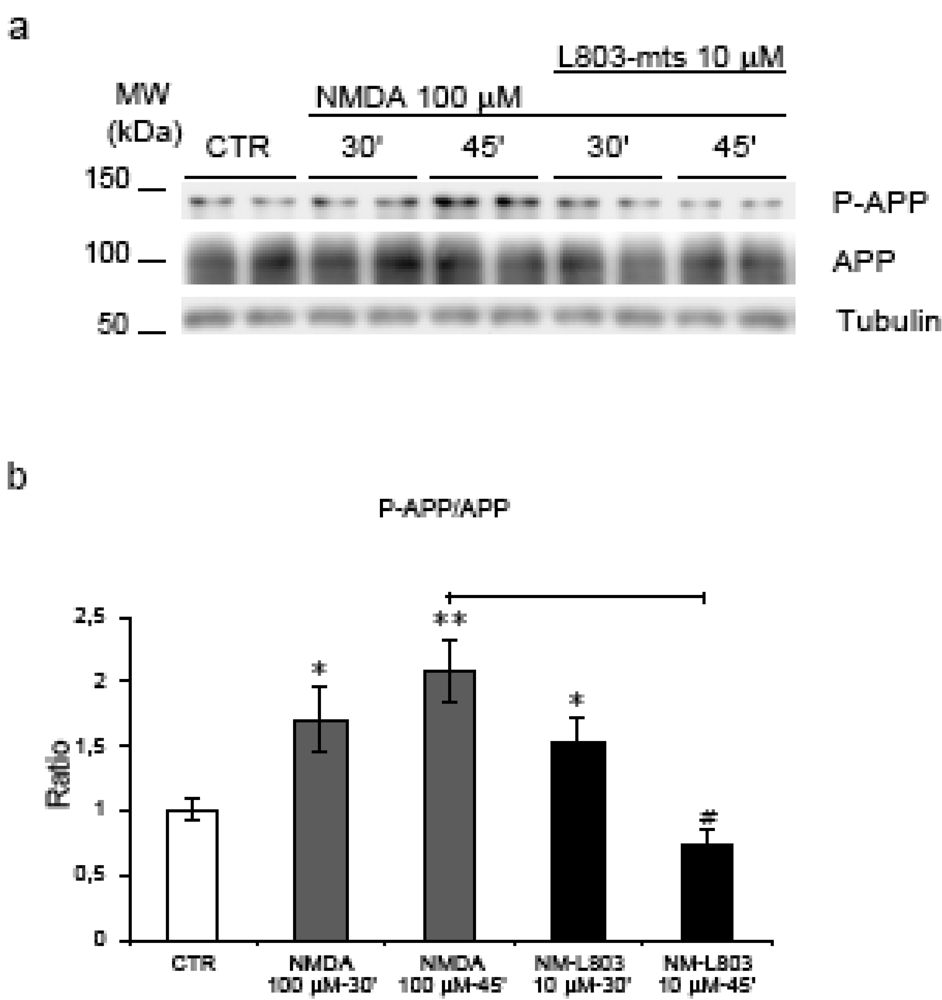
2.5. GSK-3β regulates NMDA induced APP hyperphosphorylation
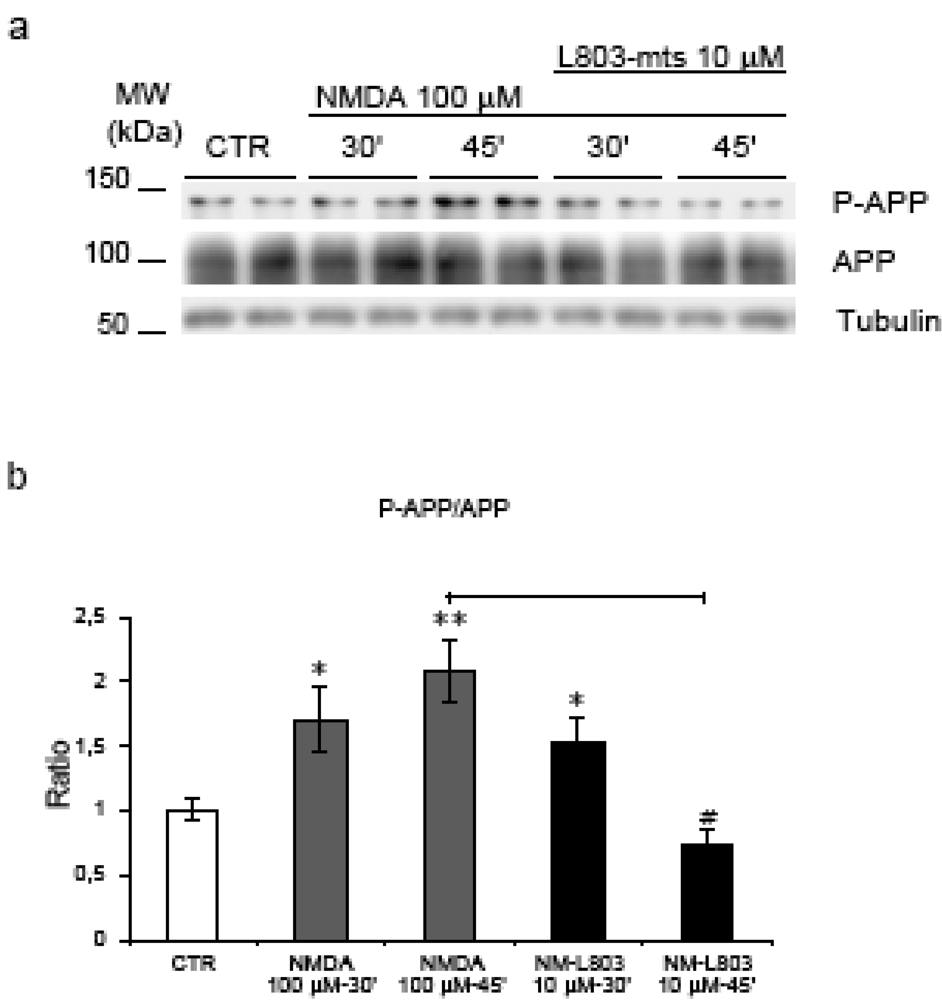
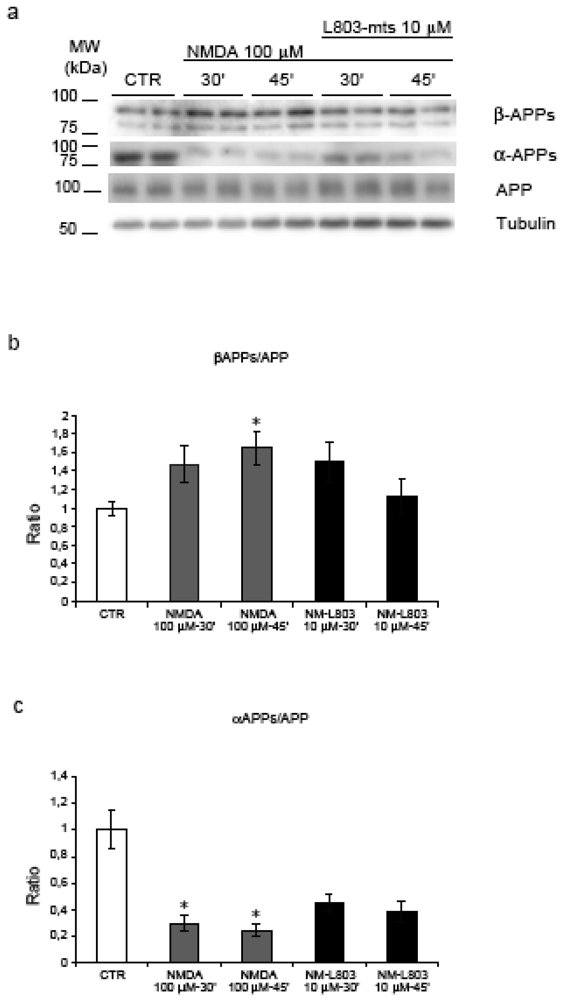
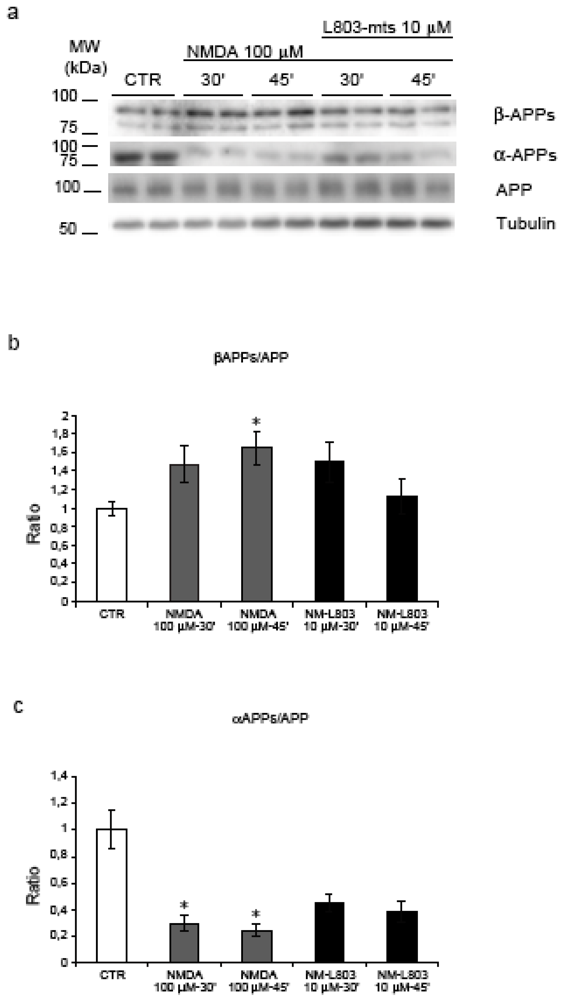
2.6. GSK-3β regulates APP amyloidogenic processing induced by NMDA stimulation
2.7. Discussion
3. Experimental Section
3.1. Cortical Neuronal Culture
3.2. Cytotoxicity Assay
3.3. Cellular Lysis
3.4. Media Proteins Precipitation
3.5. Western Blot Analysis
3.6. Cdk5 Kinase Assay
3.7. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Da Cruz e Silva, E.F.; da Cruz e Silva, O.A. Proteinphosphorylation and APP metabolism. Neurochem. Res. 2003, 28, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Kirino, Y.; Suzuki, T. A basic amino acid in the cytoplasmic domain of Alzheimer's beta-amyloid precursor protein (APP) is essential for cleavage of APP at the alpha-site. J. Biol. Chem. 1998, 273, 19304–19310. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kao, S.C.; Lemere, C.A.; Xia, W.; Tseng, H.C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.H. APP processing is regulated by cytoplasmicphosphorylation. J. Cell. Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.E.; Gibb, G.M.; Jacobsen, J.S.; Gallo, J.M.; Anderton, B.H. In vitrophosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J. Neurochem. 1996, 67, 699–707. [Google Scholar] [PubMed]
- Iijima, K.; Ando, K.; Takeda, S.; Satoh, Y.; Seki, T.; Itohara, S.; Greengard, P.; Kirino, Y.; Nairn, A.C.; Suzuki, T. Neuron-specific phosphorylation of Alzheimer's beta-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 2000, 75, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Standen, C.L.; Brownlees, J.; Grierson, A.J.; Kesavapany, S.; Lau, K.F.; McLoughlin, D.M.; Miller, C.C. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer's disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J. Neurochem. 2001, 76, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Muresan, Z.; Muresan, V. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-beta precursor protein. J. Neurosci. 2005, 25, 3741–3751. [Google Scholar] [CrossRef] [PubMed]
