

Investigation of the ABCB1 Gene Polymorphism and Food Effects on the Avatrombopag Pharmacokinetics in Chinese Individuals: A Population Pharmacokinetic/Pharmacodynamic Analysis

 ,
,  , and
, and
Abstract

1. Introduction
2. Results
2.1. Demographic Characteristics
2.2. PK Assessment
2.2.1. Food and Sex Effects on PK
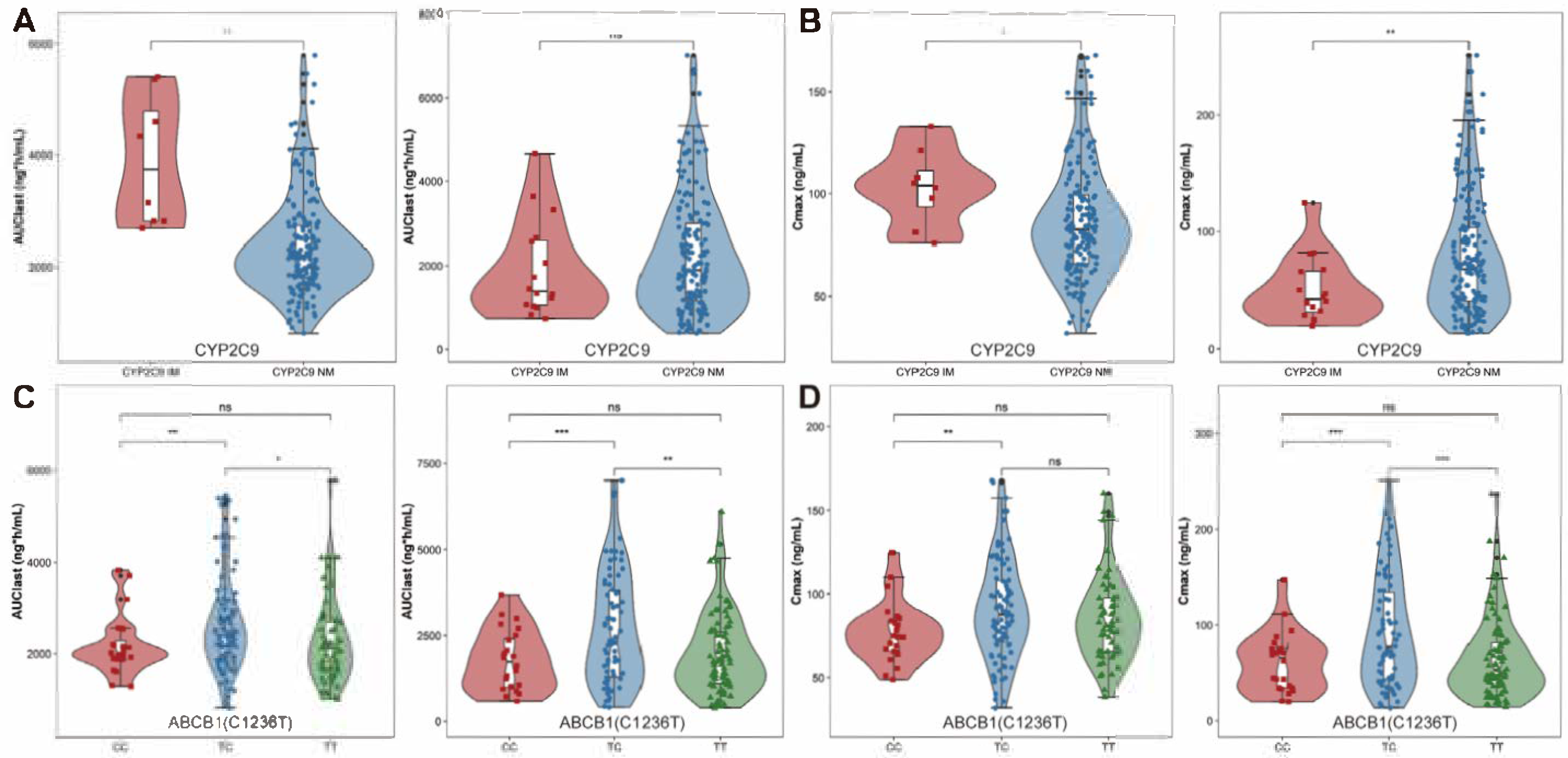
2.2.2. Genotypes Effects on PK
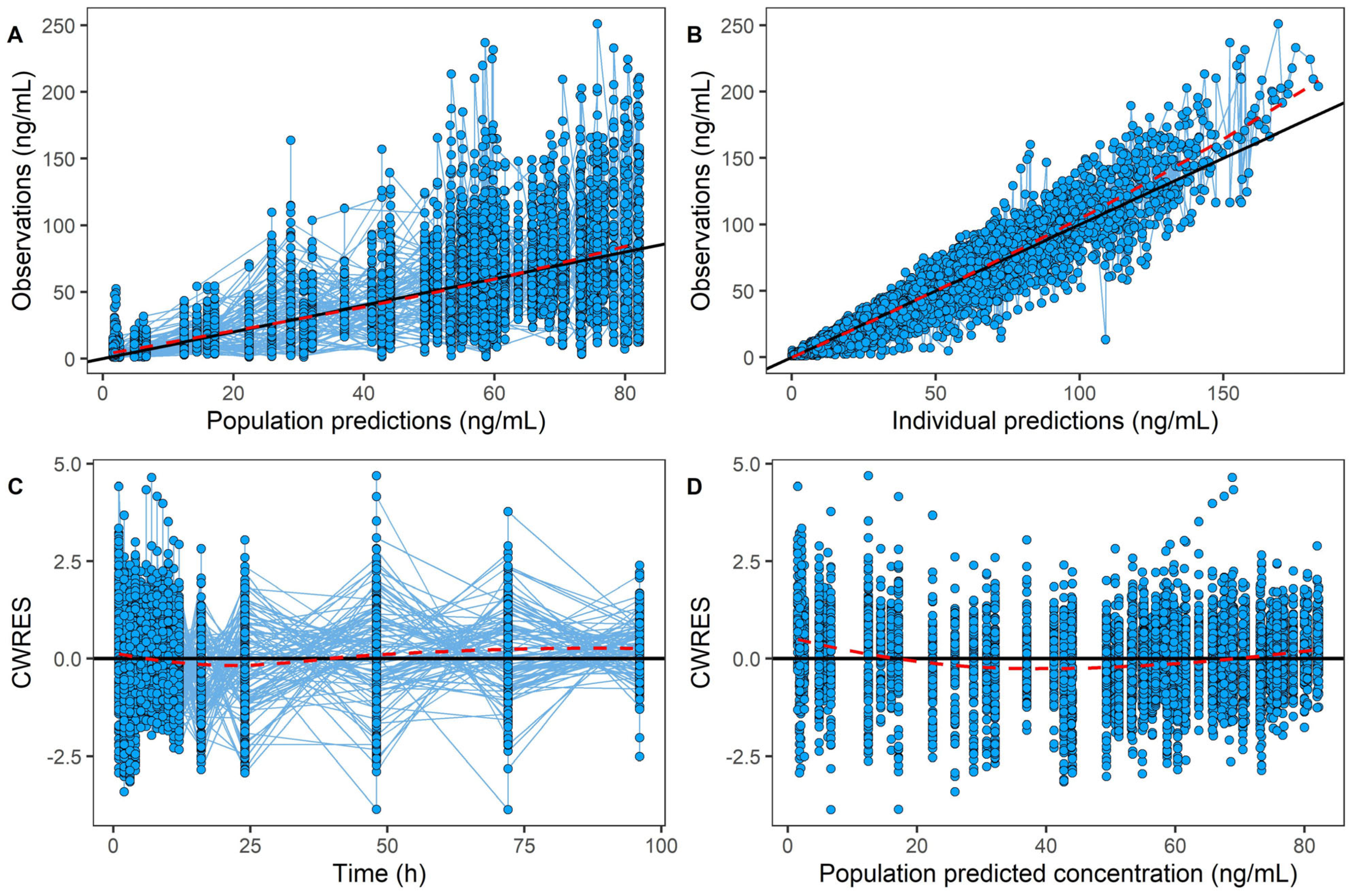
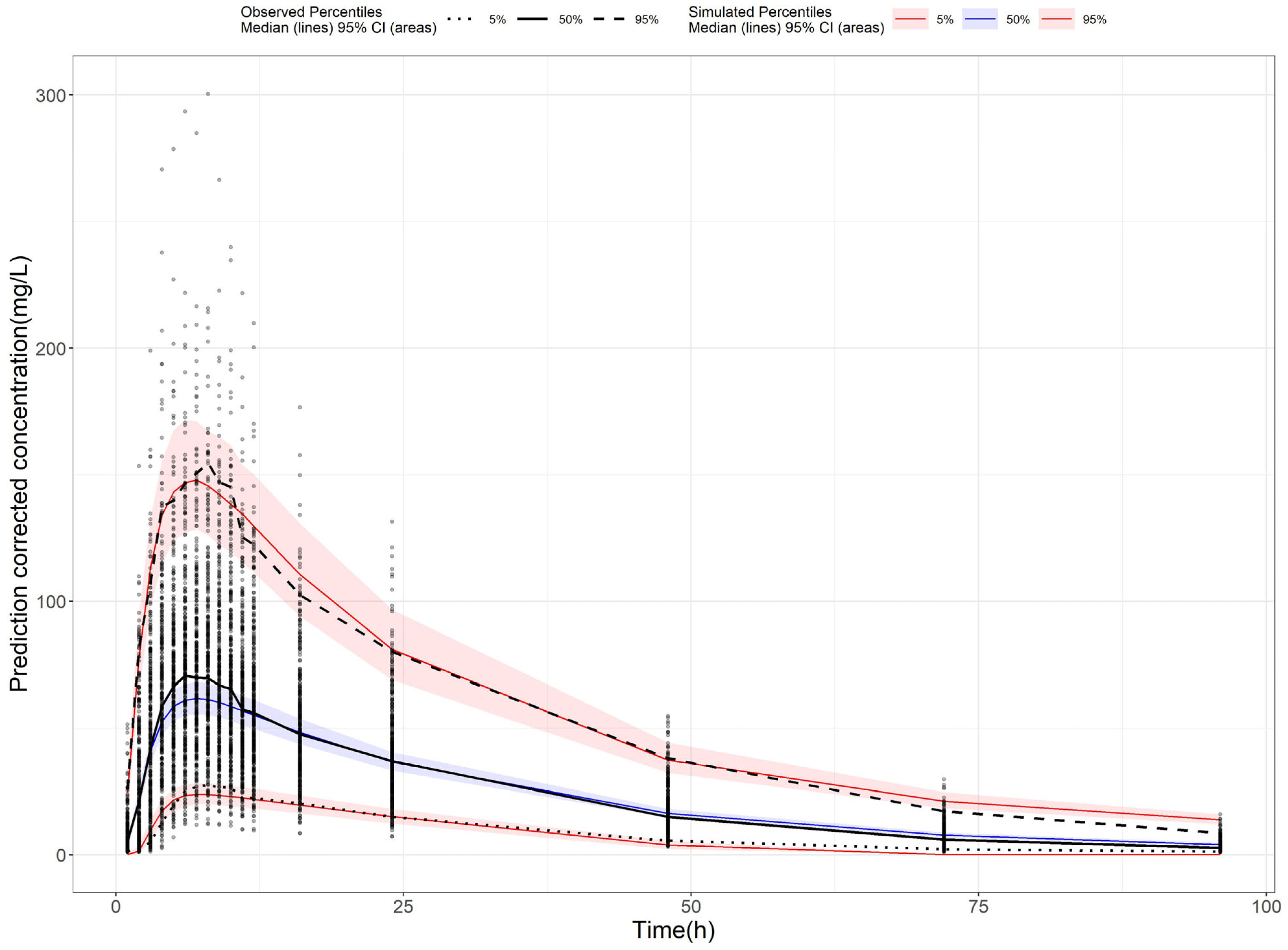
2.3. Population Pharmacokinetic Model of AVA
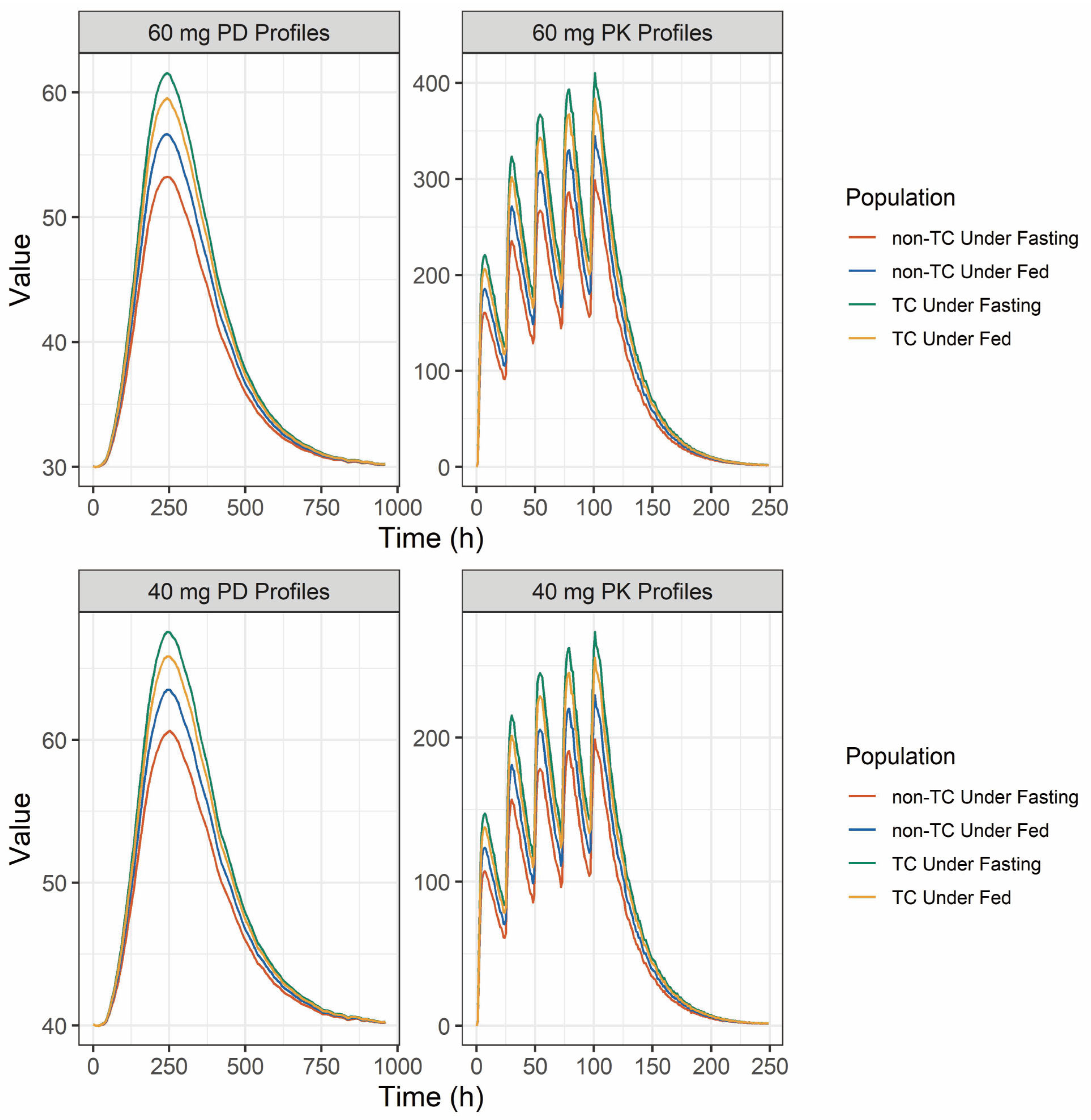
2.4. Population Pharmacokinetic and Pharmacodynamic Simulation
