

In Silico Investigation of TATA-Binding Protein as a Therapeutic Target for Chagas Disease: Insights into FDA Drug Repositioning


 , and
, and 
Abstract

1. Introduction
2. Results
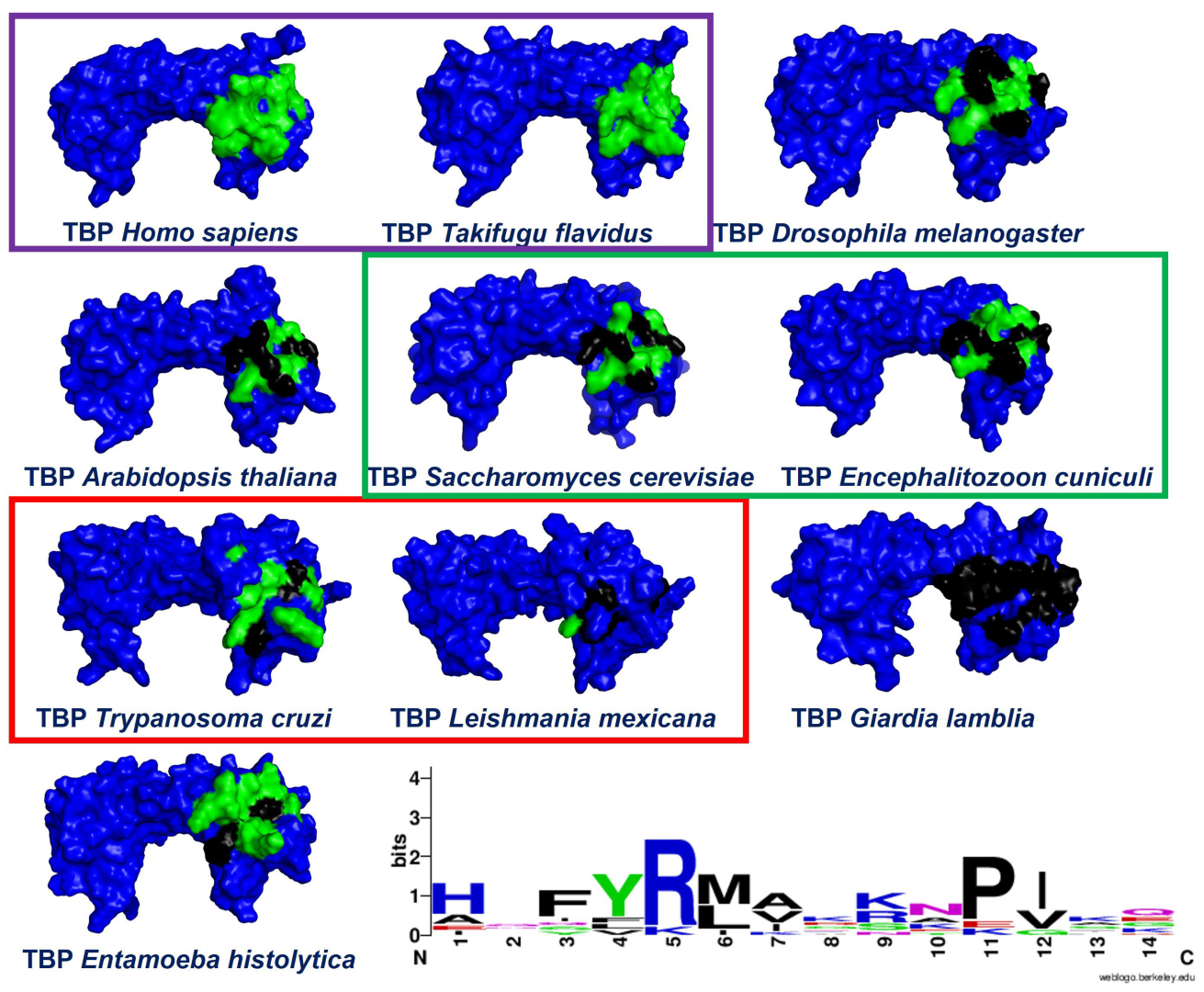
2.1. Multiple Sequence Alignments
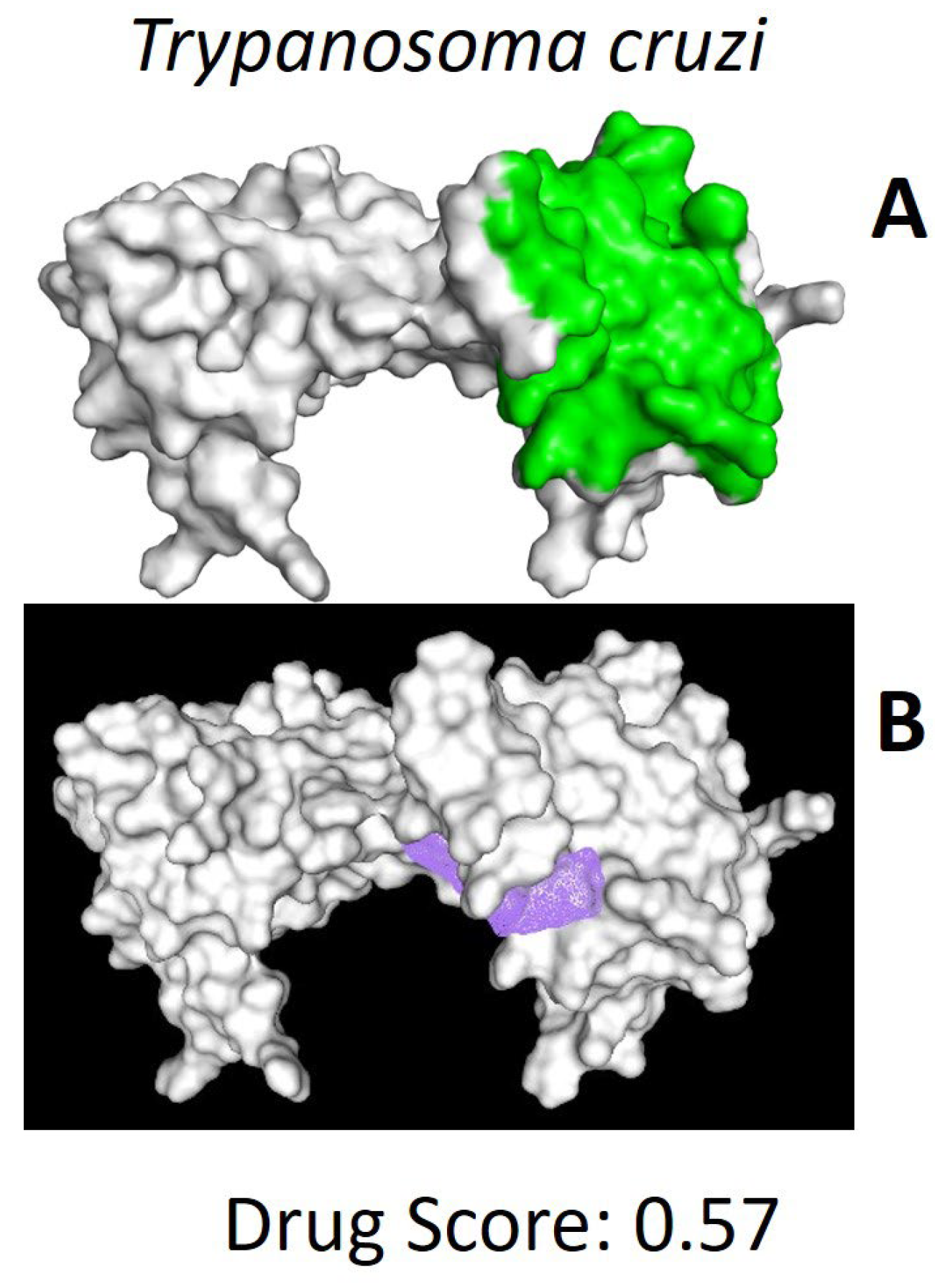
2.2. In Silico Analysis of TcTBP and HsTBP
2.2.1. TcTBP Molecular Docking
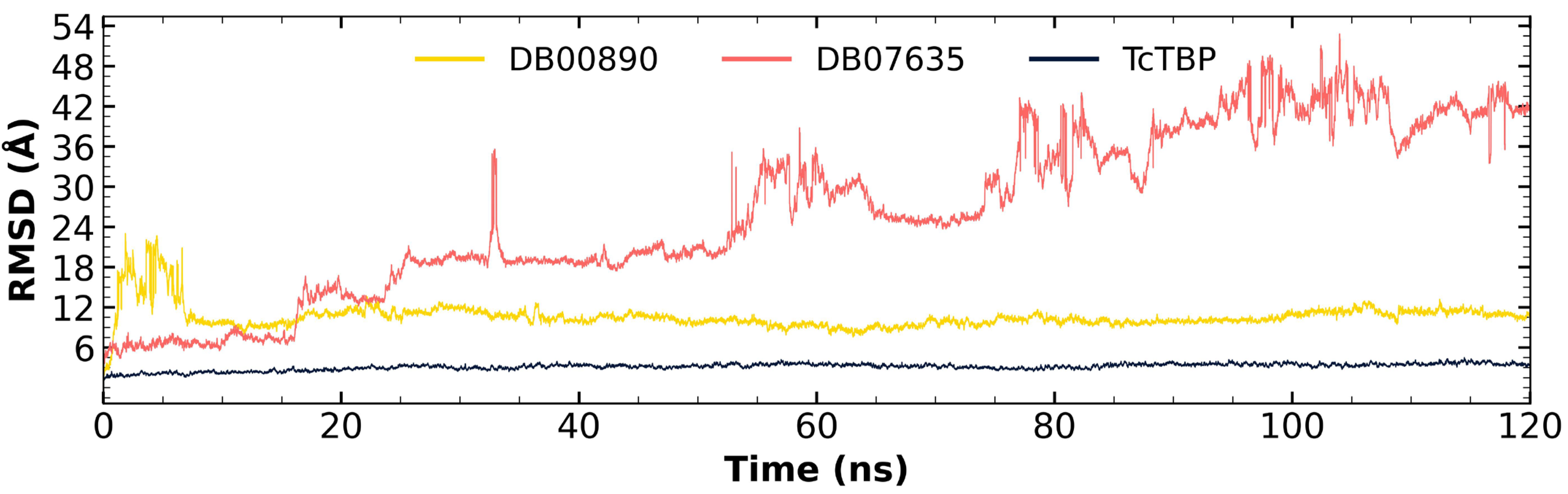
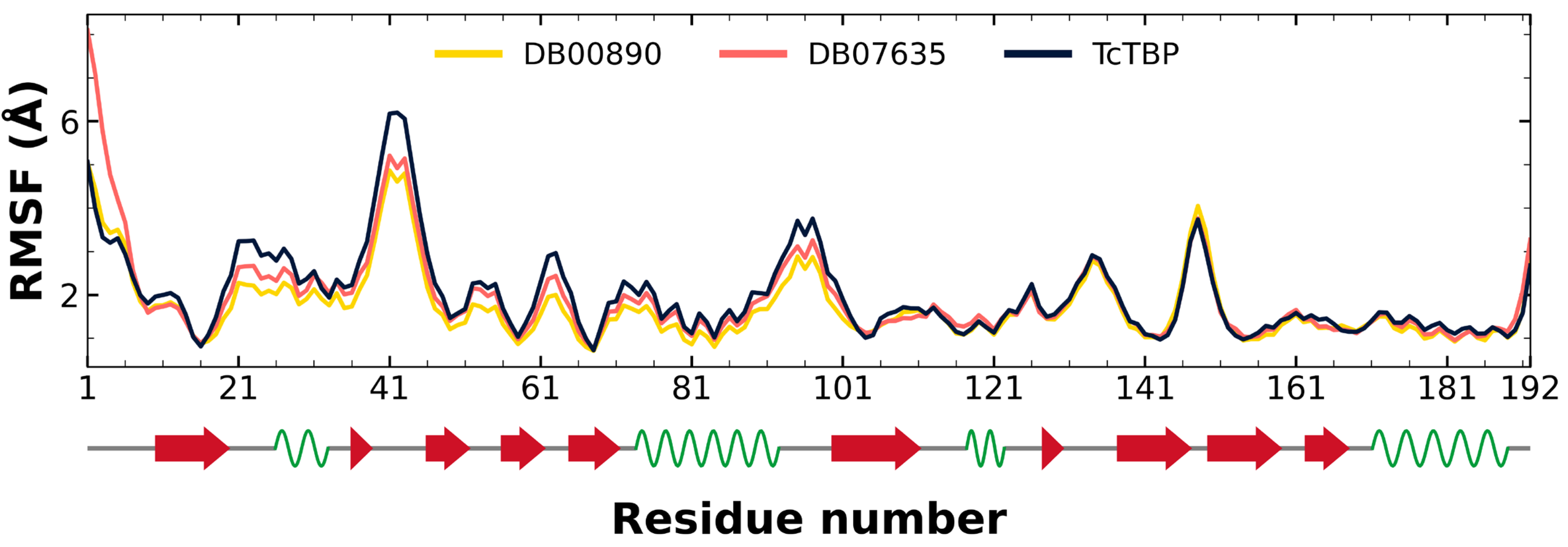
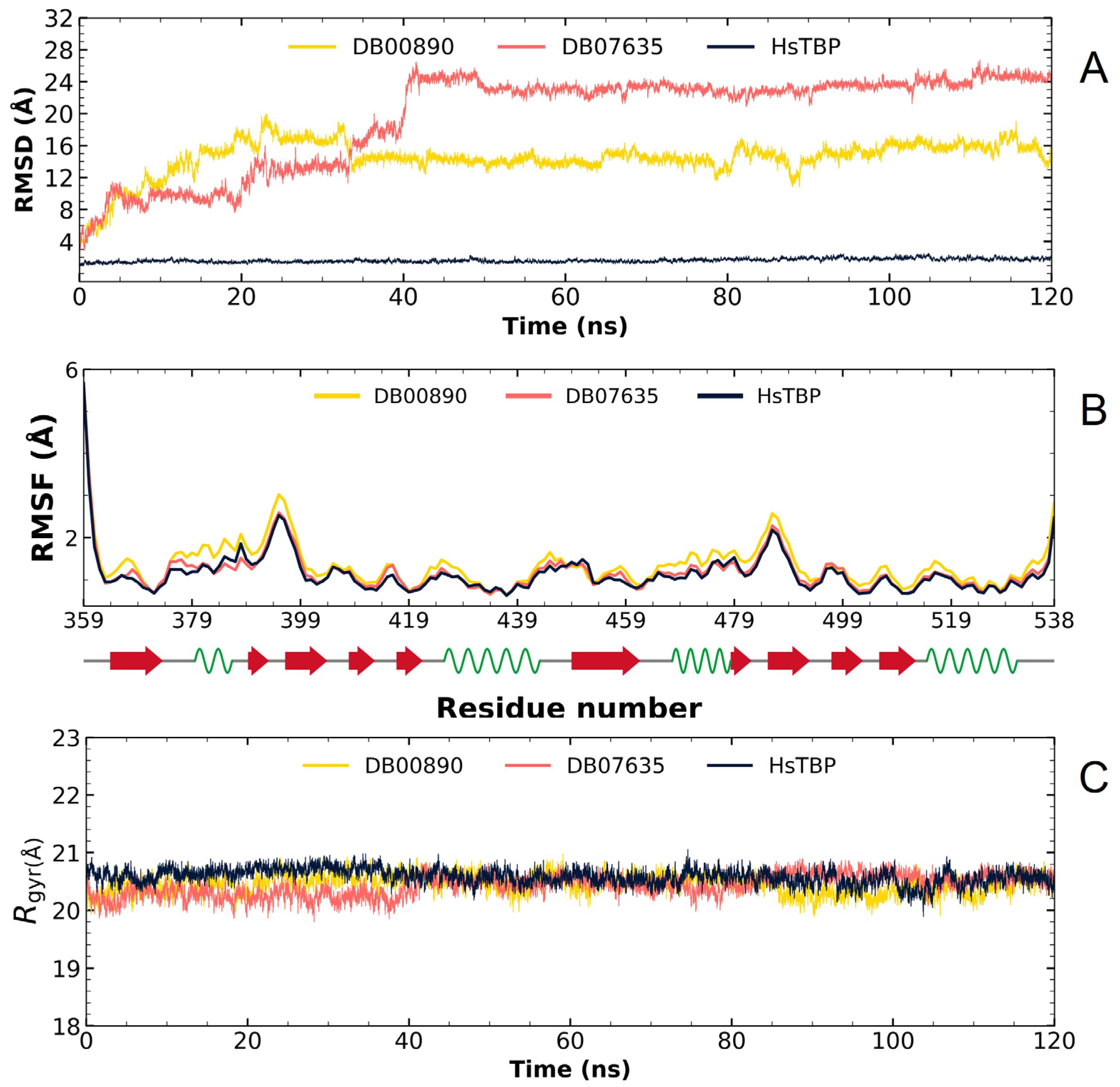
2.2.2. TcTBP Molecular Dynamics
2.3. In Vitro Assays
3. Discussion
3.1. In Silico Evaluation
3.2. In Vitro Evaluation
4. Materials and Methods
4.1. Download of Protein Sequences
4.2. Multiple Sequence Alignments
4.3. Identification of the Ligand Interaction Region
4.4. Docking Simulation
4.4.1. Receptor Preparation
4.4.2. Ligand Configuration for Docking Simulations
4.4.3. Grid Parameter Configuration
4.5. MD Simulations
4.5.1. Trypanocidal Activity In Vitro
4.5.2. Cytotoxicity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A. thaliana | Arabidopsis thaliana |
| BFE | Binding free energy |
| Bzn | Benznidazole |
| CC50 | Half-maximal cytotoxic concentration |
| C-terminal | Carbon terminal |
| DHEA | Dehydroepiandrosterone |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| E. cuniculi | Encephalitozoon cuniculi |
| E. histolytica | Entamoeba histolytica |
| FBS | Fetal bovine serum |
| FDA | Food and Drug Administration |
| G. lamblia | Giardia lamblia |
| GAFF | General Amber Force Field |
| GlTBP | Giardia lamblia TATA Binding Protein |
