
Pharmacokinetics, Safety, and Tolerability of (R)-Ketamine Hydrochloride Injection, a Novel Rapid-Acting Antidepressant, in Healthy Chinese Subjects


Abstract
1. Introduction
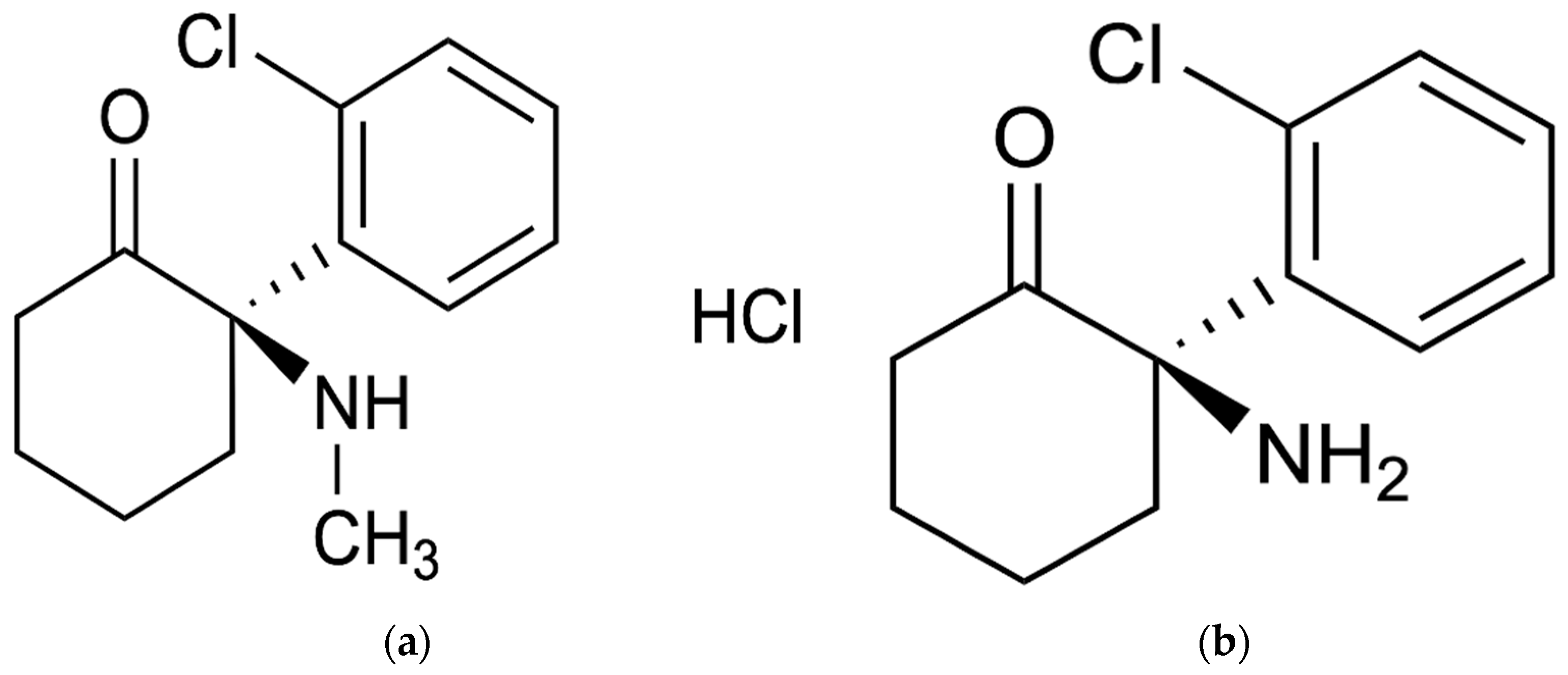
2. Results
2.1. Study Population

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Dose Cohorts | ||||||
|---|---|---|---|---|---|---|---|
| 10.0 mg | 30.0 mg | 60.0 mg | 120 mg | 180 mg | Placebo | Total | |
| n | 8 | 8 | 8 | 8 | 8 | 10 | 50 |
| Sex n, Male/female | 4/4 | 4/4 | 3/5 | 3/5 | 4/4 | 7/3 | 25/25 |
| Age, years | |||||||
| Mean (SD) | 33.4 (4.5) | 26.6 (8.2) | 34.9 (7.3) | 35.5 (6.4) | 34.1 (5.7) | 29.9 (7.5) | 32.3 (7.1) |
| Max, Min | 26, 41 | 20, 44 | 25, 44 | 25, 44 | 28, 42 | 19, 41 | 19, 44 |
| Median | 33.0 | 24.0 | 35.5 | 37.0 | 32.0 | 32.0 | 32.0 |
| Ethnic group n, Han/others | 8/0 | 8/0 | 7/1 | 8/0 | 8/0 | 10/0 | 49/1 |
| Height, cm | |||||||
| Mean (SD) | 164.88 (6.95) | 166.00 (5.32) | 162.75 (5.81) | 165.00 (6.77) | 162.25 (5.71) | 164.85 (10.65) | 164.31 (7.04) |
| Max, Min | 151.5, 174.0 | 160.0, 176.5 | 155.0, 171.0 | 158.5, 177.5 | 153.0, 168.5 | 148.5, 179.5 | 148.5, 179.5 |
| Weight, kg | |||||||
| Mean (SD) | 64.70 (10.31) | 57.93 (5.21) | 61.65 (4.61) | 64.58 (7.23) | 59.71 (7.74) | 64.99 (8.87) | 62.37 (7.75) |
| Max, Min | 47.2, 77.9 | 50.2, 65.3 | 54.6, 69.9 | 54.7, 77.2 | 49.1, 71.4 | 54.0, 83.3 | 47.2, 83.3 |
| BMI, kg/m2 | |||||||
| Mean (SD) | 23.76 (2.25) | 21.06 (1.77) | 23.31 (2.55) | 23.83 (1.95) | 22.58 (2.03) | 23.98 (1.57) | 23.12 (2.17) |
| Max, Min | 20.3, 25.9 | 19.0, 24.9 | 20.5, 27.0 | 20.6, 26.5 | 18.9, 25.3 | 20.4, 25.5 | 18.9, 27.0 |
2.2. Pharmacokinetics
2.2.1. (R)-Ketamine
| Dose Cohorts | Cmax (ng/mL) | Tmax (h) | AUC0–t (h·ng/mL) | AUC0–∞ (h·ng/mL) | CL (L/h) | Vd (L) | λz (/h) | t1/2 (h) | MRT0–t (h) | MRT0–∞ (h) | AUMC0–t (h2·ng/mL) | AUMC0–∞ (h2·ng/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10.0 mg (n = 8) | 56.0 (9.9, 17.7) | 0.67 (0.67–0.67) | 121 (18, 15.3) | 129 (21, 16.3) | 79.2 (11.6, 14.6) | 817 (209, 25.5) | 0.104 (0.040, 38.4) | 7.19 (1.63, 22.6) | 3.97 (0.63, 15.8) | 5.46 (1.06, 19.4) | 432 (117, 27.0) | 688 (209, 30.5) |
| 30.0 mg (n = 8) | 222 (104, 46.9) | 0.67 (0.66–0.75) | 468 (131, 28.0) | 478 (134, 28.0) | 67.3 (18.6, 27.7) | 828 (234, 28.3) | 0.0882 (0.0426, 48.3) | 9.11 (3.07, 33.7) | 5.27 (0.85, 16.1) | 6.26 (1.21, 19.3) | 2337 (879, 37.6) | 2888 (1170, 40.5) |
| 60.0 mg (n = 7) | 448 (85, 19.0) | 0.67 (0.67–0.67) | 1031 (75, 7.3) | 1047 (76, 7.2) | 57.5 (4.0, 6.9) | 1066 (251, 23.6) | 0.0573 (0.0180, 31.4) | 12.9 (3.1, 23.9) | 7.48 (2.25, 30.1) | 8.55 (2.47, 28.8) | 7411 (2661, 35.9) | 8659 (2912, 33.6) |
