

PARP Inhibition in Colorectal Cancer—A Comparison of Potential Predictive Biomarkers for Therapy


Abstract
1. Introduction
2. Results
2.1. PARP1 and PARP2 Expression Display Correlations with the Clinico-Molecular and Genomic Features of CRC
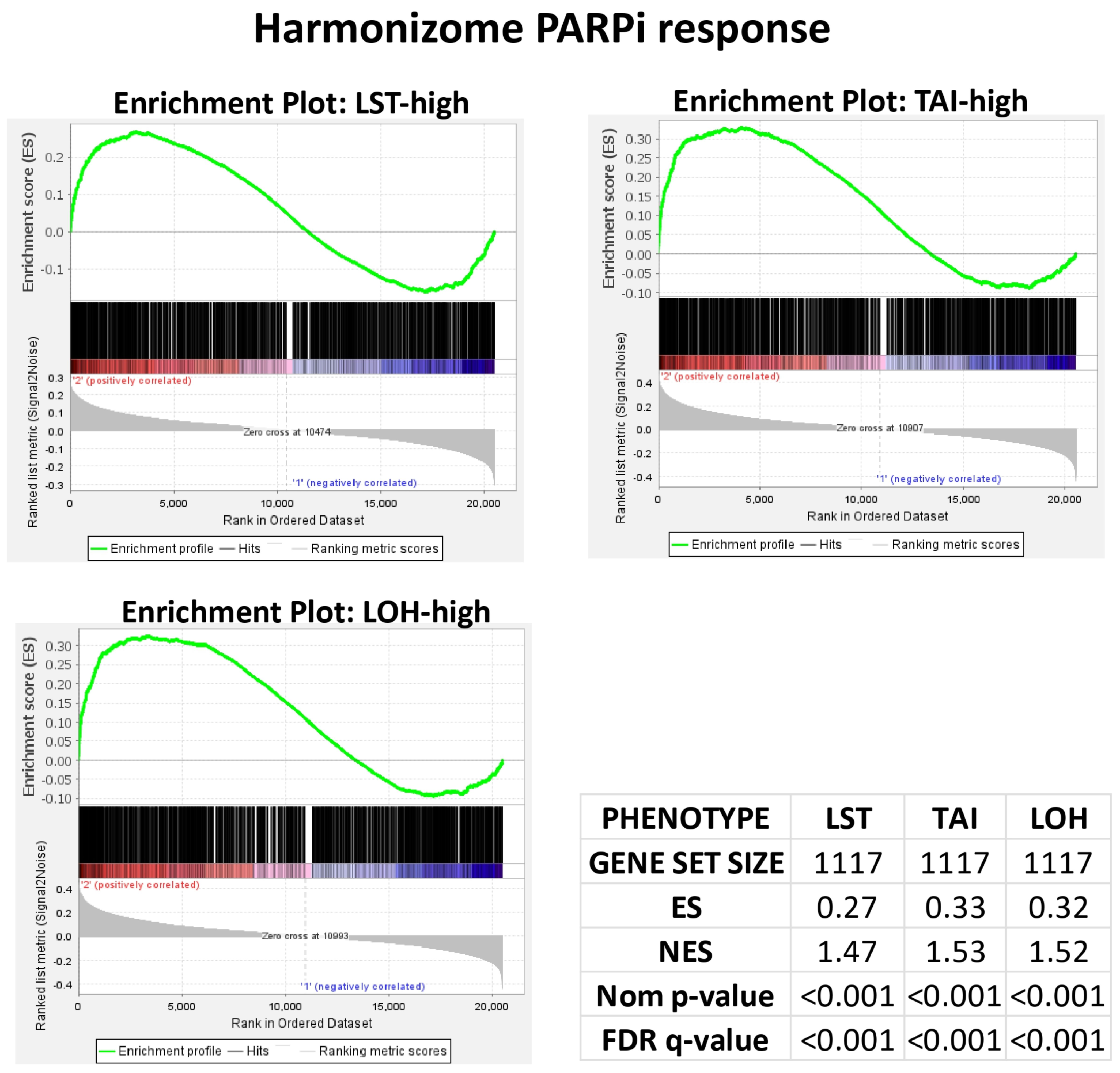
2.2. PARPi Response Gene Set Validation in Using Breast Cancer (BC) Cohort
2.3. PARPi Response Enrichment in CRC Cases with Low Chromosomal Instability (CIN)
2.4. PARP1 Expression, PARP2 Expression and MSI Status Exhibit Non-Inferior Associations with PARPi Response in CRC
2.5. Drug Ontology Enrichment Analysis Confirms the Relative Magnitude of the Relationships Between PARPi Response Biomarkers
3. Discussion
4. Materials and Methods
4.1. Study Approach
4.2. Cancer Cohorts
4.3. Data Retrieval and Processing
4.4. Genomic Indices
4.5. Gene Set Enrichment Analysis
4.6. Differential Enrichment Score (ES) Analysis
4.7. Differential PARPi Response Ontology
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TCGA | The cancer genome atlas |
| Sidra-LUMC | Sidra-Leiden University Medical Center’s Atlas and Compass of Immune–Cancer–Microbiome |
| GDC | Genome Data Commons |
| GSEA | Gene set enrichment analysis |
| DOEA | Drug ontology enrichment analysis |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Pierantoni, C.; Cosentino, L.; Ricciardiello, L. Molecular Pathways of Colorectal Cancer Development: Mechanisms of Action and Evolution of Main Systemic Therapy Compounds. Dig. Dis. 2024, 42, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cotan, H.T.; Emilescu, R.A.; Iaciu, C.I.; Orlov-Slavu, C.M.; Olaru, M.C.; Popa, A.M.; Jinga, M.; Nitipir, C.; Schreiner, O.D.; Ciobanu, R.C. Prognostic and Predictive Determinants of Colorectal Cancer: A Comprehensive Review. Cancers 2024, 16, 3928. [Google Scholar] [CrossRef] [PubMed]
- Koncina, E.; Haan, S.; Rauh, S.; Letellier, E. Prognostic and Predictive Molecular Biomarkers for Colorectal Cancer: Updates and Challenges. Cancers 2020, 12, 319. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, Y.; Chen, S.; Weng, X.; Rao, Y.; Fang, H. Mechanism and Current Progress of Poly ADP-Ribose Polymerase (PARP) Inhibitors in the Treatment of Ovarian Cancer. Biomed. Pharmacother. 2019, 123, 109661. [Google Scholar] [CrossRef]
- Bondar, D.; Karpichev, Y. Poly(ADP-Ribose) Polymerase (PARP) Inhibitors for Cancer Therapy: Advances, Challenges, and Future Directions. Biomolecules 2024, 14, 1269. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.-Y. Recent Advancements in PARP Inhibitors-Based Targeted Cancer Therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef]