| HsTBP | Homo sapiens TATA Binding Protein |
| IC50 | Half-maximal inhibitory concentration |
| kcal/mol | Kilocalories per mole |
| L. mexicana | Leishmania mexicana |
| LIT medium | Liver infusion tryptose medium |
| mRNA | Messenger RNA |
| µM | Micromolar |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NC2 | Negative cofactor 2 |
| NCBI | National Center for Biotechnology Information |
| Nfx | Nifurtimox |
| ns | Nanoseconds |
| NTD | Neglected Tropical Disease |
| P. falciparum | Plasmodium falciparum |
| PDBe | Protein Data Bank Europe |
| PLIP | Protein–Ligand Interaction Profiler |
| RCSB PDB | Research Collaboratory for Structural Bioinformatics Protein Data Bank |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| RNA | Ribonucleic acid |
| RPMI medium | Roswell Park Memorial Institute medium |
| rRNA | Ribosomal RNA |
| S. cerevisae | Saccharomyces cerevisae |
| SI | Selective index |
| snRNA | Small nuclear RNA |
| T. cruzi | Trypanosoma cruzi |
| TBP | TATA Binding Protein |
| TcTBP | Trypanosoma cruzi TATA Binding Protein |
| TFIIA | Transcription factor IIA |
| TFIIB | Transcription factor IIB |
| tRNA | Transcription RNA |
| UCSF Chimera | University of California San Francisco Chimera |
| W | Watts |
| WHO | World Health Organization |
References
- Anisuzzaman; Hossain, M.S.; Hatta, T.; Labony, S.S.; Kwofie, K.D.; Kawada, H.; Tsuji, N.; Alim, M.A. Food- and Vector-Borne Parasitic Zoonoses: Global Burden and Impacts. Adv. Parasitol. 2023, 120, 87–136. [Google Scholar]
