
Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union


 , ,
, ,  and
and 
Abstract

1. Introduction
2. Challenges in the Research and Development of Medicinal Products (Drugs)
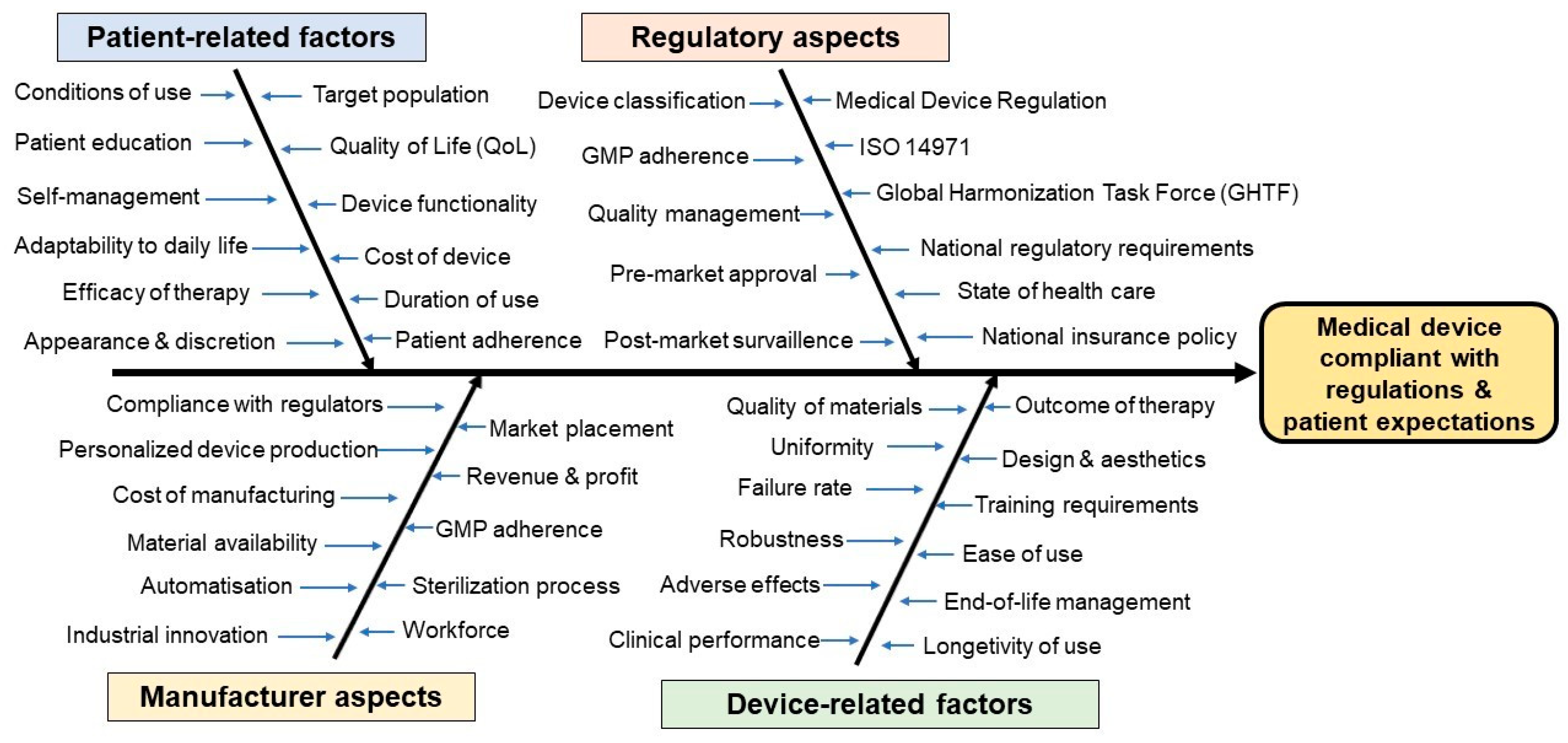
3. Challenges in the Research and Development of Medical Devices
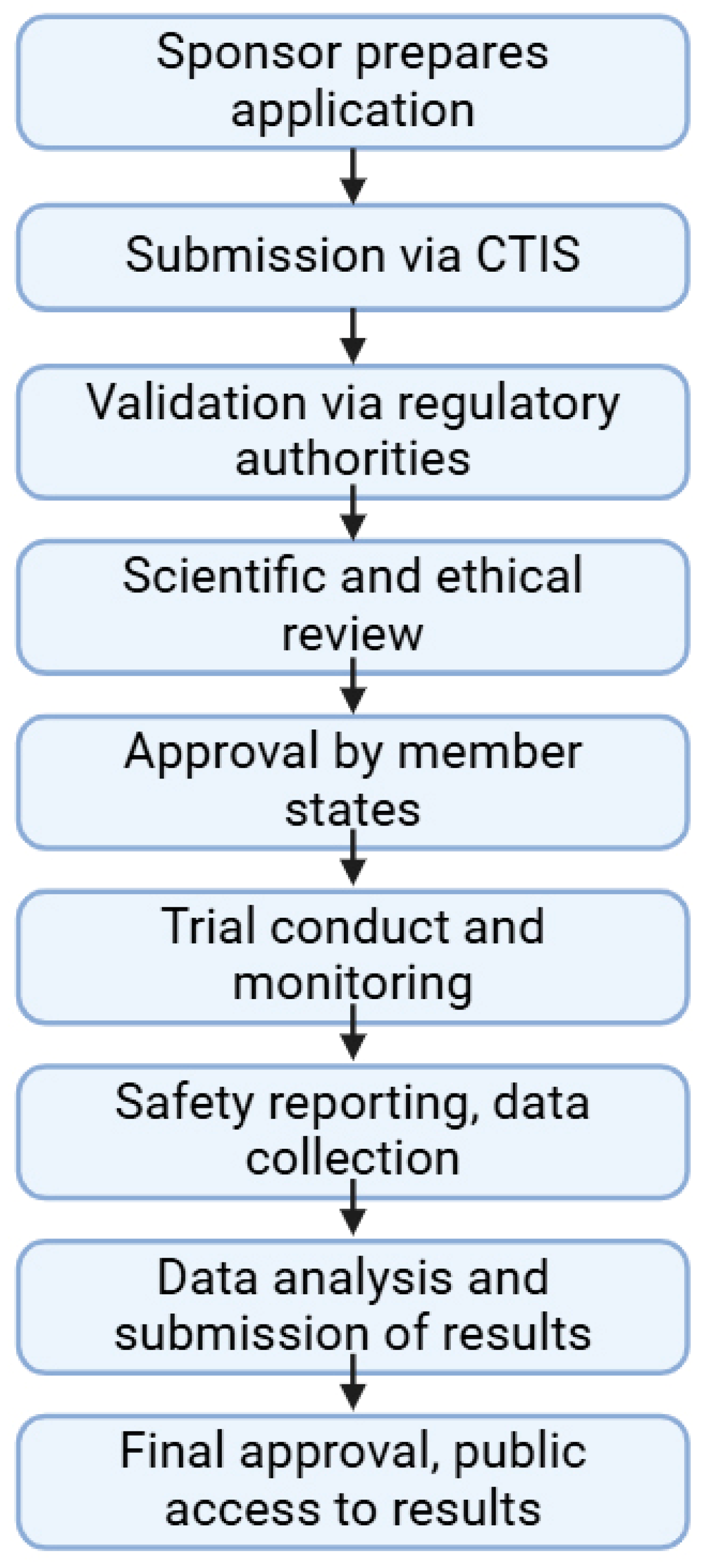
4. Regulation of Clinical Trials for Drugs
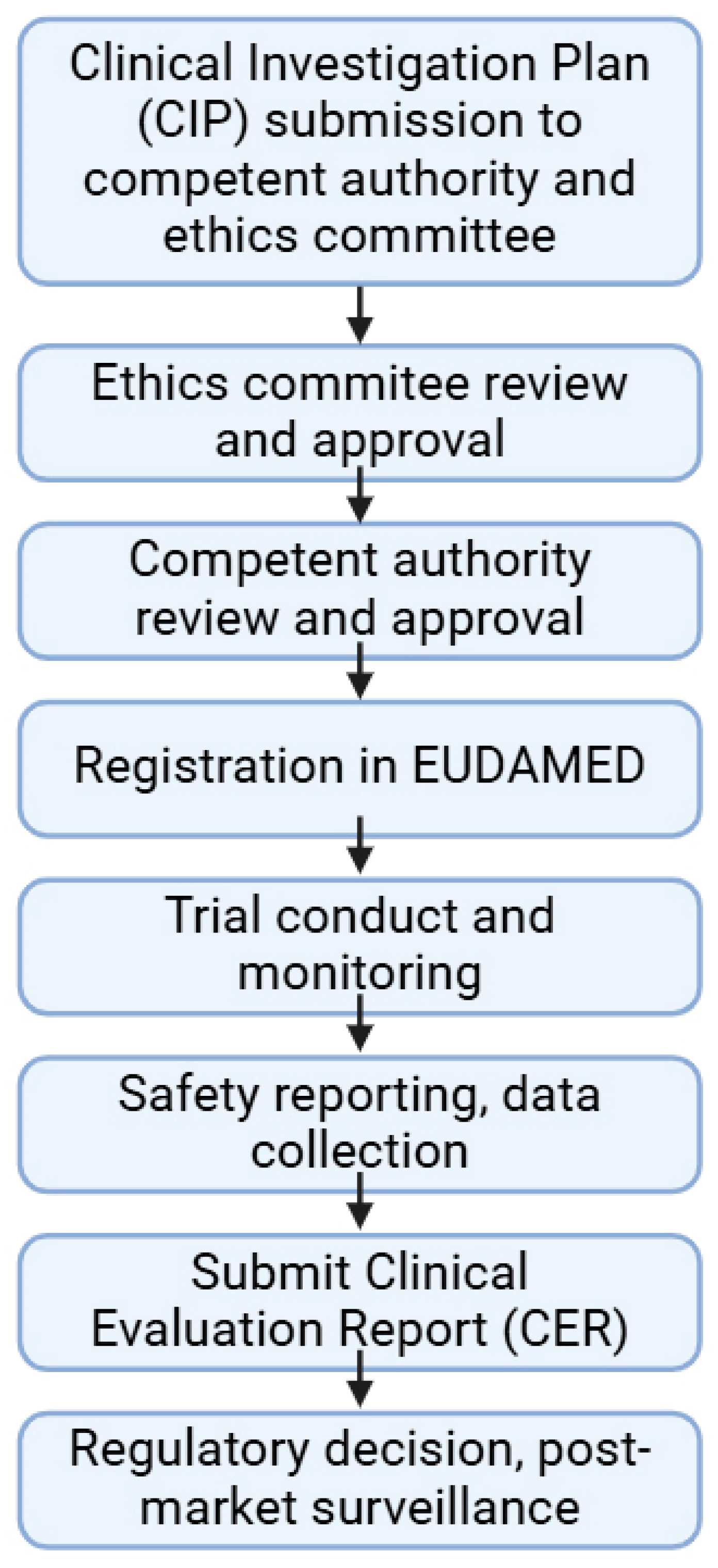
5. Regulation of Clinical Trials for Medical Devices
6. Outlook of Non-EU Clinical Trials
6.1. Clinical Trials for Drugs
6.2. Clinical Trials for Medical Devices
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CER | Clinical Evaluation Report |
| CIP | Clinical Investigation Plan |
| CTIS | Clinical Trials Information System |
| EU | European Union |
| GCP | Good Clinical Practice |
| GDPR | General Data Protection Regulation |
| GMP | Good Manufacturing Practice |
| ICH | The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use |
| ISO | International Organization for Standardization |
| MDD | Medical Device Directive |
| MDR | Medical Device Regulation |
| R&D | Research and development |
| RCT | Randomized controlled trial |
References
- Asgardoon, M.H.; Amirzade-Iranaq, M.H.; Mehri, A.; Piri, S.M.; Jalali, P.; Ghodsi, Z.; Dehghan, H.R.; Rahimi-Movaghar, V.; Salamati, P. Adverse Impacts of Imposing International Economic Sanctions on Health. Arch. Iran. Med. 2022, 25, 182–190. [Google Scholar] [PubMed]
- Sawyer, T.; Gray, M.M.; Umoren, R. The Global Healthcare Simulation Economy: A Scoping Review. Cureus 2022, 14, e22629. [Google Scholar] [CrossRef] [PubMed]
- Mahlangu, J. Emicizumab and Unmet Needs of Patients with Hemophilia a Who Are Managed with Replacement Therapies. Expert Rev. Hematol. 2024, 17, 741–748. [Google Scholar] [CrossRef]
- DeGroot, L.; Pavlovic, N.; Perrin, N.; Dy, S.M.; Szanton, S.; Gilotra, N.; Denfeld, Q.; Miller, H.; McIlvennan, C.; Davidson, P.; et al. The Impact of Unmet Palliative Care Needs and Physical Frailty on Clinical Outcomes: A Prospective Study of Adults with Heart Failure. J. Card. Fail. 2024, 30, 129. [Google Scholar] [CrossRef]
- Demirci, E.; Knicley, J.; Fiorentino, L. Clinical Development and Marketing Application Review Times for Novel Orphan-Designated Drugs. Front. Med. 2024, 11, 1404922. [Google Scholar] [CrossRef]