| * 60.0 mg (n = 8, including subject 3-001) | 463 (90, 19.4) | 0.67 (0.67–0.67) | 1069 (126, 11.8) | 1086 (127, 11.7) | 55.9 (5.9, 10.6) | 1050 (237, 22.5) | 0.0560 (0.0170, 30.4) | 13.1 (2.9, 22.2) | 7.78 (2.25, 29.0) | 8.89 (2.47, 27.8) | 8070 (3090, 38.3) | 9398 (3410, 36.3) |
| 120 mg (n = 8) | 805 (166, 20.6) | 0.67 (0.67–0.75) | 1774 (364, 20.5) | 1815 (392, 21.6) | 68.7 (14.4, 20.9) | 1327 (285, 21.5) | 0.0529 (0.0115, 21.8) | 13.6 (2.7, 20.0) | 7.45 (1.44, 19,3) | 8.16 (1.53, 18.7) | 12,955 (4474, 34.5) | 14,849 (4957, 33.4) |
| 180 mg (n = 8) | 1424 (350, 24.6) | 0.67 (0.67–0.68) | 2997 (333, 11.1) | 3018 (331, 11.0) | 60.3 (6.5, 10.7) | 1185 (304, 25.7) | 0.0527 (0.0099, 18.8) | 13.6 (2.7, 19.6) | 7.38 (1.36, 18.4) | 8.00 (1.52, 18.9) | 20,974 (4087, 19.5) | 22,953 (4434, 19.3) |
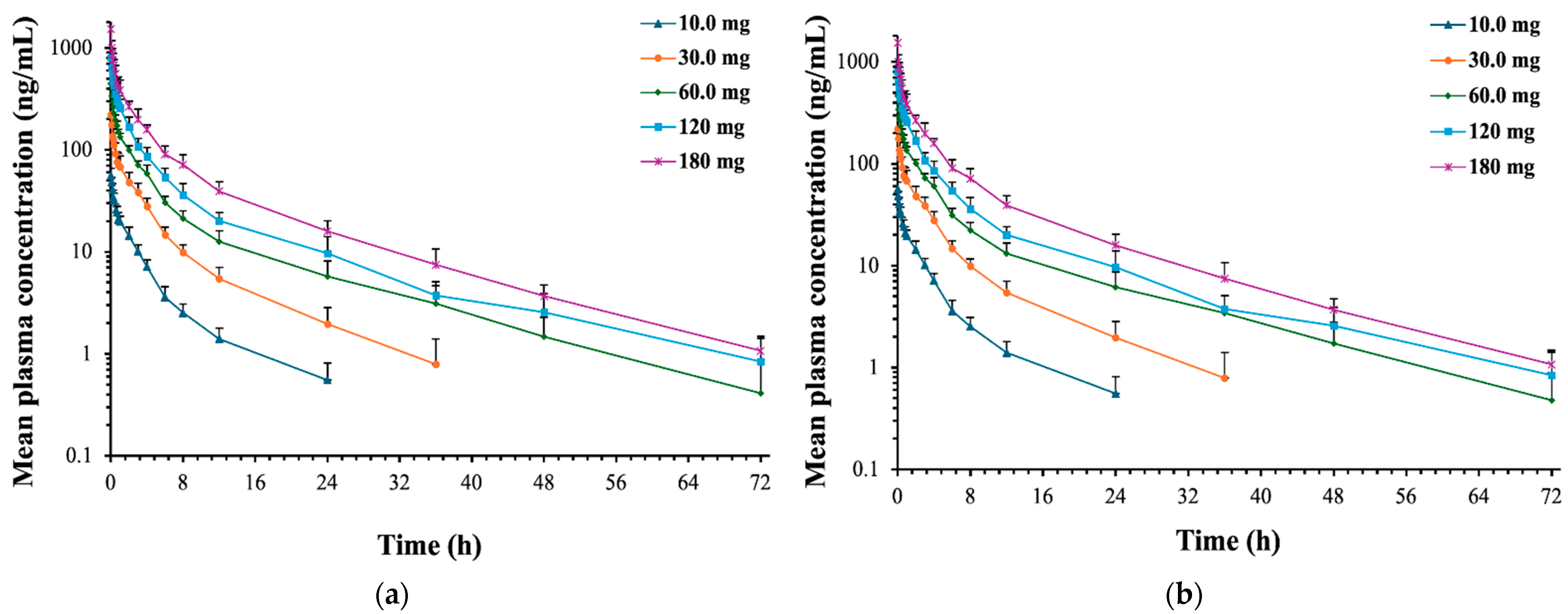
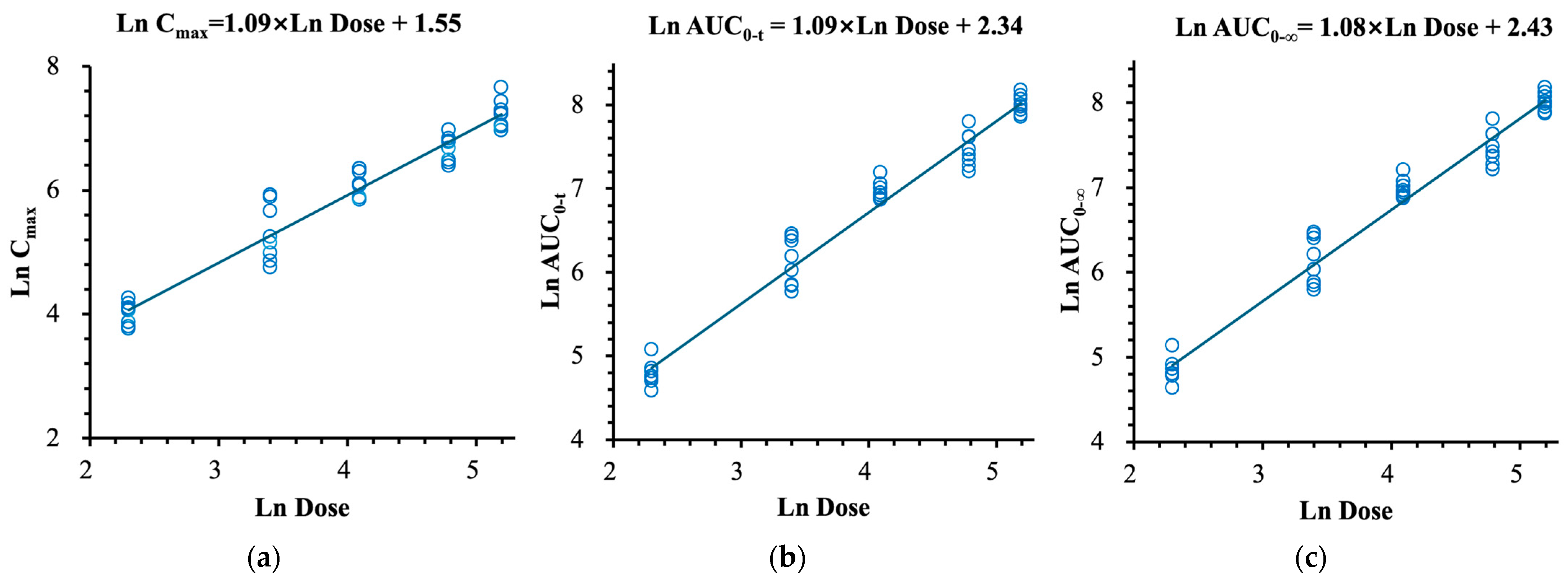
2.2.2. (R)-Norketamine
| Dose Cohorts | Cmax (ng/mL) | Tmax (h) | AUC0–t (h·ng/mL) | AUC0–∞ (h·ng/mL) | λz (h) | t1/2 (h) | MRT0–t (h) | MRT0–∞ (h) | AUMC0–t (h2·ng/mL) | AUMC0–∞ (h2·ng/mL) | MPratio Cmax | MPratio_AUC0–∞ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10.0 mg (n = 8) | 27.7 (6.5, 23.5) | 0.92 (0.75–1.34) | 222 (61, 27.4) | 236 (64, 26.9) | 0.0524 (0.0162, 30.9) | 14.3 (4.2, 29.6) | 12.0 (2.9, 23.7) | 15.6 (4.0, 25.5) | 2674 (1365, 51.1) | 3739 (1872, 50.1) | 0.534 (0.124, 23.2) | 1.95 (0.46, 23.8) |
| 30.0 mg (n = 8) | 115 (16, 13.9) | 0.92 (0.75–1.17) | 972 (239, 24.5) | 990 (249, 25.1) | 0.0636 (0.0234, 36.9) | 12.0 (3.6, 29.8) | 12.9 (2.7, 20.9) | 14.2 (3.5, 24.5) | 12,457 (5379, 43.2) | 14,080 (6673, 47.4) | 0.642 (0.241, 37.6) | 2.31 (0.63, 27.3) |
| 60.0 mg (n = 7) | 153 (28, 18.4) | 1.19 (0.83–1.50) | 1574 (395, 25.1) | 1617 (407, 25.2) | 0.0535 (0.0179, 33.4) | 13.8 (3.1, 22.5) | 14.0 (2.9, 20.8) | 15.8 (4.0, 25.2) | 21,700 (8315, 38.3) | 25,365 (10356, 40.8) | 0.375 (0.113, 30.2) | 1.65 (0.43, 26.4) |
| * 60.0mg (n = 8, including subject 3-001) | 156 (28, 17.7) | 1.13 (0.83–1.50) | 1775 (673, 37.9) | 1868 (810, 43.3) | 0.0503 (0.0190, 37.7) | 15.3 (5.0, 32.7) | 15.1 (4.2, 27.7) | 18.1 (7.4, 41.1) | 27,870 (19,074, 68.4) | 37,461 (35,530, 94.8) | 0.369 (0.106, 28.7) | 1.80 (0.59, 32.8) |
| 120 mg (n = 8) | 285 (51, 17.9) | 0.92 (0.67–1.50) | 2566 (397, 15.5) | 2615 (418, 16.0) | 0.0518 (0.0084, 16.2) | 13.7 (2.0, 14.9) | 13.1 (2.0, 15.0) | 14.5 (2.8, 19.4) | 32,860 (8898, 27.1) | 37,472 (12,006, 32.0) | 0.392 (0.110, 27.9) | 1.53 (0.35, 22.6) |