- Pisarski, K. The Global Burden of Disease of Zoonotic Parasitic Diseases: Top 5 Contenders for Priority Consideration. Trop. Med. Infect. Dis. 2019, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Balaña-Fouce, R.; Álvarez-Velilla, R.; Fernández-Prada, C.; García-Estrada, C.; Reguera, R.M. Trypanosomatids Topoisomerase Re-Visited. New Structural Findings and Role in Drug Discovery. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The Spreading of Parasites by Human Migratory Activities. Virulence 2020, 11, 1177–1191. [Google Scholar] [CrossRef]
- Hotez, P.J.; Bottazzi, M.E.; Franco-Paredes, C.; Ault, S.K.; Periago, M.R. The Neglected Tropical Diseases of Latin America and the Caribbean: A Review of Disease Burden and Distribution and a Roadmap for Control and Elimination. PLoS Negl. Trop. Dis. 2008, 2, e300. [Google Scholar] [CrossRef]
- Lee, S.-M.; Kim, M.-S.; Hayat, F.; Shin, D. Recent Advances in the Discovery of Novel Antiprotozoal Agents. Molecules 2019, 24, 3886. [Google Scholar] [CrossRef]
- Brindha, J.; Balamurali, M.M.; Chanda, K. An Overview on the Therapeutics of Neglected Infectious Diseases—Leishmaniasis and Chagas Diseases. Front. Chem. 2021, 9, 622286. [Google Scholar] [CrossRef]
- Capela, R.; Moreira, R.; Lopes, F. An Overview of Drug Resistance in Protozoal Diseases. Int. J. Mol. Sci. 2019, 20, 5748. [Google Scholar] [CrossRef]
- Hotez, P.J.; Aksoy, S.; Brindley, P.J.; Kamhawi, S. World Neglected Tropical Diseases Day. PLoS Negl. Trop. Dis. 2020, 14, e0007999. [Google Scholar] [CrossRef]