- Kalland, M.E.; Pose-Boirazian, T.; Palomo, G.M.; Naumann-Winter, F.; Costa, E.; Matusevicius, D.; Duarte, D.M.; Malikova, E.; Vitezic, D.; Larsson, K.; et al. Advancing Rare Disease Treatment: EMA’s Decade-Long Insights into Engineered Adoptive Cell Therapy for Rare Cancers and Orphan Designation. Gene Ther. 2024, 31, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, L.; Sepodes, B.; Leufkens, H.; Torre, C. Trends in Orphan Medicinal Products Approvals in the European Union between 2010–2022. Orphanet J. Rare Dis. 2024, 19, 91. [Google Scholar] [CrossRef]
- Holmes, D.R.; Geoffrion, R.; Hunt, J.; Hance, R.C.; Leon, M.B.; Mack, M.J.; Kaplan, A.V. Regulatory Strategies for Early Device Development and Approval. Catheter. Cardiovasc. Interv. 2022, 99, 1784–1788. [Google Scholar] [CrossRef]
- Kallio, M.J.; Starokozhko, V.; Agricola, E.; Burggraf, M.; Heß, A.; Ballensiefen, W.; Löbker, W.; Nuevo, Y.; Pasmooij, A.M.G.; Mol, P.G.M.; et al. Translating Academic Drug Discovery into Clinical Development: A Survey of the Awareness of Regulatory Support and Requirements Among Stakeholders in Europe. Clin. Pharmacol. Ther. 2023, 113, 349–359. [Google Scholar] [CrossRef]
- Farlow, A.; Torreele, E.; Gray, G.; Ruxrungtham, K.; Rees, H.; Prasad, S.; Gomez, C.; Sall, A.; Magalhães, J.; Olliaro, P.; et al. The Future of Epidemic and Pandemic Vaccines to Serve Global Public Health Needs. Vaccines 2023, 11, 690. [Google Scholar] [CrossRef]
- Pettitt, D.; Arshad, Z.; Davies, B.; Smith, J.; French, A.; Cole, D.; Bure, K.; Dopson, S.; DiGiusto, D.; Karp, J.; et al. An Assessment of the Factors Affecting the Commercialization of Cell-Based Therapeutics: A Systematic Review Protocol. Syst. Rev. 2017, 6, 120. [Google Scholar] [CrossRef] [PubMed]
- Kokudeva, M.; Vichev, M.; Naseva, E.; Miteva, D.G.; Velikova, T. Artificial Intelligence as a Tool in Drug Discovery and Development. World J. Exp. Med. 2024, 14, 96042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mastouri, M.; Zhang, Y. Accelerating Drug Discovery, Development, and Clinical Trials by Artificial Intelligence. Med 2024, 5, 1050–1070. [Google Scholar] [CrossRef]
- Rowan, N.J. Digital Technologies to Unlock Safe and Sustainable Opportunities for Medical Device and Healthcare Sectors with a Focus on the Combined Use of Digital Twin and Extended Reality Applications: A Review. Sci. Total Environ. 2024, 926, 171672. [Google Scholar] [CrossRef] [PubMed]
- Hasselgren, C.; Oprea, T.I. Artificial Intelligence for Drug Discovery: Are We There Yet? Annu. Rev. Pharmacol. Toxicol. 2024, 64, 527–550. [Google Scholar] [CrossRef]