- Smeby, J.; Kryeziu, K.; Berg, K.C.G.; Eilertsen, I.A.; Eide, P.W.; Johannessen, B.; Guren, M.G.; Nesbakken, A.; Bruun, J.; Lothe, R.A.; et al. Molecular Correlates of Sensitivity to PARP Inhibition Beyond Homologous Recombination Deficiency in Pre-Clinical Models of Colorectal Cancer Point to Wild-Type TP53 Activity. EBioMedicine 2020, 59, 102923. [Google Scholar] [CrossRef]
- Jarrar, A.; Lotti, F.; DeVecchio, J.; Ferrandon, S.; Gantt, G.; Mace, A.; Karagkounis, G.; Orloff, M.; Venere, M.; Hitomi, M.; et al. Poly(ADP-Ribose) Polymerase Inhibition Sensitizes Colorectal Cancer-Initiating Cells to Chemotherapy. Stem Cells 2019, 37, 42–53. [Google Scholar] [CrossRef]
- Genther Williams, S.M.; Kuznicki, A.M.; Andrade, P.; Dolinski, B.M.; Elbi, C.; O’Hagan, R.C.; Toniatti, C. Treatment with the PARP Inhibitor, Niraparib, Sensitizes Colorectal Cancer Cell Lines to Irinotecan Regardless of MSI/MSS Status. Cancer Cell Int. 2015, 15, 14. [Google Scholar] [CrossRef]
- Incorvaia, L.; Perez, A.; Marchetti, C.; Brando, C.; Gristina, V.; Cancelliere, D.; Pivetti, A.; Contino, S.; Di Giovanni, E.; Barraco, N.; et al. Theranostic Biomarkers and PARP-Inhibitors Effectiveness in Patients with Non-BRCA Associated Homologous Recombination Deficient Tumors: Still Looking Through a Dirty Glass Window? Cancer Treat. Rev. 2023, 121, 102650. [Google Scholar] [CrossRef] [PubMed]
- Phan, Z.; Ford, C.E.; Caldon, C.E. DNA Repair Biomarkers to Guide Usage of Combined PARP Inhibitors and Chemotherapy: A Meta-Analysis and Systematic Review. Pharmacol. Res. 2023, 196, 106927. [Google Scholar] [CrossRef]
- Dibitetto, D.; Widmer, C.A.; Rottenberg, S. PARPi, BRCA, and Gaps: Controversies and Future Research. Trends Cancer 2024, 10, 857–869. [Google Scholar] [CrossRef]
- Kupfer, S.S.; Gupta, S.; Weitzel, J.N.; Samadder, J. AGA Clinical Practice Update on Colorectal and Pancreatic Cancer Risk and Screening in BRCA1 and BRCA2 Carriers: Commentary. Gastroenterology 2020, 159, 760–764. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.L.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals with Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO Recommendations on Predictive Biomarker Testing for Homologous Recombination Deficiency and PARP Inhibitor Benefit in Ovarian Cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Streel, S.; Salmon, A.; Dheur, A.; Bours, V.; Leroi, N.; Habran, L.; Delbecque, K.; Goffin, F.; Pleyers, C.; Kakkos, A.; et al. Diagnostic Performance of Immunohistochemistry Compared to Molecular Techniques for Microsatellite Instability and p53 Mutation Detection in Endometrial Cancer. Int. J. Mol. Sci. 2023, 24, 4866. [Google Scholar] [CrossRef]