| 180 mg (n = 8) | 491 (119, 24.3) | 1.08 (0.83–4.67) | 5506 (2495, 45.3) | 5643 (2646, 46.9) | 0.0523 (0.0079, 15.1) | 13.5 (2.1, 15.3) | 14.1 (3.0, 21.5) | 15.6 (4.0, 25.4) | 81,382 (54,954, 67.5) | 94,261 (69,617, 73.9) | 0.380 (0.117, 30.8) | 1.98 (0.85, 42.8) |
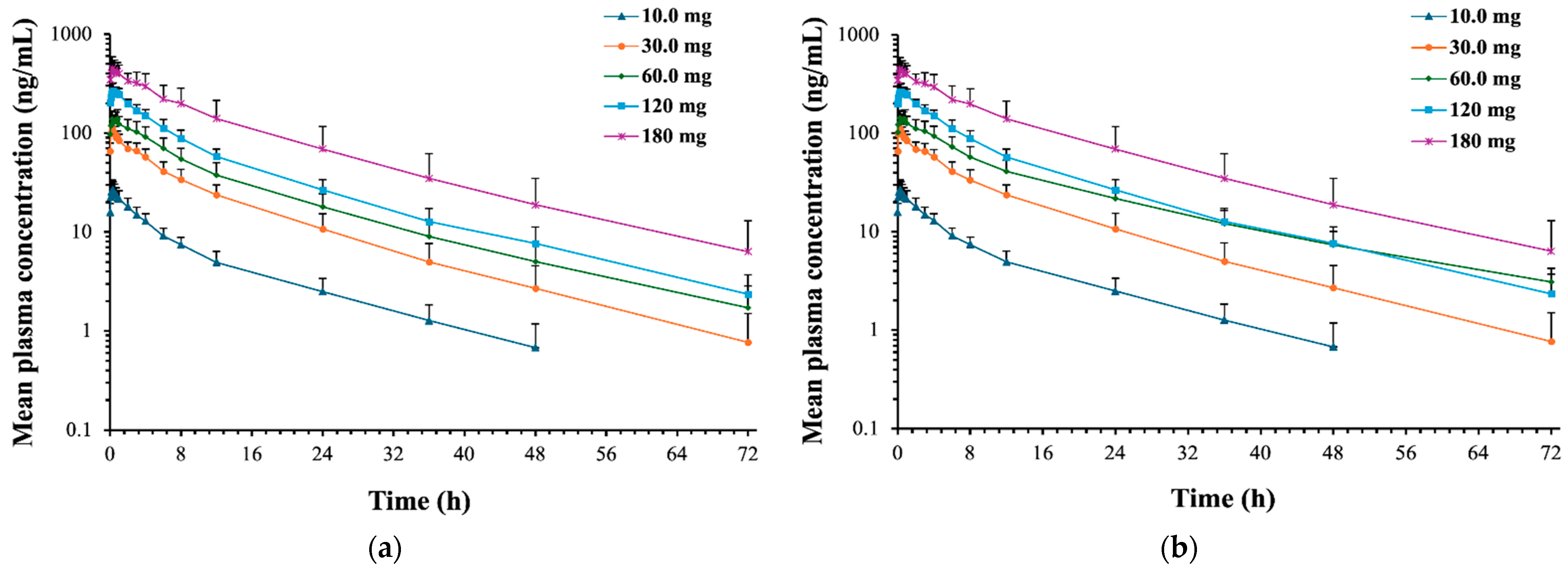
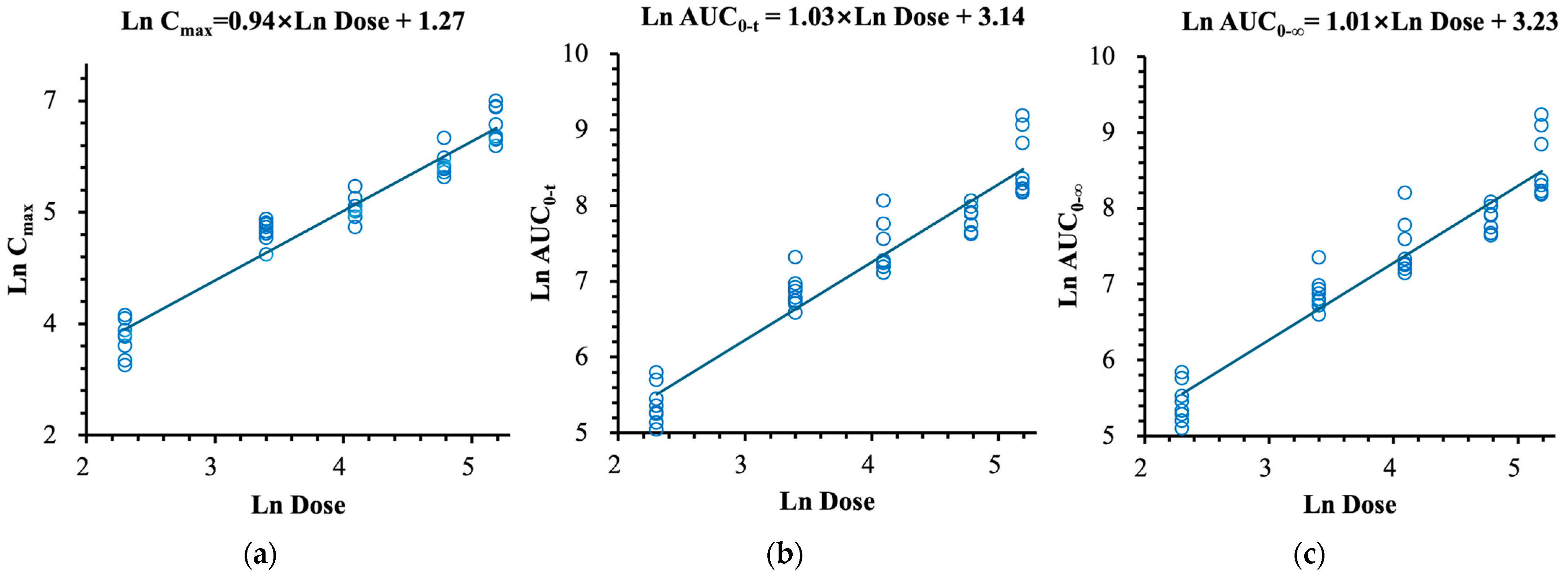
2.3. Safety and Tolerability
| Adverse Event | Dose Cohorts | ||||||
|---|---|---|---|---|---|---|---|
| 10.0 mg | 30.0 mg | 60.0 mg | 120 mg | 180 mg | Placebo | Total (No Placebo) | |
| n | 8 | 8 | 8 | 8 | 8 | 10 | 40 |
| TEAE | |||||||
| mild | 3 (37.5%) [6] | 7 (87.5%) [21] | 8 (100.0%) [32] | 8 (100.0%) [22] | 8 (100.0%) [37] | 4 (40.0%) [4] | 34 (85.0%) [118] |
| moderate | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ADR | |||||||
| mild | 2 (25.0%) [4] | 7 (87.5%) [18] | 7 (87.5%) [30] | 7 (87.5%) [18] | 8 (100.0%) [36] | 2 (20.0%) [2] | 31 (77.5%) [106] |
| moderate | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SAE | |||||||
| Suspicious and unexpected severe ADRs | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs leading to study termination | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ADRs leading to study termination | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe ADRs leading to study termination | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SAEs leading to study termination | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs | Dose Cohorts | ||||||
|---|---|---|---|---|---|---|---|
| 10.0 mg (n = 8) | 30.0 mg (n = 8) | 60.0 mg (n = 8) | 120 mg (n = 8) | 180 mg (n = 8) | Placebo (n = 10) | Total (No Placebo) (n = 40) | |
| Dizziness | 0 | 5 (62.5%) [5] | 5 (62.5%) [6] | 5 (62.5%) [5] | 6 (75.0%) [6] | 0 | 21 (52.5%) [22] |
| Somnolence | 0 | 0 | 0 | 0 | 8 (100.0%) [9] | 0 | 8 (20.0%) [9] |
| Decreased sensation | 0 | 1 (12.5%) [1] | 2 (25.0%) [2] | 1 (12.5%) [1] | 0 | 0 | 4 (10.0%) [4] |
| Decreased level of consciousness | 0 | 1 (12.5%) [1] | 0 | 0 | 1 (12.5%) [1] | 1 (10.0%) [1] | 2 (5.0%) [2] |
| Sleepiness | 0 | 1 (12.5%) [1] | 0 | 0 | 1 (12.5%) [1] | 0 | 2 (5.0%) [2] |
| Hypertonia | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Head discomfort | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Headache | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Tremor | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Blood pressure reduction | 1 (12.5%) [2] | 2 (25.0%) [2] | 1 (12.5%) [1] | 0 | 0 | 0 | 4 (10.0%) [5] |