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Study Design and Pharmacokinetic Assessments
4.3. Genotyping of CYP2C9 and ABCB1
4.4. Population Pharmacokinetic Model
4.5. Simulations of PPK-PD
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nomoto, M.; Pastino, G.; Rege, B.; Aluri, J.; Ferry, J.; Han, D. Pharmacokinetics, Pharmacodynamics, Pharmacogenomics, Safety, and Tolerability of Avatrombopag in Healthy Japanese and White Subjects. Clin. Pharmacol. Drug Dev. 2018, 7, 188–195. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Kaplan, Z.S. Platelets Are Not Just for Clots. Transfus. Med. Rev. 2015, 29, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Fustolo-Gunnink, S.F.; Roehr, C.C.; Lieberman, L.; Christensen, R.D.; Van Der Bom, J.G.; Dame, C.; Del Vecchio, A.; Keir, A.K.; Curley, A.; Stanworth, S.J.; et al. Platelet and Red Cell Transfusions for Neonates: Lifesavers or Trojan Horses? Expert. Rev. Hematol. 2019, 12, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, I.S.; Hafer, M.; Sangkhae, V.; Tucker, J.A. The Thrombopoietin Receptor: Revisiting the Master Regulator of Platelet Production. Platelets 2021, 32, 770–778. [Google Scholar] [CrossRef]
- Erickson-Miller, C.L.; Delorme, E.; Tian, S.-S.; Hopson, C.B.; Landis, A.J.; Valoret, E.I.; Sellers, T.S.; Rosen, J.; Miller, S.G.; Luengo, J.I.; et al. Preclinical Activity of Eltrombopag (SB-497115), an Oral, Nonpeptide Thrombopoietin Receptor Agonist. Stem Cells 2009, 27, 424–430. [Google Scholar] [CrossRef]
- Broudy, V.C.; Lin, N.L. AMG531 Stimulates Megakaryopoiesis in Vitro by Binding to Mpl. Cytokine 2004, 25, 52–60. [Google Scholar] [CrossRef]
- DOPTELET® (Avatrombopag) Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210238s009lbl.pdf (accessed on 26 May 2025).