- Binch, A.; Snuggs, J.; Le Maitre, C.L. Immunohistochemical Analysis of Protein Expression in Formalin Fixed Paraffin Embedded Human Intervertebral Disc Tissues. JOR Spine 2020, 3, e1098. [Google Scholar] [CrossRef]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing Whole Genomes Using DNA Microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor Mutational Burden Is Predictive of Response to Immune Checkpoint Inhibitors in MSI-High Metastatic Colorectal Cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Chang, M.; Chang, H.M.; Chang, F. Microsatellite Instability: A Predictive Biomarker for Cancer Immunotherapy. Appl. Immunohistochem. Mol. Morphol. 2018, 26, e15–e21. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s Not All One and the Same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Koppensteiner, R.; Samartzis, E.P.; Noske, A.; von Teichman, A.; Dedes, I.; Gwerder, M.; Imesch, P.; Ikenberg, K.; Moch, H.; Fink, D.; et al. Effect of MRE11 Loss on PARP-Inhibitor Sensitivity in Endometrial Cancer In Vitro. PLoS ONE 2014, 9, e100041. [Google Scholar] [CrossRef] [PubMed]
- Miquel, C.; Jacob, S.; Grandjouan, S.; Aimé, A.; Viguier, J.; Sabourin, J.C.; Sarasin, A.; Duval, A.; Praz, F. Frequent Alteration of DNA Damage Signalling and Repair Pathways in Human Colorectal Cancers with Microsatellite Instability. Oncogene 2007, 26, 5919–5926. [Google Scholar] [CrossRef]
- Jank, P.; Leichsenring, J.; Kolb, S.; Hoffmann, I.; Bischoff, P.; Kunze, C.A.; Dragomir, M.P.; Gleitsmann, M.; Jesinghaus, M.; Schmitt, W.D.; et al. High EVI1 and PARP1 Expression as Favourable Prognostic Markers in High-Grade Serous Ovarian Carcinoma. J. Ovarian Res. 2023, 16, 150. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef]
- Dehdashti, F.; Reimers, M.A.; Shoghi, K.I.; Chen, D.L.; Luo, J.; Rogers, B.; Pachynski, R.K.; Sreekumar, S.; Weimholt, C.; Zhou, D. Pilot Study: PARP1 Imaging in Advanced Prostate Cancer. Mol. Imaging Biol. 2022, 24, 853–861. [Google Scholar] [CrossRef]
- O’Connor, K.W.; Dejsuphong, D.; Park, E.; Nicolae, C.M.; Kimmelman, A.C.; D’Andrea, A.D.; Moldovan, G.L. PARI Overexpression Promotes Genomic Instability and Pancreatic Tumorigenesis. Cancer Res. 2013, 73, 2529–2539. [Google Scholar] [CrossRef]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef] [PubMed]