- Pfob, A.; Hillen, C.; Seitz, K.; Griewing, S.; Becker, S.; Bayer, C.; Wagner, U.; Fasching, P.; Wallwiener, M.; Kommission Digitale Medizin, Deutsche Gesellschaft für Gynäkologie und Gebursthilfe (DGGG). Status Quo and Future Directions of Digitalization in Gynecology and Obstetrics in Germany: A Survey of the Commission Digital Medicine of the German Society for Gynecology and Obstetrics. Arch. Gynecol. Obstet. 2024, 309, 195–204. [Google Scholar] [CrossRef]
- Knitza, J.; Gupta, L.; Hügle, T. Rheumatology in the Digital Health Era: Status Quo and Quo Vadis? Nat. Rev. Rheumatol. 2024, 20, 747–759. [Google Scholar] [CrossRef]
- Kandi, V.; Vadakedath, S. Clinical Trials and Clinical Research: A Comprehensive Review. Cureus 2023, 15, e35077. [Google Scholar] [CrossRef]
- Goetz, L.H.; Schork, N.J. Personalized Medicine: Motivation, Challenges, and Progress. Fertil. Steril. 2018, 109, 952–963. [Google Scholar] [CrossRef]
- Makurvet, F.D. Biologics vs. Small Molecules: Drug Costs and Patient Access. Med. Drug Discov. 2021, 9, 100075. [Google Scholar] [CrossRef]
- Wouters, O.J.; Vogel, M.; Feldman, W.B.; Beall, R.F.; Kesselheim, A.S.; Tu, S.S. Differential Legal Protections for Biologics vs Small-Molecule Drugs in the US. JAMA 2024, 332, 2101. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, W.S.; Lee, K.H. Advances in Cardiovascular Care. JACC Basic Transl. Sci. 2018, 3, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, V.V.; Patel, N.H.; Smith, E.E.; Barnes, C.L.; Gustafson, M.P.; Rao, R.R.; Samsonraj, R.M. Clinical Utility of Mesenchymal Stem/Stromal Cells in Regenerative Medicine and Cellular Therapy. J. Biol. Eng. 2023, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Wang, Y.; Liang, S.; Han, R.; Toumi, M. Partnership Agreements for Regenerative Medicines: A Database Analysis and Implications for Future Innovation. Regen. Med. 2021, 16, 733–755. [Google Scholar] [CrossRef]
- Hofstädter-Thalmann, E.; Largier, D. EFPIA Guideline on a Quality Framework of Principles in Lifelong Learning in Healthcare. J. Eur. CME 2022, 11, 2068216. [Google Scholar] [CrossRef]
- Herrero-Martinez, E.; Hussain, N.; Roux, N.L.; MacDonald, J.; Mayer, M.; Palacios, R.; Kühler, T.C. Dynamic Regulatory Assessment: Evolving the European Regulatory Framework for the Benefit of Patients and Public Health—An EFPIA View. Clin. Ther. 2022, 44, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, A.K. Noncovalent PEGylation of Protein and Peptide Therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, 15, e1897. [Google Scholar] [CrossRef]