- Deng, J.; Hu, H.; Huang, F.; Huang, C.; Huang, Q.; Wang, L.; Wu, A.; Yang, J.; Qin, D.; Zou, W.; et al. Comparative Efficacy and Safety of Thrombopoietin Receptor Agonists in Adults With Thrombocytopenia: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trial. Front. Pharmacol. 2021, 12, 704093. [Google Scholar] [CrossRef]
- Nomoto, M.; Zamora, C.A.; Schuck, E.; Boyd, P.; Chang, M.-K.; Aluri, J.; Siu, Y.A.; Lai, W.G.; Yasuda, S.; Ferry, J.; et al. Pharmacokinetic/Pharmacodynamic Drug-Drug Interactions of Avatrombopag When Coadministered with Dual or Selective CYP2C9 and CYP3A Interacting Drugs. Br. J. Clin. Pharmacol. 2018, 84, 952–960. [Google Scholar] [CrossRef]
- Nomoto, M.; Ferry, J.; Hussein, Z. Population Pharmacokinetic/Pharmacodynamic Analyses of Avatrombopag in Patients With Chronic Liver Disease and Optimal Dose Adjustment Guide With Concomitantly Administered CYP3A and CYP2C9 Inhibitors. J. Clin. Pharmacol. 2018, 58, 1629–1638. [Google Scholar] [CrossRef]
- Kuter, D.J.; Allen, L.F. Avatrombopag, an Oral Thrombopoietin Receptor Agonist: Results of Two Double-Blind, Dose-Rising, Placebo-Controlled Phase 1 Studies. Br. J. Haematol. 2018, 183, 466–478. [Google Scholar] [CrossRef]
- Jurczak, W.; Chojnowski, K.; Mayer, J.; Krawczyk, K.; Jamieson, B.D.; Tian, W.; Allen, L.F. Phase 3 Randomised Study of Avatrombopag, a Novel Thrombopoietin Receptor Agonist for the Treatment of Chronic Immune Thrombocytopenia. Br. J. Haematol. 2018, 183, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Uttamsingh, V.; Lu, C.; Miwa, G.; Gan, L.-S. Relative Contributions of the Five Major Human Cytochromes P450, 1A2, 2C9, 2C19, 2D6, and 3A4, to the Hepatic Metabolism of the Proteasome Inhibitor Bortezomib. Drug Metab. Dispos. 2005, 33, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Lyseng-Williamson, K.A. Avatrombopag in the Treatment of Thrombocytopenia in Adults with Chronic Liver Disease Scheduled to Undergo a Procedure: A Profile of Its Use in the USA. Drugs Ther. Perspect. 2019, 35, 1–6. [Google Scholar] [CrossRef]
- Skinner, K.T.; Palkar, A.M.; Hong, A.L. Genetics of ABCB1 in Cancer. Cancers 2023, 15, 4236. [Google Scholar] [CrossRef] [PubMed]
- Wolking, S.; Schaeffeler, E.; Lerche, H.; Schwab, M.; Nies, A.T. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin. Pharmacokinet. 2015, 54, 709–735. [Google Scholar] [CrossRef]
- Clemons Bankston, P.; Al-Horani, R.A. New Small Molecule Drugs for Thrombocytopenia: Chemical, Pharmacological, and Therapeutic Use Considerations. Int. J. Mol. Sci. 2019, 20, 3013. [Google Scholar] [CrossRef]
- Zuo, W.; Liu, B.; Chen, M.; Zhang, B.; Han, B. Relationship between CYP2C8, UGT1A1, and ABCG2 Gene Polymorphisms and the Exposure, Efficacy, and Toxicity of Eltrombopag in the Treatment of Refractory Aplastic Anemia. Eur. J. Clin. Pharmacol. 2022, 78, 1657–1666. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Hoffmeister, K.M.; Hu, Z.; Mager, D.E.; Ait-Oudhia, S.; Debrincat, M.A.; Pleines, I.; Josefsson, E.C.; Kile, B.T.; Italiano, J.; et al. Expansion of the Neonatal Platelet Mass Is Achieved via an Extension of Platelet Lifespan. Blood 2014, 123, 3381–3389. [Google Scholar] [CrossRef]
- Markham, A. Avatrombopag: A Review in Thrombocytopenia. Drugs 2021, 81, 1905–1913. [Google Scholar] [CrossRef]