- Schonhoft, J.D.; Zhao, J.L.; Jendrisak, A.; Carbone, E.A.; Barnett, E.S.; Hullings, M.A.; Gill, A.; Sutton, R.; Lee, J.; Dago, A.E.; et al. Morphology-Predicted Large-Scale Transition Number in Circulating Tumor Cells Identifies a Chromosomal Instability Biomarker Associated with Poor Outcome in Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 4892–4903. [Google Scholar] [CrossRef] [PubMed]
- Manié, E.; Popova, T.; Battistella, A.; Tarabeux, J.; Caux-Moncoutier, V.; Golmard, L.; Smith, N.K.; Mueller, C.R.; Mariani, O.; Sigal-Zafrani, B.; et al. Genomic Hallmarks of Homologous Recombination Deficiency in Invasive Breast Carcinomas. Int. J. Cancer 2016, 138, 891–900. [Google Scholar] [CrossRef]
- Wagener-Ryczek, S.; Merkelbach-Bruse, S.; Siemanowski, J. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Pers. Med. 2021, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Mitchell, D.C.; Kuljanin, M.; Li, J.; Van Vranken, J.G.; Bulloch, N.; Schweppe, D.K.; Huttlin, E.L.; Gygi, S.P. A Proteome-Wide Atlas of Drug Mechanism of Action. Nat. Biotechnol. 2023, 41, 845–857. [Google Scholar] [CrossRef]
- Qi, X.; Zhao, L.; Tian, C.; Li, Y.; Chen, Z.L.; Huo, P.; Chen, R.; Liu, X.; Wan, B.; Yang, S.; et al. Predicting Transcriptional Responses to Novel Chemical Perturbations Using Deep Generative Model for Drug Discovery. Nat. Commun. 2024, 15, 9256. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Garana, B.B.; Joly, J.H.; Delfarah, A.; Hong, H.; Graham, N.A. Drug Mechanism Enrichment Analysis Improves Prioritization of Therapeutics for Repurposing. BMC Bioinf. 2023, 24, 215. [Google Scholar] [CrossRef]
- Thomas, P.D.; Hill, D.P.; Mi, H.; Osumi-Sutherland, D.; Van Auken, K.; Carbon, S.; Balhoff, J.P.; Albou, L.P.; Good, B.; Gaudet, P.; et al. Gene Ontology Causal Activity Modeling (GO-CAM) Moves Beyond GO Annotations to Structured Descriptions of Biological Functions and Systems. Nat. Genet. 2019, 51, 1429–1433. [Google Scholar] [CrossRef]
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology Knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [PubMed]
- Roelands, J.; Kuppen, P.J.K.; Ahmed, E.I.; Mall, R.; Masoodi, T.; Singh, P.; Monaco, G.; Raynaud, C.; de Miranda, N.F.C.C.; Ferraro, L.; et al. An Integrated Tumor, Immune and Microbiome Atlas of Colon Cancer. Nat. Med. 2023, 29, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Templeton, G.F. A Two-Step Approach for Transforming Continuous Variables to Normal: Implications and Recommendations for IS Research. Commun. Assoc. Inf. Syst. 2011, 28, 4. [Google Scholar] [CrossRef]