- Muresan, Z.; Muresan, V. The amyloid-beta precursor protein is phosphorylatedvia distinct pathways during differentiation, mitosis, stress, and degeneration. Mol. Biol. Cell. 2007, 18, 3835–3844. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Tang, X.; Wiedmann, M.; Wang, X.; Peng, J.; Zheng, D.; Blair, L.A.; Marshall, J.; Mao, Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 2003, 38, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Oishi, M.; Marshak, D.R.; Czernik, A.J.; Nairn, A.C.; Greengard, P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. Embo. J. 1994, 13, 1114–1122. [Google Scholar] [PubMed]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK regulates APP cleavage and degradation in a model of Alzheimer's disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincon, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Centeno, C.; Repici, M.; Chatton, J.Y.; Riederer, B.M.; Bonny, C.; Nicod, P.; Price, M.; Clarke, P.G.; Papa, S.; Franzoso, G.; Borsello, T. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. 2007, 14, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Iwakami, N.; Takeuchi, S.; Waragai, M.; Suzuki, M.; Kanazawa, I.; Lippa, C.F.; Ono, S.; Okazawa, H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res. Mol. Brain Res. 2000, 85, 221–233. [Google Scholar] [CrossRef]
- Colombo, A.; Repici, M.; Pesaresi, M.; Santambrogio, S. The TAT-JNK inhibitor peptide interferes with beta amyloid protein stability. Cell Death Differ. 2007, 14, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- O'Hare, M.J.; Kushwaha, N.; Zhang, Y.; Aleyasin, H.; Callaghan, S.M.; Slack, R.S.; Albert, P.R.; Vincent, I.; Park, D.S. Differential roles of nuclear and cytoplasmiccyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J. Neurosci. 2005, 25, 8954–8966. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.C.; Kim, D.; Moy, L.Y.; Dobbin, M.M.; Sun, X.; Bronson, R.T.; Tsai, L.H. p25/Cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J. Neurosci. 2006, 26, 10536–10541. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Toro, R.; Caceres, A.; Maccioni, R.B. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999, 459, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, F.; Hsu, S.C.; Cordell, B.; McCarthy, J.V. Substitution of a glycogen synthase kinase-3beta phosphorylation site in presenilin 1 separates presenilin function from beta-catenin signaling. J. Biol. Chem. 2001, 276, 7366–7375. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthasekinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.V.; Budd Haeberlein, S.L.; Avila, J. Glycogen synthasekinase 3: A drug target for CNS therapies. J. Neurochem. 2004, 89, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Noguchi, K.; Michel, G.; Mercken, M.; Hoshi, M.; Ishiguro, K.; Imahori, K. Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3 beta. Neurosci. Lett. 1996, 203, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Zhao, H.; Hua Sun, G.; Cheng, D.; Qiao, Y.; Luo, J.; Martin, K.; Steinberg, G.K.; Harrison, S.D.; Yenari, M.A. Glycogen synthasekinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp. Neurol. 2004, 188, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Ali, C.; Gabriel, C.; Croci, N.; MacKenzie, E.T.; Glabe, C.G.; Plotkine, M.; Marchand-Verrecchia, C.; Vivien, D.; Buisson, A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J. Neurosci. 2005, 25, 9367–9377. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, B.; Kaidanovich, O.; Talior, I.; Eldar-Finkelman, H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 2003, 305, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. A hundred years of Alzheimer's disease research. Neuron 2006, 52, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Yang, L.L.; Cotman, C.W. Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain. Res. 1990, 533, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [PubMed]
- Wu, J.; Anwyl, R.; Rowan, M.J. [beta]-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport 1995, 6, 2409–2413. [Google Scholar] [PubMed]