- Kuter, D.J. The Structure, Function, and Clinical Use of the Thrombopoietin Receptor Agonist Avatrombopag. Blood Rev. 2022, 53, 100909. [Google Scholar] [CrossRef]
- Kirchheiner, J.; Brockmöller, J. Clinical Consequences of Cytochrome P450 2C9 Polymorphisms. Clin. Pharmacol. Ther. 2005, 77, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kurata, Y.; Ieiri, I.; Kimura, M.; Morita, T.; Irie, S.; Urae, A.; Ohdo, S.; Ohtani, H.; Sawada, Y.; Higuchi, S.; et al. Role of Human MDR1 Gene Polymorphism in Bioavailability and Interaction of Digoxin, a Substrate of P-Glycoprotein. Clin. Pharmacol. Ther. 2002, 72, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Lown, K.S.; Mayo, R.R.; Leichtman, A.B.; Hsiao, H.L.; Turgeon, D.K.; Schmiedlin-Ren, P.; Brown, M.B.; Guo, W.; Rossi, S.J.; Benet, L.Z.; et al. Role of Intestinal P-Glycoprotein (Mdr1) in Interpatient Variation in the Oral Bioavailability of Cyclosporine. Clin. Pharmacol. Ther. 1997, 62, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I.; Takane, H.; Otsubo, K. The MDR1 (ABCB1) Gene Polymorphism and Its Clinical Implications. Clin. Pharmacokinet. 2004, 43, 553–576. [Google Scholar] [CrossRef]
- Fellay, J.; Marzolini, C.; Meaden, E.R.; Back, D.J.; Buclin, T.; Chave, J.P.; Decosterd, L.A.; Furrer, H.; Opravil, M.; Pantaleo, G.; et al. Response to Antiretroviral Treatment in HIV-1-Infected Individuals with Allelic Variants of the Multidrug Resistance Transporter 1: A Pharmacogenetics Study. Lancet 2002, 359, 30–36. [Google Scholar] [CrossRef]
- Barusrux, S.; Koomdee, N.; Sukprasong, R.; Manosuthi, W.; Chamnanphon, M.; Puangpetch, A.; Sukasem, C.; Nissapatorn, V.; Pereira, M. Effect of ABCB1 3435C>T Transporter Gene Polymorphism on Plasma Efavirenz Concentration in HIV-1 Infected Thai Adults. Asian Pac. J. Trop. Med. 2020, 13, 266–271. [Google Scholar] [CrossRef]
- Balram, C.; Sharma, A.; Sivathasan, C.; Lee, E.J.D. Frequency of C3435T Single Nucleotide MDR1 Genetic Polymorphism in an Asian Population: Phenotypic-Genotypic Correlates. Br. J. Clin. Pharmacol. 2003, 56, 78–83. [Google Scholar] [CrossRef]
- Sai, K.; Kaniwa, N.; Itoda, M.; Saito, Y.; Hasegawa, R.; Komamura, K.; Ueno, K.; Kamakura, S.; Kitakaze, M.; Shirao, K.; et al. Haplotype Analysis of ABCB1/MDR1 Blocks in a Japanese Population Reveals Genotype-Dependent Renal Clearance of Irinotecan. Pharmacogenetics 2003, 13, 741–757. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakaeda, T.; Horinouchi, M.; Tamura, T.; Aoyama, N.; Shirakawa, T.; Matsuo, M.; Kasuga, M.; Okumura, K. Effect of the Mutation (C3435T) at Exon 26 of the MDR1 Gene on Expression Level of MDR1 Messenger Ribonucleic Acid in Duodenal Enterocytes of Healthy Japanese Subjects. Clin. Pharmacol. Ther. 2002, 71, 297–303. [Google Scholar] [CrossRef]
- Prasad, B.; Evers, R.; Gupta, A.; Hop, C.E.C.A.; Salphati, L.; Shukla, S.; Ambudkar, S.V.; Unadkat, J.D. Interindividual Variability in Hepatic Organic Anion-Transporting Polypeptides and P-Glycoprotein (ABCB1) Protein Expression: Quantification by Liquid Chromatography Tandem Mass Spectroscopy and Influence of Genotype, Age, and Sex. Drug Metab. Dispos. 2014, 42, 78–88. [Google Scholar] [CrossRef]