- Tsang, E.S.; Csizmok, V.; Williamson, L.M.; Pleasance, E.; Topham, J.T.; Karasinska, J.M.; Titmuss, E.; Schrader, I.; Yip, S.; Tessier-Cloutier, B.; et al. Homologous Recombination Deficiency Signatures in Gastrointestinal and Thoracic Cancers Correlate with Platinum Therapy Duration. npj Precis. Oncol. 2023, 7, 31. [Google Scholar] [CrossRef]
- Ebili, H.O.; Omenai, S.; Ezenkwa, U.S. Insights into the Molecular and Clinical Significances of NEIL2 Expression in Colorectal Cancer. NJGH 2025, accepted. [Google Scholar]
- Mouillet-Richard, S.; Cazelles, A.; Sroussi, M.; Gallois, C.; Taieb, J.; Laurent-Puig, P. Clinical Challenges of Consensus Molecular Subtype CMS4 Colon Cancer in the Era of Precision Medicine. Clin. Cancer Res. 2024, 30, 2351–2358. [Google Scholar] [CrossRef]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal Cancer Intrinsic Subtypes Predict Chemotherapy Benefit, Deficient Mismatch Repair and Epithelial-to-Mesenchymal Transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef]
- Diamant, I.; Clarke, D.J.B.; Evangelista, J.E.; Lingam, N.; Ma’ayan, A. Harmonizome 3.0: Integrated Knowledge about Genes and Proteins from Diverse Multi-Omics Resources. Nucleic Acids Res. 2025, 53, D1016–D1028. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- VassarStats: Website for Statistical Computation. Available online: http://vassarstats.net/rdiff.html (accessed on 2 May 2025).
- Free Statistics Calculator Version 4.0. Available online: https://www.danielsoper.com/statcalc/calculator.aspx?id=104 (accessed on 2 May 2025).
- Roder, J.; Linstid, B.; Oliveira, C. Improving the Power of Gene Set Enrichment Analyses. BMC Bioinf. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Welz, T.; Doebler, P.; Pauly, M. Fisher Transformation Based Confidence Intervals of Correlations in Fixed- and Random-Effects Meta-Analysis. Br. J. Math. Stat. Psychol. 2022, 75, 1–22. [Google Scholar] [CrossRef]
- Bishara, A.J.; Hittner, J.B. Confidence Intervals for Correlations When Data Are Not Normal. Behav. Res. Methods 2017, 49, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarkers | Gene Set Size | ES | NES | Nominal p Value | FDR q Value |
|---|---|---|---|---|---|
| LST | 849 | 0.591 | 2.908 | <0.001 | <0.001 |
| MSI status | 849 | 0.550 | 2.582 | <0.001 | <0.001 |
| TP53 mutation status | 849 | 0.344 | 1.781 | <0.001 | <0.001 |
| PARP1 Expression | 849 | 0.665 | 2.438 | <0.001 | <0.001 |
| PARP2 Expression | 849 | 0.653 | 2.334 | <0.001 | <0.001 |
| TP53 Expression | 849 | −0.262 | −1.012 | 0.423 | 0.423 |
| ATM Expression | 849 | 0.172 | 0.512 | 0.982 | 0.982 |
| FGA | 849 | 0.545 | 2.683 | <0.001 | <0.001 |