- Hotez, P.J.; Aksoy, S.; Brindley, P.J.; Kamhawi, S. What Constitutes a Neglected Tropical Disease? PLoS Negl. Trop. Dis. 2020, 14, e0008001. [Google Scholar] [CrossRef]
- Shu Kurizky, P.; dos Santos Neto, L.L.; Barbosa Aires, R.; Henrique da Mota, L.M.; Martins Gomes, C. Opportunistic Tropical Infections in Immunosuppressed Patients. Best. Pract. Res. Clin. Rheumatol. 2020, 34, 101509. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Molina, J.A.; Molina, I. Chagas Disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, L.E.; Morillo, C.A. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2019, 33, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, J.; Davies, C.; Simonazzi, A.; Pablo Real, J.; Palma, S. Current Drug Therapy and Pharmaceutical Challenges for Chagas Disease. Acta Trop. 2016, 156, 1–16. [Google Scholar] [CrossRef]
- Castro, J.A.; deMecca, M.M.; Bartel, L.C. Toxic Side Effects of Drugs Used to Treat Chagas’ Disease (American Trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Kelly, J.M. Trypanocidal Drugs: Mechanisms, Resistance and New Targets. Expert. Rev. Mol. Med. 2009, 11, e31. [Google Scholar] [CrossRef]
- Clare Vinaud, M.; De Souza Lino Junior, R. Mode of action of the main anti-parasitic drugs. Rev. De Patol. Trop. 2017, 46, 121. [Google Scholar] [CrossRef]
- Best, A.A.; Morrison, H.G.; McArthur, A.G.; Sogin, M.L.; Olsen, G.J. Evolution of Eukaryotic Transcription: Insights From the Genome of Giardia lamblia. Genome Res. 2004, 14, 1537–1547. [Google Scholar] [CrossRef]