- Siegmund, W.; Ludwig, K.; Giessmann, T.; Dazert, P.; Schroeder, E.; Sperker, B.; Warzok, R.; Kroemer, H.K.; Cascorbi, I. The Effects of the Human MDR1 Genotype on the Expression of Duodenal P-Glycoprotein and Disposition of the Probe Drug Talinolol. Clin. Pharmacol. Ther. 2002, 72, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Owen, A.; Goldring, C.; Morgan, P.; Chadwick, D.; Park, B.K.; Pirmohamed, M. Relationship between the C3435T and G2677T(A) Polymorphisms in the ABCB1 Gene and P-Glycoprotein Expression in Human Liver. Br. J. Clin. Pharmacol. 2005, 59, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional Polymorphisms of the Human Multidrug-Resistance Gene: Multiple Sequence Variations and Correlation of One Allele with P-Glycoprotein Expression and Activity in Vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-Y.; Lim, H.-S.; Yoo, Y.-K.; Shin, E.S.; Park, Y.H.; Lee, S.Y.; Lee, J.-E.; Lee, D.H.; Kim, H.T.; Lee, J.S. Associations of ABCB1, ABCC2, and ABCG2 Polymorphisms with Irinotecan-Pharmacokinetics and Clinical Outcome in Patients with Advanced Non-Small Cell Lung Cancer. Cancer 2007, 110, 138–147. [Google Scholar] [CrossRef]
- Puranik, Y.G.; Birnbaum, A.K.; Marino, S.E.; Ahmed, G.; Cloyd, J.C.; Remmel, R.P.; Leppik, I.E.; Lamba, J.K. Association of Carbamazepine Major Metabolism and Transport Pathway Gene Polymorphisms and Pharmacokinetics in Patients with Epilepsy. Pharmacogenomics 2013, 14, 35–45. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Fasting (n = 47) | Fed (n = 45) |
|---|---|---|
| Mean ± SD | Mean ± SD | |
| Age (years) | 28.98 ± 6.27 | 28.48 ± 7.55 |
| Body weight (kg) | 63.22 ± 7.88 | 60.95 ± 6.32 |
| Height (cm) | 167.62 ± 6.82 | 167.16 ± 5.67 |
| Sex (Male/Female) | 41/6 | 42/3 |
| Alanine Aminotransferase (U/L) | 23.04 ± 12.35 | 19.46 ± 12.31 |
| Aspartate Aminotransferase (U/L) | 21.2 ± 4.71 | 18.97 ± 4.5 |
| Albumin (g/L) | 47.06 ± 2.25 | 47.19 ± 2.07 |
| Total Protein (g/L) | 74.6 ± 4.18 | 74.26 ± 3.54 |
| Total Bilirubin (µmol/L) | 14.76 ± 4.42 | 14.38 ± 6.92 |
| Direct Bilirubin (µmol/L) | 3.89 ± 1.27 | 3.63 ± 1.77 |
| Prothrombin Time (s) | 13.65 ± 0.73 | 12.4 ± 0.74 |
| Activated Partial Thromboplastin Time (s) | 30.36 ± 3.35 | 35.8 ± 3.39 |
| International Normalized Ratio | 1.12 ± 0.07 | 1.03 ± 0.06 |
| Fasting Blood Glucose (mmol/L) | 2.7 ± 0.49 | 2.41 ± 0.34 |
| Creatinine (µmol/L) | 76.26 ± 11.03 | 76.79 ± 10.56 |
| Blood Urea Nitrogen (mmol/L) | 4.06 ± 0.69 | 4.33 ± 0.83 |
| Uric Acid (µmol/L) | 387.44 ± 79.85 | 360.76 ± 76.24 |
| CYP2C9 (NM/IM) | 43/4 | 43/2 |
| ABCB1 (C1236T) (CC/TC/TT) | 7/28/12 | 6/19/20 |
| ABCB1 (C3435T) (CC/TC/TT) | 22/19/6 | 15/24/6 |
| ABCB1 (G2677T/A) (GA/GG/TA/TG/TT) | 3/9/4/23/8 | 10/9/5/17/4 |
| PK Parameters | Fasting | Fed | ||||
|---|---|---|---|---|---|---|
| ALL (N = 47) | Male (N = 41) | Female (N = 6) | ALL (N = 45) | Male (N = 42) | Female (N = 3) | |
| t1/2 (h) | 18.92 ± 3.56 | 19.27 ± 3.60 | 16.55 ± 2.09 | 18.18 ± 2.91 | 18.14 ± 3.02 | 19.51 ± 1.69 |
| tmax (h) | 7.02(3.00~16.00) | 7.02(3.00~10.02) | 6.51(4.00~16.00) | 6.00(2.00~12.00) | 5.00(2.00~12.00) | 5.01(4.00~10.02) |
| Cmax (ng/mL) | 80.44 ± 52.70 | 77.22 ± 49.15 | 101.90 ± 71.02 | 85.06 ± 27.99 | 86.00 ± 28.51 | 73.92 ± 25.77 |