- Butterfield, D.A.; Pocernich, C.B. The glutamatergic system and Alzheimer's disease: Therapeutic implications. CNS Drugs 2003, 17, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Rowan, M.J.; Klyubin, I.; Wang, Q.; Anwyl, R. Mechanisms of the inhibitory effects of amyloid beta-protein on synaptic plasticity. Exp. Gerontol. 2004, 39, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Dunah, A.W.; Yasuda, R.P.; Luo, J.; Wang, Y.; Prybylowski, K.L.; Wolfe, B.B. Biochemical studies of the structure and function of the N-methyl-D-aspartate subtype of glutamate receptors. Mol. Neurobiol. 1999, 19, 151–179. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Doody, R.; Stoffler, A.; Schmitt, F.; Ferris, S.; Mobius, H.J. Memantine in moderate-to-severe Alzheimer's disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Zoladz, P.R.; Campbell, A.M.; Park, C.R.; Schaefer, D.; Danysz, W.; Diamond, D.M. Enhancement of long-term spatial memory in adult rats by the noncompetitive NMDA receptor antagonists, memantine and neramexane. Pharmacol. Biochem. Behav. 2006, 85, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, E.; Habas, A.; Yang, P.; Zheng, J.J.; Hagg, T.; Hetman, M. A positive feedback loop between glycogen synthasekinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J. Biol. Chem. 2005, 280, 37526–37535. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Lee, J.E.; Kim, M.J.; Cho, E.G.; Cho, S.G.; Choi, E.J. Glycogen synthasekinase 3 beta is a natural activator of mitogen-activated protein kinase/extracellular signal-regulated kinasekinasekinase 1 (MEKK1). J. Biol. Chem. 2003, 278, 13995–14001. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Szebenyi, G.; Brown, H.; Pant, H.C.; Pigino, G.; DeBoer, S.; Beffert, U.; Brady, S.T. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. Embo. J. 2004, 23, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Fornoni, A.; Pileggi, A.; Molano, R.D.; Sanabria, N.Y.; Tejada, T.; Gonzalez-Quintana, J.; Ichii, H.; Inverardi, L.; Ricordi, C.; Pastori, R.L. Inhibition of c-jun N terminal kinase (JNK) improves functional beta cell mass in human islets and