- Parra-Marín, O.; López-Pacheco, K.; Hernández, R.; López-Villaseñor, I. The Highly Diverse TATA Box-Binding Proteins among Protists: A Review. Mol. Biochem. Parasitol. 2020, 239, 111312. [Google Scholar] [CrossRef]
- Santiago, Á.; Razo-Hernández, R.S.; Pastor, N. The TATA-binding Protein DNA-binding Domain of Eukaryotic Parasites Is a Potentially Druggable Target. Chem. Biol. Drug Des. 2020, 95, 130–149. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. Ribosomal RNA Transcription Machineries in Intestinal Protozoan Parasites: A Bioinformatic Analysis. Acta Parasitol. 2022, 67, 1788–1799. [Google Scholar] [CrossRef]
- Drummond, D.A.; Bloom, J.D.; Adami, C.; Wilke, C.O.; Arnold, F.H. Why Highly Expressed Proteins Evolve Slowly. Proc. Natl. Acad. Sci. USA 2005, 102, 14338–14343. [Google Scholar] [CrossRef]
- Hoshiyama, D.; Kuma, K.; Miyata, T. Extremely Reduced Evolutionary Rate of TATA-Box Binding Protein in Higher Vertebrates and Its Evolutionary Implications. Gene 2001, 280, 169–173. [Google Scholar] [CrossRef]
- Walters, H.A.; Temesvari, L.A. Target Acquired: Transcriptional Regulators as Drug Targets for Protozoan Parasites. Int. J. Parasitol. 2021, 51, 599–611. [Google Scholar] [CrossRef]
- Gaona-López, C.; Méndez-Álvarez, D.; Moreno-Rodríguez, A.; Bautista-Martínez, J.L.; De Fuentes-Vicente, J.A.; Nogueda-Torres, B.; García-Torres, I.; López-Velázquez, G.; Rivera, G. TATA-Binding Protein-Based Virtual Screening of FDA Drugs Identified New Anti-Giardiasis Agents. Int. J. Mol. Sci. 2024, 25, 6238. [Google Scholar] [CrossRef]