| Serum bilirubin elevation | 0 | 0 | 0 | 0 | 2 (25.0%) [2] | 1 (10.0%) [1] | 2 (5.0%) [2] |
| Positive urine leukocytes | 0 | 1 (12.5%) [1] | 0 | 1 (12.5%) [1] | 0 | 0 | 2 (5.0%) [2] |
| Blood glucose elevation | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (10.0%) [1] | 1 (2.5%) [1] |
| Decreased neutrophil count | 0 | 0 | 0 | 1 (12.5%) [1] | 1 (12.5%) [1] | 0 | 2 (5.0%) [2] |
| Decreased lymphocyte count | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Decreased heart rate | 0 | 0 | 1 (12.5%) [2] | 0 | 0 | 0 | 1 (2.5%) [2] |
| Serum phosphorus elevation | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 0 | 1 (2.5%) [1] |
| Blood pressure elevation | 1 (12.5%) [1] | 0 | 0 | 0 | 0 | 0 | 1 (2.5%) [1] |
| Dissociation | 0 | 1 (12.5%) [1] | 3 (37.5%) [3] | 3 (37.5%) [3] | 8 (100.0%) [8] | 0 | 15 (37.5%) [15] |
| Derealization | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 1 (2.5%) [1] |
| Drunkenness | 0 | 1 (12.5%) [1] | 5 (62.5%) [5] | 2 (25.0%) [2] | 1 (12.5%) [1] | 0 | 9 (22.5%) [9] |
| Fatigue | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Feel hot | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 0 | 1 (2.5%) [1] |
| Thirst | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Nausea | 0 | 2 (25.0%) [2] | 1 (12.5%) [1] | 2 (25.0%) [2] | 2 (25.0%) [2] | 0 | 7 (17.5%) [7] |
| Vomiting | 0 | 2 (25.0%) [2] | 0 | 2 (25.0%) [2] | 0 | 0 | 4 (10.0%) [4] |
| Dry mouth | 0 | 0 | 3 (37.5%) [3] | 0 | 0 | 0 | 3 (7.5%) [3] |
| Blurred vision | 0 | 0 | 2 (25.0%) [2] | 0 | 3 (37.5%) [3] | 0 | 5 (12.5%) [5] |
| Sinus bradycardia | 1 (12.5%) [1] | 1 (12.5%) [1] | 2 (25.0%) [2] | 0 | 0 | 0 | 4 (10.0%) [4] |
| Anemia | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (12.5%) [1] | 1 (10.0%) [1] | 2 (5.0%) [2] |
| Urinary tract infections | 1 (12.5%) [1] | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 2 (5.0%) [2] |
| Excessive sweating | 0 | 0 | 0 | 2 (25.0%) [2] | 0 | 0 | 2 (5.0%) [2] |
| Dry nose | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Hematuria | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 0 | 1 (2.5%) [1] |
| ADRs | Dose Cohorts | ||||||
|---|---|---|---|---|---|---|---|
| 10.0 mg (n = 8) | 30.0 mg (n = 8) | 60.0 mg (n = 8) | 120 mg (n = 8) | 180 mg (n = 8) | Placebo (n = 10) | Total (No Placebo) (n = 40) | |
| Dizziness | 0 | 5 (62.5%) [5] | 5 (62.5%) [6] | 5 (62.5%) [5] | 6 (75.0%) [6] | 0 | 21 (52.5%) [22] |
| Somnolence | 0 | 0 | 0 | 0 | 8 (100.0%) [9] | 0 | 8 (20.0%) [9] |
| Decreased sensation | 0 | 1 (12.5%) [1] | 2 (25.0%) [2] | 1 (12.5%) [1] | 0 | 0 | 4 (10.0%) [4] |