- Årdal, C.; Baraldi, E.; Theuretzbacher, U.; Outterson, K.; Plahte, J.; Ciabuschi, F.; Røttingen, J.-A. Insights into Early Stage of Antibiotic Development in Small- and Medium-Sized Enterprises: A Survey of Targets, Costs, and Durations. J. Pharm. Policy Pract. 2018, 11, 8. [Google Scholar] [CrossRef]
- Mulcahy, A.; Rennane, S.; Schwam, D.; Dickerson, R.; Baker, L.; Shetty, K. Use of Clinical Trial Characteristics to Estimate Costs of New Drug Development. JAMA Netw. Open 2025, 8, e2453275. [Google Scholar] [CrossRef]
- Ngum, N.; Ndomondo-Sigonda, M.; Habonimana, R.; Siyoi, F.; Irasabwa, C.; Ojukwu, J.; Apolinary, F.; Okello, A.; Ahmada, S.; Walker, S.; et al. Evaluation of the Review Models and Approval Timelines of Authorities Participating in the East African Medicine Regulatory Harmonisation Initiative: Alignment and Strategies for Moving Forward. Front. Med. 2024, 11, 1438041. [Google Scholar] [CrossRef]
- MacPherson, A.; Hutchinson, N.; Schneider, O.; Oliviero, E.; Feldhake, E.; Ouimet, C.; Sheng, J.; Awan, F.; Wang, C.; Papenburg, J.; et al. Probability of Success and Timelines for the Development of Vaccines for Emerging and Reemerged Viral Infectious Diseases. Ann. Intern. Med. 2021, 174, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; Anderson-Smits, C.; Rubinstein, R.; Thai, S.T.; Purcell, R.; Girman, C. A Framework for the Use and Likelihood of Regulatory Acceptance of Single-Arm Trials. Ther. Innov. Regul. Sci. 2024, 58, 1214–1232. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.R.; Mani, K. Improving Post-Market Surveillance for New Endovascular Devices. Eur. J. Vasc. Endovasc. Surg. 2024, 67, 754–755. [Google Scholar] [CrossRef]
- Exner, H.M.; Gregorchuk, B.S.J.; Castor, A.-G.; Crisostomo, L.; Kolsun, K.; Giesbrecht, S.; Dust, K.; Alexander, D.C.; Bolaji, A.; Quill, Z.; et al. Post-Market Surveillance of Six COVID-19 Point-of-Care Tests Using Pre-Omicron and Omicron SARS-CoV-2 Variants. Microbiol. Spectr. 2024, 12, e0016324. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllidi, E.; Triantafillidis, J.K. Systematic Review on the Use of Biosimilars of Trastuzumab in HER2+ Breast Cancer. Biomedicines 2022, 10, 2045. [Google Scholar] [CrossRef]
- Bischoff, H.; O’Connor, N.K.; Kim, J.; Popescu, B.V.; Bigot, C.; Pradhan, S.; Chakraborty, R.; Jaison, L.; Majeed, F.; Park, L.S.; et al. Comparative Preclinical Evaluation of Tuznue Versus Referent Herceptin: A Registered Trastuzumab Biosimilar. Drugs R D 2025, 25, 67–77. [Google Scholar] [CrossRef]
- Garattini, S.; Natsis, Y.; Banzi, R. Pharmaceutical Strategy for Europe: Reflections on Public Health-Driven Drug Development, Regulation, and Policies. Front. Pharmacol. 2021, 12, 685604. [Google Scholar] [CrossRef]