- Kramm, K.; Engel, C.; Grohmann, D. Transcription Initiation Factor TBP: Old Friend New Questions. Biochem. Soc. Trans. 2019, 47, 411–423. [Google Scholar] [CrossRef]
- Ravarani, C.N.J.; Flock, T.; Chavali, S.; Anandapadamanaban, M.; Babu, M.M.; Balaji, S. Molecular Determinants Underlying Functional Innovations of TBP and Their Impact on Transcription Initiation. Nat. Commun. 2020, 11, 2384. [Google Scholar] [CrossRef]
- Ferreira, R.; Schneekloth, J.S.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef]
- Mars, J.-C.; Tremblay, M.G.; Valere, M.; Sibai, D.S.; Sabourin-Felix, M.; Lessard, F.; Moss, T. The Chemotherapeutic Agent CX-5461 Irreversibly Blocks RNA Polymerase I Initiation and Promoter Release to Cause Nucleolar Disruption, DNA Damage and Cell Inviability. NAR Cancer 2020, 2, zcaa032. [Google Scholar] [CrossRef]
- Mishal, R.; Luna-Arias, J.P. Role of the TATA-Box Binding Protein (TBP) and Associated Family Members in Transcription Regulation. Gene 2022, 833, 146581. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]
- Hasegawa, H.; Holm, L. Advances and Pitfalls of Protein Structural Alignment. Curr. Opin. Struct. Biol. 2009, 19, 341–348. [Google Scholar] [CrossRef]
- Herman, J.L. Enhancing Statistical Multiple Sequence Alignment and Tree Inference Using Structural Information. In Computational Methods in Protein Evolution; Springer: Berlin/Heidelberg, Germany, 2019; pp. 183–214. [Google Scholar]
- Tokuriki, N.; Tawfik, D.S. Protein Dynamism and Evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef]
- DrugBank Online. Available online: https://go.drugbank.com/ (accessed on 20 December 2024).