| Decreased level of consciousness | 0 | 1 (12.5%) [1] | 0 | 0 | 1 (12.5%) [1] | 1 (10.0%) [1] | 2 (5.0%) [2] |
| Sleepiness | 0 | 1 (12.5%) [1] | 0 | 0 | 1 (12.5%) [1] | 0 | 2 (5.0%) [2] |
| Hypertonia | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Head discomfort | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Headache | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Tremor | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
| Dissociation | 0 | 1 (12.5%) [1] | 3 (37.5%) [3] | 3 (37.5%) [3] | 8 (100.0%) [8] | 0 | 15 (37.5%) [15] |
| Derealization | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 1 (2.5%) [1] |
| Drunkenness | 0 | 1 (12.5%) [1] | 5 (62.5%) [5] | 2 (25.0%) [2] | 1 (12.5%) [1] | 0 | 9 (22.5%) [9] |
| Fatigue | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Feel hot | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 0 | 1 (2.5%) [1] |
| Thirst | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Nausea | 0 | 2 (25.0%) [2] | 1 (12.5%) [1] | 2 (25.0%) [2] | 2 (25.0%) [2] | 0 | 7 (17.5%) [7] |
| Vomiting | 0 | 2 (25.0%) [2] | 0 | 2 (25.0%) [2] | 0 | 0 | 4 (10.0%) [4] |
| Dry mouth | 0 | 0 | 3 (37.5%) [3] | 0 | 0 | 0 | 3 (7.5%) [3] |
| Blood pressure reduction | 1 (12.5%) [2] | 2 (25.0%) [2] | 1 (12.5%) [1] | 0 | 0 | 0 | 4 (10.0%) [5] |
| Serum bilirubin elevation | 0 | 0 | 0 | 0 | 2 (25.0%) [2] | 1 (10.0%) [1] | 2 (5.0%) [2] |
| Decreased heart rate | 0 | 0 | 1 (12.5%) [2] | 0 | 0 | 0 | 1 (2.5%) [2] |
| Blood pressure elevation | 1 (12.5%) [1] | 0 | 0 | 0 | 0 | 0 | 1 (2.5%) [1] |
| Decreased neutrophil count | 0 | 0 | 0 | 0 | 1 (12.5%) [1] | 0 | 1 (2.5%) [1] |
| Blurred vision | 0 | 0 | 2 (25.0%) [2] | 0 | 3 (37.5%) [3] | 0 | 5 (12.5%) [5] |
| sinus bradycardia | 1 (12.5%) [1] | 1 (12.5%) [1] | 1 (12.5%) [1] | 0 | 0 | 0 | 3 (7.5%) [3] |
| Excessive sweating | 0 | 0 | 0 | 2 (25.0%) [2] | 0 | 0 | 2 (5.0%) [2] |
| Dry nose | 0 | 0 | 1 (12.5%) [1] | 0 | 0 | 0 | 1 (2.5%) [1] |
3. Discussion
3.1. Safety and Tolerability
3.2. Dose Range Design
3.2.1. Starting Dose Design
| Species | Sex | NOAEL (mg/kg/day) | HED 1 (mg) | AUC0–t (h·ng/mL) | Predicted Human CL (L/h) | HED 2(mg) | SF | MRSD (mg) |
|---|---|---|---|---|---|---|---|---|
| SD rats | female/male | 15.0 | 144 | NA | NA | NA | 10 | 14.4 |
| Beagle | female/male | 20.0 | 648 | NA | NA | NA | 64.8 | |
| SD rats | female | 15.0 | NA | 2907.931 | 193.4 | 562.39 | 56.24 | |
| male | NA | 2079.141 | 402.11 | 40.21 | ||||
| Beagle | female | 20.0 | NA | 3296.135 | 637.47 | 63.75 | ||