| Aneuploidy | 849 | 0.598 | 2.731 | <0.001 | <0.001 |
| Biomarkers | Gene Set Size | ES | NES | Nominal p Value | FDR q Value |
|---|---|---|---|---|---|
| LST | 667 | 0.419 | 1.595 | <0.001 | <0.001 |
| MSI status | 667 | 0.536 | 1.975 | <0.001 | <0.001 |
| TP53 mutation status | 667 | 0.390 | 1.851 | <0.001 | <0.001 |
| PARP1 Expression | 667 | 0.407 | 2.182 | <0.001 | <0.001 |
| PARP2 Expression | 667 | 0.355 | 1.758 | <0.001 | <0.001 |
| TP53 Expression | 667 | 0.384 | 1.541 | 0.053 | 0.053 |
| ATM Expression | 667 | 0.262 | 1.052 | 0.406 | 0.406 |
| FGA | 667 | 0.490 | 1.812 | <0.001 | <0.001 |
| Aneuploidy | 667 | 0.494 | 1.851 | <0.001 | <0.001 |
| Biomarker Pairs | N (LST vs. Other) | LST ES | Other ES | ∆ ES z-Score | Adjusted p Value |
|---|---|---|---|---|---|
| LST vs. MSI | 527 vs. 397 | 0.591 | 0.550 | 0.905 | 0.439 |
| LST vs. TP53 mutation status | 527 vs. 491 | 0.591 | 0.343 | 5.103 | 1.98 × 10−6 |
| LST vs. PARP2 expression | 527 vs. 537 | 0.591 | 0.653 | −1.646 | 0.199 |
| LST vs. PARP1 expression | 527 vs. 537 | 0.591 | 0.665 | −1.985 | 0.141 |
| LST vs. FGA | 527 vs. 522 | 0.591 | 0.545 | 1.097 | 0.409 |
| LST vs. Aneuploidy | 527 vs. 524 | 0.591 | 0.598 | −0.161 | 0.872 |
| Biomarker Pairs | N (LST vs. Other) | LST ES | Other ES | ∆ ES z-Score | Adjusted p Value |
|---|---|---|---|---|---|
| LST vs. MSI | 281 vs. 348 | 0.419 | 0.536 | −1.887 | 0.354 |
| LST vs. TP53 mutation status | 281 vs. 281 | 0.419 | 0.390 | 0.297 | 0.858 |
| LST vs. PARP2 expression | 281 vs. 348 | 0.419 | 0.355 | 0.935 | 0.525 |
| LST vs. PARP1 expression | 281 vs. 348 | 0.419 | 0.407 | 0.180 | 0.858 |
| LST vs. FGA | 281 vs. 280 | 0.419 | 0.490 | −1.055 | 0.525 |
| LST vs. Aneuploidy | 281 vs. 281 | 0.419 | 0.494 | −1.118 | 0.525 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfahed, A. PARP Inhibition in Colorectal Cancer—A Comparison of Potential Predictive Biomarkers for Therapy. Pharmaceuticals 2025, 18, 905. https://doi.org/10.3390/ph18060905
Alfahed A. PARP Inhibition in Colorectal Cancer—A Comparison of Potential Predictive Biomarkers for Therapy. Pharmaceuticals. 2025; 18(6):905. https://doi.org/10.3390/ph18060905
Chicago/Turabian StyleAlfahed, Abdulaziz. 2025. "PARP Inhibition in Colorectal Cancer—A Comparison of Potential Predictive Biomarkers for Therapy" Pharmaceuticals 18, no. 6: 905. https://doi.org/10.3390/ph18060905
APA StyleAlfahed, A. (2025). PARP Inhibition in Colorectal Cancer—A Comparison of Potential Predictive Biomarkers for Therapy. Pharmaceuticals, 18(6), 905. https://doi.org/10.3390/ph18060905