- Pattanaik, P.; Bethel, C.R.; Hujer, A.M.; Hujer, K.M.; Distler, A.M.; Taracila, M.; Anderson, V.E.; Fritsche, T.R.; Jones, R.N.; Pagadala, S.R.R.; et al. Strategic Design of an Effective β-Lactamase Inhibitor. J. Biol. Chem. 2009, 284, 945–953. [Google Scholar] [CrossRef]
- Docampo, R.; Moreno, S.N.; Gadelha, F.R.; de Souza, W.; Cruz, F.S. Prevention of Chagas’ Disease Resulting from Blood Transfusion by Treatment of Blood: Toxicity and Mode of Action of Gentian Violet. Biomed. Environ. Sci. 1988, 1, 406–413. [Google Scholar]
- Allgardsson, A.; David Andersson, C.; Akfur, C.; Worek, F.; Linusson, A.; Ekström, F. An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet. Molecules 2017, 22, 1433. [Google Scholar] [CrossRef]
- Pung, O.J.; Luster, M.I.; Hayes, H.T.; Rader, J. Influence of Steroidal and Nonsteroidal Sex Hormones on Host Resistance in Mice: Increased Susceptibility to Listeria Monocytogenes after Exposure to Estrogenic Hormones. Infect. Immun. 1984, 46, 301–307. [Google Scholar] [CrossRef]
- Mankau, S.K. Host Sex and Sex Hormones as a Factor Affecting Trypanosoma Lewisi Population in White Rats. Jpn. J. Parasitol. 1975, 24, 379–384. [Google Scholar]
- Vaubourgeix, J.; Bardou, F.; Boissier, F.; Julien, S.; Constant, P.; Ploux, O.; Daffé, M.; Quémard, A.; Mourey, L. S-Adenosyl-N-Decyl-Aminoethyl, a Potent Bisubstrate Inhibitor of Mycobacterium Tuberculosis Mycolic Acid Methyltransferases. J. Biol. Chem. 2009, 284, 19321–19330. [Google Scholar] [CrossRef]
- Eddine, A.N.; von Kries, J.P.; Podust, M.V.; Warrier, T.; Kaufmann, S.H.E.; Podust, L.M. X-Ray Structure of 4,4′-Dihydroxybenzophenone Mimicking Sterol Substrate in the Active Site of Sterol 14α-Demethylase (CYP51). J. Biol. Chem. 2008, 283, 15152–15159. [Google Scholar] [CrossRef]