| male | NA | 3954.165 | 764.74 | 76.47 |
3.2.2. Ascending Dose Design
3.3. Pharmacokinetics
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Study Population
4.3. Study Design
4.4. Sample Collection
4.5. Pharmacokinetic Assessments
4.6. Safety and Tolerability Assessments
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, S.C.; Kim, Y.K. Challenges and Strategies for Current Classifications of Depressive Disorders: Proposal for Future Diagnostic Standards. Adv. Exp. Med. Biol. 2021, 1305, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Kverno, K.S.; Mangano, E. Treatment-Resistant Depression: Approaches to Treatment. J. Psychosoc. Nurs. Ment. Health Serv. 2021, 59, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.; Cars, T.; Lööv, S.; Söderling, J.; Sundström, J.; Tiihonen, J.; Leval, A.; Gannedahl, A.; Björkholm, C.; Själin, M.; et al. Association of Treatment-Resistant Depression With Patient Outcomes and Health Care Resource Utilization in a Population-Wide Study. JAMA Psychiatry 2023, 80, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Reutfors, J.; Andersson, T.M.; Brenner, P.; Brandt, L.; DiBernardo, A.; Li, G.; Hägg, D.; Wingård, L.; Bodén, R. Mortality in treatment-resistant unipolar depression: A register-based cohort study in Sweden. J. Affect. Disord. 2018, 238, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Olivier, B.; Olivier, J.D.A. Efficacy, Safety, and Tolerability of Psychedelics in Treatment-Resistant Depression (TRD). Adv. Exp. Med. Biol. 2024, 1456, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Kellner, C.H.; Lisanby, S.H.; Weiner, R.; Prudic, J.; Rudorfer, M.V.; Young, R.C.; Petrides, G.; McCall, W.V.; Husain, M.; Greenberg, R.M.; et al. Speed of response to electroconvulsive therapy compared with ketamine. Psychiatry Res. 2015, 225, 215. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Kazemi, M.H.; Yoosefi, A.; Ghasemi, A.; Paragomi, P.; Amini, H.; Afzali, M.H. Rapid antidepressant effects of repeated doses of ketamine compared with electroconvulsive therapy in hospitalized patients with major depressive disorder. Psychiatry Res. 2014, 215, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Basso, L.; Bönke, L.; Aust, S.; Gärtner, M.; Heuser-Collier, I.; Otte, C.; Wingenfeld, K.; Bajbouj, M.; Grimm, S. Antidepressant and neurocognitive effects of serial ketamine administration versus ECT in depressed patients. J. Psychiatr. Res. 2020, 123, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.; Mikkelsen, S.; Thorkildsen, C.; Borgbjerg, F.M. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur. J. Pharmacol. 1997, 333, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Scotton, E.; Antqueviezc, B.; Vasconcelos, M.F.; Dalpiaz, G.; Paul Géa, L.; Ferraz Goularte, J.; Colombo, R.; Ribeiro Rosa, A. Is (R)-ketamine a potential therapeutic agent for treatment-resistant depression with less detrimental side effects? A review of molecular mechanisms underlying ketamine and its enantiomers. Biochem. Pharmacol. 2022, 198, 114963. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Molecular mechanisms of the rapid-acting and long-lasting antidepressant actions of (R)-ketamine. Biochem. Pharmacol. 2020, 177, 113935. [Google Scholar] [CrossRef] [PubMed]
- Mathisen, L.C.; Skjelbred, P.; Skoglund, L.A.; Øye, I. Effect of ketamine, an NMDA receptor inhibitor, in acute and chronic orofacial pain. Pain 1995, 61, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Leenders, K.L.; Oye, I.; Hell, D.; Angst, J. Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET). Eur. Neuropsychopharmacol. 1997, 7, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kobayashi, S.; Nakao, K.; Dong, C.; Han, M.; Qu, Y.; Ren, Q.; Zhang, J.C.; Ma, M.; Toki, H.; et al. AMPA Receptor Activation-Independent Antidepressant Actions of Ketamine Metabolite (S)-Norketamine. Biol. Psychiatry 2018, 84, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.C.; Bandeira, I.D.; Correia-Melo, F.S.; Telles, M.; Mello, R.P.; Vieira, F.; Lima, C.S.; Jesus-Nunes, A.P.; Guerreiro-Costa, L.N.F.; Marback, R.F.; et al. Intravenous arketamine for treatment-resistant depression: Open-label pilot study. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.C.; Souza-Marques, B.; Mello, R.P.; Bandeira, I.D.; Caliman-Fontes, A.T.; Carneiro, B.A.; Faria-Guimarães, D.; Guerreiro-Costa, L.N.F.; Jesus-Nunes, A.P.; Silva, S.S.; et al. Arketamine as adjunctive therapy for treatment-resistant depression: A placebo-controlled pilot study. J. Affect. Disord. 2023, 330, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Pae, C.U.; Masand, P.S. Key considerations for the use of ketamine and esketamine for the treatment of depression: Focusing on administration, safety, and tolerability. Expert Opin. Drug Saf. 2022, 21, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv. Drug Deliv. Rev. 2007, 59, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.A.; Reddy, M.B.; Heikkinen, A.T.; Lukacova, V.; Parrott, N. Physiologically Based Pharmacokinetic Modelling for First-In-Human Predictions: An Updated Model Building Strategy Illustrated with Challenging Industry Case Studies. Clin. Pharmacokinet. 