- Geiger, S.; Bourgeron, T. In the Name of Transparency: Organizing European Pharmaceutical Markets through Struggles over Transparency Devices. Organ. Stud. 2023, 44, 1751–1773. [Google Scholar] [CrossRef]
- Hemmerling, T.M.; Hofer, I.S. Protecting Intellectual Property While Satisfying Scientific Transparency. Anesth. Analg. 2022, 135, 239–240. [Google Scholar] [CrossRef]
- Gilbert, P.; Fawcett, R.; Coles, J.; Hillson, W. Intellectual Property Rights and Vaccines. Methods Mol. Biol. 2022, 2412, 505–518. [Google Scholar] [CrossRef]
- Gaspar, R.S.; Silva-Lima, B.; Magro, F.; Alcobia, A.; da Costa, F.L.; Feio, J. Non-Biological Complex Drugs (NBCDs): Complex Pharmaceuticals in Need of Individual Robust Clinical Assessment Before Any Therapeutic Equivalence Decision. Front. Med. 2020, 7, 590527. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-H.; Chen, Y.-S.; Tseng, T.; Jiang, M.-L.; Gau, C.-S.; Chang, L.-C. Regulatory Considerations for Generic Products of Non-Biological Complex Drugs. J. Food Drug Anal. 2023, 31, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Malheiro, V.; Duarte, J.; Veiga, F.; Mascarenhas-Melo, F. Exploiting Pharma 4.0 Technologies in the Non-Biological Complex Drugs Manufacturing: Innovations and Implications. Pharmaceutics 2023, 15, 2545. [Google Scholar] [CrossRef] [PubMed]
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on Medical Devices, Amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and Repealing Council Directives 90/385/EEC and 93/42/EEC 2017. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0745 (accessed on 6 May 2025).
- Schmitz, A.A.; Font-Nieves, M.; Doucouré, T.; Podhaisky, H.-P. Impact of Rule 11 on the European Medical Software Landscape: Analysis of EUDAMED and Further Databases Three Years After MDR Implementation. Ther. Innov. Regul. Sci. 2025, 59, 365–378. [Google Scholar] [CrossRef]
- Antal, A.; Ganho-Ávila, A.; Assecondi, S.; Barbour, T.; Bjekić, J.; Blumberger, D.M.; Bolognini, N.; Brunelin, J.; Chanes, L.; Dale, M.; et al. The Consequences of the New European Reclassification of Non-Invasive Brain Stimulation Devices and the Medical Device Regulations Pose an Existential Threat to Research and Treatment: An Invited Opinion Paper. Clin. Neurophysiol. 2024, 163, 280–291. [Google Scholar] [CrossRef]
- ISO 14971:2019; Medical Devices—Application of Risk Management to Medical Devices. ISO: Geneva, Switzerland, 2019.