| AUC0-t (h×ng/mL) | 2335.67 ± 1456.90 | 2302.61 ± 1430.75 | 2556.05 ± 1672.36 | 2389.39 ± 933.23 | 2415.02 ± 970.48 | 2018.66 ± 615.38 |
| AUC0-∞ (h×ng/mL) | 2428.42 ± 1502.16 | 2399.72 ± 1479.64 | 2619.79 ± 1702.32 | 2478.66 ± 1014.60 | 2506.96 ± 1058.14 | 2095.01 ± 625.05 |
| Parameters | Final Model | Bootstrap | |
|---|---|---|---|
| Estimates | RSE% | Median [2.5th–97.5th] | |
| CL/F (L/h) | 7.85 | 14% | 7.81 [5.92–10.62] |
| Vd/F (L) | 199 | 13% | 198.01 [149.77–267.64] |
| ka (/h) | 0.582 | 13% | 0.581 [0.4723–0.785] |
| MTT (h) | 1.33 | 5% | 1.33 [1.22–1.45] |
| COV1 * | −0.0653 | 111% | −0.0702 [−0.3069–0.2698] |
| COV2 * | −0.272 | 15% | −0.28 [−0.47–0.01] |
| COV3 * | 0.177 | 56% | 0.186 [−0.216–0.431] |
| Between-subject variability | |||
| CL/F | 0.182 | 35% | 0.174 [0.126–0.235] |
| Vd/F | 0.159 | 32% | 0.153 [0.106–0.204] |
| KA | 0.504 | 35% | 0.48 [0.29–0.78] |
| MTT | 0.131 | 23% | 0.128 [0.093–0.169] |
| OCC [KA] | 0.913 | 12% | 0.922 [0.681–1.346] |
| Residual unexplained variability | |||
| σprop (%) | 23.90% | 4% | 23.79% [22.28–25.74%] |
| σadd (ng/mL) | 1.18 | 3.00% | 1.17 [0.86–1.48] |
| Dosage Regimen | Group | PK | PD | ||
|---|---|---|---|---|---|
| Cmax (ng/mL) | AUC (ng × h/mL) | Cmax (×109) | AUC (×109) | ||
| 60 mg | Non-TC under Fasting State | 160.92 | 5480.21 | 53.23 | 35,907.80 |
| Non-TC under Fed State | 185.54 | 6321.18 | 56.67 | 36,911.66 | |
| TC under Fasting State | 220.88 | 7527.41 | 61.56 | 38,331.20 | |
| TC under Fed State | 206.50 | 7035.84 | 59.55 | 37,750.24 | |
| 40 mg | Non-TC under Fasting State | 107.24 | 3653.12 | 60.64 | 44,904.43 |
| Non-TC under Fed State | 123.76 | 4214.01 | 63.51 | 45,807.92 | |
| TC under Fasting State | 147.41 | 5018.61 | 67.58 | 47,086.80 | |
| TC under Fed State | 137.80 | 4691.04 | 65.85 | 46,569.38 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Chen, L.; Ju, G.; Li, C.; Liu, B.; Fei, Y.; Wang, X.; Gao, Y.; He, Q.; Zhu, X.; et al. Investigation of the ABCB1 Gene Polymorphism and Food Effects on the Avatrombopag Pharmacokinetics in Chinese Individuals: A Population Pharmacokinetic/Pharmacodynamic Analysis. Pharmaceuticals 2025, 18, 903. https://doi.org/10.3390/ph18060903
Liu X, Chen L, Ju G, Li C, Liu B, Fei Y, Wang X, Gao Y, He Q, Zhu X, et al. Investigation of the ABCB1 Gene Polymorphism and Food Effects on the Avatrombopag Pharmacokinetics in Chinese Individuals: A Population Pharmacokinetic/Pharmacodynamic Analysis. Pharmaceuticals. 2025; 18(6):903. https://doi.org/10.3390/ph18060903
Chicago/Turabian StyleLiu, Xin, Lulu Chen, Gehang Ju, Chao Li, Bijue Liu, Yunzhou Fei, Xintong Wang, Yang Gao, Qingfeng He, Xiao Zhu, and et al. 2025. "Investigation of the ABCB1 Gene Polymorphism and Food Effects on the Avatrombopag Pharmacokinetics in Chinese Individuals: A Population Pharmacokinetic/Pharmacodynamic Analysis" Pharmaceuticals 18, no. 6: 903. https://doi.org/10.3390/ph18060903
APA StyleLiu, X., Chen, L., Ju, G., Li, C., Liu, B., Fei, Y., Wang, X., Gao, Y., He, Q., Zhu, X., & Ouyang, D. (2025). Investigation of the ABCB1 Gene Polymorphism and Food Effects on the Avatrombopag Pharmacokinetics in Chinese Individuals: A Population Pharmacokinetic/Pharmacodynamic Analysis. Pharmaceuticals, 18(6), 903. https://doi.org/10.3390/ph18060903