2019, 58, 727–746. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.; Führer, F.; Menz, S.; Diedam, H.; Göller, A.H.; Schneckener, S. Prediction of human pharmacokinetics from chemical structure: Combining mechanistic modeling with machine learning. J. Pharm. Sci. 2023, 113, 55–63. [Google Scholar] [CrossRef] [PubMed]
- China National Medical Products Administration. Guidance for Estimating the Maximum Recommended Starting Dose (MRSD) of Drugs in the First Clinical Trial of Healthy Adult Volunteers. 2012. Available online: https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20120515120001975.html (accessed on 15 May 2012).
- U.S. Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. Available online: https://www.fda.gov/media/72309/download (accessed on 6 July 2005).
- Hummel, J.; McKendrick, S.; Brindley, C.; French, R. Exploratory assessment of dose proportionality: Review of current approaches and proposal for a practical criterion. Pharm. Stat. 2009, 8, 38–49. [Google Scholar] [CrossRef] [PubMed]
- GuangDong Pharmaceutical Association, China. Guangdong Consensus on Safety Evaluation of Drug Clinical Trials (2020 Edition). 2020. Available online: http://www.sinopharmacy.com.cn/notification/2003.html (accessed on 1 August 2020).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Yang, Y.; Zhou, T.; Zou, B.; Ding, L. Pharmacokinetics, Safety, and Tolerability of (R)-Ketamine Hydrochloride Injection, a Novel Rapid-Acting Antidepressant, in Healthy Chinese Subjects. Pharmaceuticals 2025, 18, 1079. https://doi.org/10.3390/ph18071079
Wang R, Yang Y, Zhou T, Zou B, Ding L. Pharmacokinetics, Safety, and Tolerability of (R)-Ketamine Hydrochloride Injection, a Novel Rapid-Acting Antidepressant, in Healthy Chinese Subjects. Pharmaceuticals. 2025; 18(7):1079. https://doi.org/10.3390/ph18071079
Chicago/Turabian StyleWang, Rui, Yuqian Yang, Tong Zhou, Bingjie Zou, and Li Ding. 2025. "Pharmacokinetics, Safety, and Tolerability of (R)-Ketamine Hydrochloride Injection, a Novel Rapid-Acting Antidepressant, in Healthy Chinese Subjects" Pharmaceuticals 18, no. 7: 1079. https://doi.org/10.3390/ph18071079
APA StyleWang, R., Yang, Y., Zhou, T., Zou, B., & Ding, L. (2025). Pharmacokinetics, Safety, and Tolerability of (R)-Ketamine Hydrochloride Injection, a Novel Rapid-Acting Antidepressant, in Healthy Chinese Subjects. Pharmaceuticals, 18(7), 1079. https://doi.org/10.3390/ph18071079