- Maci, J.; Marešová, P. Critical Factors and Economic Methods for Regulatory Impact Assessment in the Medical Device Industry. Risk Manag. Healthc. Policy 2022, 15, 71–91. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, L.; Feng, X.; Zhang, Z.; Huang, Y. Regulatory Reliance for Convergence and Harmonisation in the Medical Device Space in Asia-Pacific. BMJ Glob. Health 2022, 7, e009798. [Google Scholar] [CrossRef]
- Huusko, J.; Kinnunen, U.-M.; Saranto, K. Medical Device Regulation (MDR) in Health Technology Enterprises—Perspectives of Managers and Regulatory Professionals. BMC Health Serv. Res. 2023, 23, 310. [Google Scholar] [CrossRef]
- Beddoe-Rosendo, J.; Heaysman, C.L.; Hajnal, J.V.; Ourselin, S.; Vanhoestenberghe, A. Medical Device Regulatory Challenges in the UK Are Affecting Innovation and Its Potential Benefits. Proc. Inst. Mech. Eng. H 2023, 237, 1243–1247. [Google Scholar] [CrossRef]
- Petrick, N.; Chen, W.; Delfino, J.G.; Gallas, B.D.; Kang, Y.; Krainak, D.; Sahiner, B.; Samala, R.K. Regulatory Considerations for Medical Imaging AI/ML Devices in the United States: Concepts and Challenges. J. Med. Imaging 2023, 10, 051804. [Google Scholar] [CrossRef]
- Aronson, J.K.; Heneghan, C.; Ferner, R.E. Medical Devices: Definition, Classification, and Regulatory Implications. Drug Saf. 2020, 43, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Matovu, B.; Baluka, J.W.; Takuwa, M.; Namuli, L.K.; Mpaata, C.N.; Mugaga, J.; Mulindwa, B.; Nalwoga, R.; Wolters, M.K.; Ssekitoleko, R.T. Translating Medical Device Innovations to Market—A Ugandan Perspective. BMC Res. Notes 2023, 16, 262. [Google Scholar] [CrossRef] [PubMed]
- Bretthauer, M.; Gerke, S.; Hassan, C.; Ahmad, O.F.; Mori, Y. The New European Medical Device Regulation: Balancing Innovation and Patient Safety. Ann. Intern. Med. 2023, 176, 844–848. [Google Scholar] [CrossRef]
- ISO 13485:2016; Medical Devices—Quality Management Systems—Requirements for Regulatory Purposes. ISO: Geneva, Switzerland, 2016.
- Kidholm, K.; Jensen, L.K.; Johansson, M.; Montori, V.M. Telemedicine and the Assessment of Clinician Time: A Scoping Review. Int. J. Technol. Assess. Health Care 2023, 40, e3. [Google Scholar] [CrossRef]
- Fields, B.G. Regulatory, Legal, and Ethical Considerations of Telemedicine. Sleep Med. Clin. 2020, 15, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Solimini, R.; Busardò, F.P.; Gibelli, F.; Sirignano, A.; Ricci, G. Ethical and Legal Challenges of Telemedicine in the Era of the COVID-19 Pandemic. Medicina 2021, 57, 1314. [Google Scholar] [CrossRef]
- Rowe, S.P.; Pomper, M.G. Molecular Imaging in Oncology: Current Impact and Future Directions. CA Cancer J. Clin. 2022, 72, 333–352. [Google Scholar] [CrossRef]
- Rodriguez-Manzano, J.; Subramaniam, S.; Uchea, C.; Szostak-Lipowicz, K.M.; Freeman, J.; Rauch, M.; Tinto, H.; Zar, H.J.; D’Alessandro, U.; Holmes, A.H.; et al. Innovative Diagnostic Technologies: Navigating Regulatory Frameworks through Advances, Challenges, and Future Prospects. Lancet Digit. Health 2024, 6, e934–e943. [Google Scholar] [CrossRef]
- European Parliament and Council of the European Union. Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on Clinical Trials on Medicinal Products for Human Use, and Repealing Directive 2001/20/EC Text with EEA relevance. Off. J. Eur. Union 2014, L158, 1–76. [Google Scholar]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH Guideline for Good Clinical Practice E6(R2); ICH: Geneva, Switzerland, 2016. [Google Scholar]
- Patrick-Brown, T.D.J.H.; Bourner, J.; Kali, S.; Trøseid, M.; Yazdanpanah, Y.; Olliaro, P.; Olsen, I.C. Experiences and Challenges with the New European Clinical Trials Regulation. Trials 2024, 25, 3. [Google Scholar] [CrossRef]
- Yamada, O.; Chiu, S.-W.; Takata, M.; Abe, M.; Shoji, M.; Kyotani, E.; Endo, C.; Shimada, M.; Tamura, Y.; Yamaguchi, T. Clinical Trial Monitoring Effectiveness: Remote Risk-Based Monitoring versus on-Site Monitoring with 100% Source Data Verification. Clin. Trials 2021, 18, 158–167. [Google Scholar] [CrossRef]
- Gnanasakthy, A.; Levy, C.; Norcross, L.; Doward, L.; Winnette, R. A Review of Labeling Based on Patient-Reported Outcome Endpoints for New Oncology Drugs Approved by the European Medicines Agency (2017–2021). Value Health 2023, 26, 893–901. [Google Scholar] [CrossRef]
- Stans, J.; Verbandt, S.; Kromar, S.; Deleersnijder, A. Implementation of Regulation (EU) No 536/2014 as a Non-Commercial Sponsor: An Internal Survey and a Descriptive Analysis of Timelines. Ther. Innov. Regul. Sci. 2023, 57, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Guideline on Quality Documentation for Medicinal Products When Used with a Medical Device; EMA/CHMP/QWP/BWP/259165/2019; EMA: Amsterdam, The Netherlands, 2021.