leads to AKT and glycogen synthase kinase-3 (GSK-3) phosphorylation. Diabetologia 2008, 51, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthasekinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Fang, W.; Han, A.; Gallagher, L.; Davis, R.J.; Xiong, B.; Yang, W. c-Jun N-terminal kinase 1 interacts with and negatively regulates Wnt/beta-catenin signaling through GSK3beta pathway. Carcinogenesis 2008, 29, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: Novel blockers of beta-cell death. Diabetes 2001, 50, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Hoey, S.E.; Williams, R.J.; Perkinton, M.S. Synaptic NMDA receptor activation stimulates alpha-secretaseamyloid precursor protein processing and inhibits amyloid-beta production. J. Neurosci. 2009, 29, 4442–4460. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ploia, C.; Sclip, A.; Colombo, A.; Repici, M.; Gardoni, F.; Di Luca, M.; Forloni, G.; Antoniou, X.; Borsello, T. Role of Glycogen Synthase Kinase-3β in APP Hyperphosphorylation Induced by NMDA Stimulation in Cortical Neurons. Pharmaceuticals 2010, 3, 42-58. https://doi.org/10.3390/ph3010042
Ploia C, Sclip A, Colombo A, Repici M, Gardoni F, Di Luca M, Forloni G, Antoniou X, Borsello T. Role of Glycogen Synthase Kinase-3β in APP Hyperphosphorylation Induced by NMDA Stimulation in Cortical Neurons. Pharmaceuticals. 2010; 3(1):42-58. https://doi.org/10.3390/ph3010042
Chicago/Turabian StylePloia, Cristina, Alessandra Sclip, Alessio Colombo, Mariaelena Repici, Fabrizio Gardoni, Monica Di Luca, Gianluigi Forloni, Xanthi Antoniou, and Tiziana Borsello. 2010. "Role of Glycogen Synthase Kinase-3β in APP Hyperphosphorylation Induced by NMDA Stimulation in Cortical Neurons" Pharmaceuticals 3, no. 1: 42-58. https://doi.org/10.3390/ph3010042
APA StylePloia, C., Sclip, A., Colombo, A., Repici, M., Gardoni, F., Di Luca, M., Forloni, G., Antoniou, X., & Borsello, T. (2010). Role of Glycogen Synthase Kinase-3β in APP Hyperphosphorylation Induced by NMDA Stimulation in Cortical Neurons. Pharmaceuticals, 3(1), 42-58. https://doi.org/10.3390/ph3010042