- Bianchini, E.; Mayer, C.C. Medical Device Regulation: Should We Care About It? Artery Res. 2022, 28, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Beckers, R.; Kwade, Z.; Zanca, F. The EU Medical Device Regulation: Implications for Artificial Intelligence-Based Medical Device Software in Medical Physics. Phys. Med. 2021, 83, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kumoluyi, R.; Khanolkar, A. Risk Management in Drug-Device Combination Product Development. Ther. Innov. Regul. Sci. 2022, 56, 685–688. [Google Scholar] [CrossRef]
- Hofer, M.P.; Criscuolo, P.; Shah, N.; Wal, A.L.J.T.; Barlow, J. Regulatory Policy and Pharmaceutical Innovation in the United Kingdom after Brexit: Initial Insights. Front. Med. 2022, 9, 1011082. [Google Scholar] [CrossRef]
- Corbin, J.; Walker, A.J. FDA Overview. In Translational Radiation Oncology; Elsevier: Amsterdam, The Netherlands, 2023; pp. 453–457. ISBN 978-0-323-88423-5. [Google Scholar]
- Ramamoorthy, A.; Araojo, R.; Vasisht, K.P.; Fienkeng, M.; Green, D.J.; Madabushi, R. Promoting Clinical Trial Diversity: A Highlight of Select US FDA Initiatives. Clin. Pharmacol. Ther. 2023, 113, 528–535. [Google Scholar] [CrossRef]
- Turner, J.R. New FDA Guidance on General Clinical Trial Conduct in the Era of COVID-19. Ther. Innov. Regul. Sci. 2020, 54, 723–724. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Zeng, W.; Li, Z.; Zhang, J.; Wang, S.; Xu, N.; Li, Z. Re-Analysis of the Current Status of Clinical Trial Registration in China. Front. Med. 2025, 11, 1394803. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, X.-Y.; Yang, Z.-M.; Wu, Y.-L. The Changing Landscape of Clinical Trial and Approval Processes in China. Nat. Rev. Clin. Oncol. 2017, 14, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Hanaoka, H.; Uyama, Y. Potential Future Drug Development Lag in Japan Based on an Analysis of Multiregional Clinical Trials in the US, Europe, and East Asia. Ther. Innov. Regul. Sci. 2022, 56, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Eba, J.; Nakamura, K. Overview of the Ethical Guidelines for Medical and Biological Research Involving Human Subjects in Japan. Jpn. J. Clin. Oncol. 2022, 52, 539–544. [Google Scholar] [CrossRef]
- Martynova, E.; Shcherbovich, A. Digital Transformation in Russia: Turning from a Service Model to Ensuring Technological Sovereignty. Comput. Law Secur. Rev. 2024, 55, 106075. [Google Scholar] [CrossRef]
- Manoharan, K.; Jinson, J.; Ramesh, K.; George, M. Clinical Trial Trends over the Last 5 Years among the BRICS (Brazil, Russia, India, China, and South Africa) Nations. Perspect. Clin. Res. 2023, 15, 128–133. [Google Scholar] [CrossRef]
- Fraser, A.G.; Redberg, R.F.; Melvin, T. The Origins of Regulations for Pharmaceutical Products and Medical Devices—What Can Be Learned for the Governance of Medical Devices in Europe? Eur. Rev. 2025, 1–34. [Google Scholar] [CrossRef]
- Ali, F.; Nollet, L.M.L. (Eds.) Global Regulations of Medicinal, Pharmaceutical, and Food Products, 1st ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2025; ISBN 978-1-003-29649-2. [Google Scholar]
- ISO 14155:2020; Clinical Investigation of Medical Devices for Human Subjects—Good Clinical Practice. ISO: Geneva, Switzerland, 2020.
- Curle, A.J.; Barnes, J.L.; Owen, R.; Barker, R.A.; El Haj, A.; Forbes, S.J.; Ghevaert, C.; Oreffo, R.O.; Rose, F.R.; Stevens, M.M.; et al. A Decade of Progress: Achievements and Future Challenges for Regenerative Medicine Research in the United Kingdom. J. Immunol. Regen. Med. 2024, 24–25, 100078. [Google Scholar] [CrossRef]
- Muralidharan, V.; Adewale, B.A.; Huang, C.J.; Nta, M.T.; Ademiju, P.O.; Pathmarajah, P.; Hang, M.K.; Adesanya, O.; Abdullateef, R.O.; Babatunde, A.O.; et al. A Scoping Review of Reporting Gaps in FDA-Approved AI Medical Devices. npj Digit. Med. 2024, 7, 273. [Google Scholar] [CrossRef]
- Tettey, F.; Parupelli, S.K.; Desai, S. A Review of Biomedical Devices: Classification, Regulatory Guidelines, Human Factors, Software as a Medical Device, and Cybersecurity. Biomed. Mater. Devices 2024, 2, 316–341. [Google Scholar] [CrossRef]
- Han, Y.; Bergmann, J. Transforming Medical Regulations into Numbers: Vectorizing a Decade of Medical Device Regulatory Shifts in the USA, EU, and China. arXiv 2024. [Google Scholar] [CrossRef]
- Liao, X.; Yao, C.; Jin, F.; Zhang, J.; Liu, L. Barriers and Facilitators to Implementing Imaging-Based Diagnostic Artificial Intelligence-Assisted Decision-Making Software in Hospitals in China: A Qualitative Study Using the Updated Consolidated Framework for Implementation Research. BMJ Open 2024, 14, e084398. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Tanaka, M.; Tanaka, Y.; Terashima, R.; Ezura, M.; Miyazawa, H.; Ikuma, M.; Tomita, Y. Concordance Between Pharmaceuticals and Medical Devices Agency Review and Ministry of Health, Labour and Welfare Decision Among New Drug Applications in Japan. Clin. Pharmacol. Ther. 2025, 117, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Chettri, B.; Ravi, R. A Comparative Study of Medical Device Regulation between Countries Based on Their Economies. Expert Rev. Med. Devices 2024, 21, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.L. Medical Sanctions Against Russia: Arresting Aggression or Abrogating Healthcare Rights? Am. J. Bioeth. 2025, 25, 3–16. [Google Scholar] [CrossRef]
- Pogarskaya, A.S. On the Issue of Parallel Import of Medical Articles and Component Units to Them in the Russian Federation in Conditions of Sanctions Policy. Probl. Sotsialnoi Gig. Zdravookhranenniiai Istor. Med. 2024, 32, 43–51. [Google Scholar] [CrossRef]
- Pawar, H.; Patel, M. Harmonization of Regulatory Frameworks for Medical Devices in BRICS Countries: A Path to Enhanced Trade and Investment. Ann. Pharm. Françaises 2025, 83, 272–286. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Directive 2001/20/EC | Regulation No. 536/2014 |
|---|---|---|
| Ethics Committee role | national discretion | in ordinance with all member states |
| Application Submission | separately to each member state | single application via CTIS |
| Assessment timeline | varying between member states | harmonized deadlines |
| Risk-based supervision | uniform requirements | based on risk level |
| transparency | limited public access | mandatory public access |
| Safety reporting | varying formats between member states | standardized |
| Aspect | MDR 2017/745 | MDD 93/42/EEC |
|---|---|---|
| Legal framework | Binding regulation | Directive |
| Scope | Covers a broader range of devices | Gaps regarding certain products |
| Classification of devices | Stringent classification rules | Less strict classification rules |
| Clinical evidence requirements | Stronger requirements | Less stringent requirements |
| Post-market surveillance | Enhanced requirements | Basic requirements |
| Unique device identification (UDI) | Mandatory | Not mandatory |
| Notified bodies oversight | Stronger role and increased scrutiny | Less oversight and scrutiny |
| EUDAMED database | Fully integrated database with public access | Limited database and availability |
| Manufacturer responsibilities | Greater and stricter responsibilities | Fewer responsibilities |
| Transition period | The 3-year transition from May 2017 | Transitioned out as MDR came into action |
| Aspect | Medicinal Product (Drug) | Medical Device | Combined Drug–Device Product |
|---|---|---|---|
| Regulation | Clinical Trials Regulation No. 536/2014 | Medical Device Regulation 2017/745 | Dual regulation: primarily CTR or MDR depending on primary model of action |
| Approval process | Clinical Trial Application via CTIS (regulatory and ethics approval) | Clinical investigation application with a submission to the competent authority and ethics committee | Coordinated submission based on the primary mode of action |
| Risk classification | No formal trial-based classification, as all investigational drugs must undergo a strict review | Classified as I, IIa, IIb, and III based on invasiveness and risk | Risk driven by both drug safety profile and device classification (usually elevated) |
| Clinical trial phases | Following Phase I–IV | Follows feasibility and pivotal studies | May include hybrid trial phases combining tests for drug efficacy and device performance |
| Study design | Randomized controlled trials | Flexible, comparative studies | RCTs with embedded device usability/performance metrics |
| Sample size | Hundreds to thousands of patients | Dozens to hundreds | Depends on both drug efficacy and device-specific endpoints, but larger than for medical devices |
| Clinical endpoint | Pharmacokinetics, efficacy, and safety | Device performance, usability, functionality | Based on the assessment of both drug and medical device performance |
| Regulatory review time | 12–18 months until EMA approval | Faster for low-risk devices, whilst Class III requires rigorous notified body assessments | Can be prolonged due to dual evaluation |
| Post-market surveillance | Adverse drug reaction monitoring | Device failure and usability issues monitoring | Requires integrated vigilance system covering both adverse drug reactions and device incidents |
| Manufacturing requirements | Good Manufacturing Practice | ISO 13485 (Medical Device Quality Management System) [56] | Must comply with both GMP and ISO 13485 [56] |
| Investigator requirements | Medical doctor | Healthcare professional | Medical doctor |
| Interaction with the human body | Usually works systematically | Usually works locally | Usually combined systemic and localized effects |
| Need for placebo control | Standard | Rare or unethical | Standard for the drug part, but ethically it should be justified for the device part |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pannonhalmi, Á.; Sipos, B.; Kurucz, R.I.; Katona, G.; Kemény, L.; Csóka, I. Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union. Pharmaceuticals 2025, 18, 876. https://doi.org/10.3390/ph18060876
Pannonhalmi Á, Sipos B, Kurucz RI, Katona G, Kemény L, Csóka I. Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union. Pharmaceuticals. 2025; 18(6):876. https://doi.org/10.3390/ph18060876
Chicago/Turabian StylePannonhalmi, Ádám, Bence Sipos, Róbert Imre Kurucz, Gábor Katona, Lajos Kemény, and Ildikó Csóka. 2025. "Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union" Pharmaceuticals 18, no. 6: 876. https://doi.org/10.3390/ph18060876
APA StylePannonhalmi, Á., Sipos, B., Kurucz, R. I., Katona, G., Kemény, L., & Csóka, I. (2025). Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union. Pharmaceuticals, 18(6), 876. https://doi.org/10.3390/ph18060876