- Choi, M.S.; Kim, J.H.; Lee, C.Y.; Lee, Y.M.; Lee, S.; Chang, H.K.; Kim, H.J.; Heo, K. Gentian Violet Inhibits Cell Proliferation through Induction of Apoptosis in Ovarian Cancer Cells. Biomedicines 2023, 11, 1657. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, F.; Yang, H.; Wen, J.; Tang, Y.; Wan, F.; Zhang, X.; Wu, J. Gentian Violet Induces Apoptosis and Ferroptosis via Modulating P53 and MDM2 in Hepatocellular Carcinoma. Am. J. Cancer Res. 2022, 12, 3357–3372. [Google Scholar]
- Sayé, M.; Gauna, L.; Valera-Vera, E.; Reigada, C.; Miranda, M.R.; Pereira, C.A. Crystal Violet Structural Analogues Identified by in Silico Drug Repositioning Present Anti-Trypanosoma Cruzi Activity through Inhibition of Proline Transporter TcAAAP069. PLoS Negl. Trop. Dis. 2020, 14, e0007481. [Google Scholar] [CrossRef]
- Díaz, H.A.; Castro, K.E.N. Endocrine Immune Interactions in the Host-Parasite Relationship: Steroid Hormones as Immune Regulators in Parasite Infections. J. Steroids Horm. Sci. 2015, 6, 165. [Google Scholar] [CrossRef]
- Morachis, J.M.; Huang, R.; Emerson, B.M. Identification of Kinase Inhibitors That Target Transcription Initiation by RNA Polymerase II. Oncotarget 2011, 2, 18–28. [Google Scholar] [CrossRef]
- Schug, T.T. Targeting Transcription Through Inhibition of TBP. Oncotarget 2011, 2, 5–7. [Google Scholar] [CrossRef]
- Parra-Marín, O.; Rosas-Hernández, L.; López-Pacheco, K.; Franco, B.; Ibáñez-Escribano, A.; Hernández, R.; López-Villaseñor, I. An in Vitro Characterisation of the Trichomonas Vaginalis TATA Box-Binding Proteins (TBPs). Parasitol. Res. 2019, 118, 3019–3031. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A Web Server for Automatic Binding Site Prediction, Analysis and Druggability Assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef]
- Ghersi, D.; Sanchez, R. Improving Accuracy and Efficiency of Blind Protein-ligand Docking by Focusing on Predicted Binding Sites. Proteins Struct. Funct. Bioinform. 2009, 74, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Efficient Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully Automated Protein–Ligand Interaction Profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
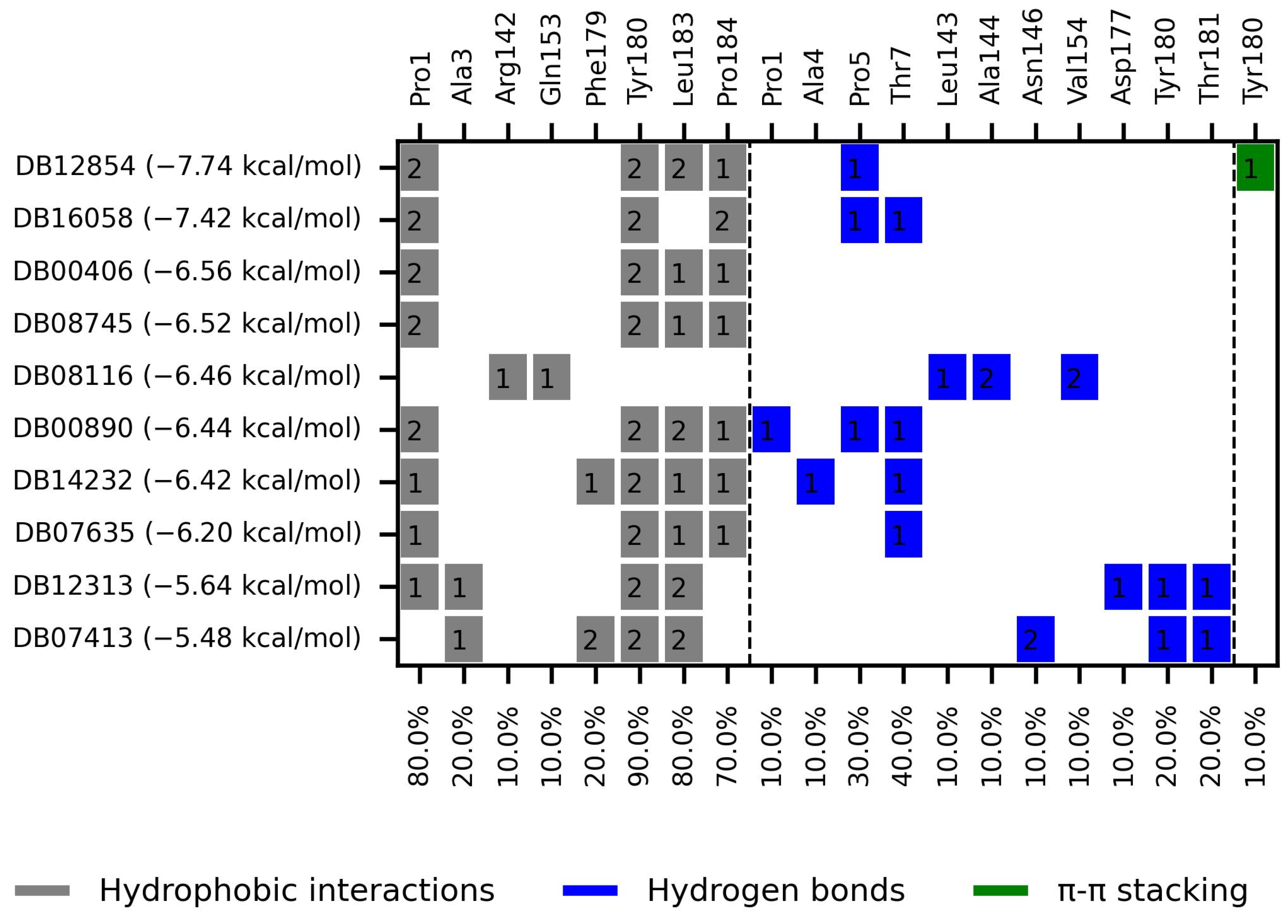
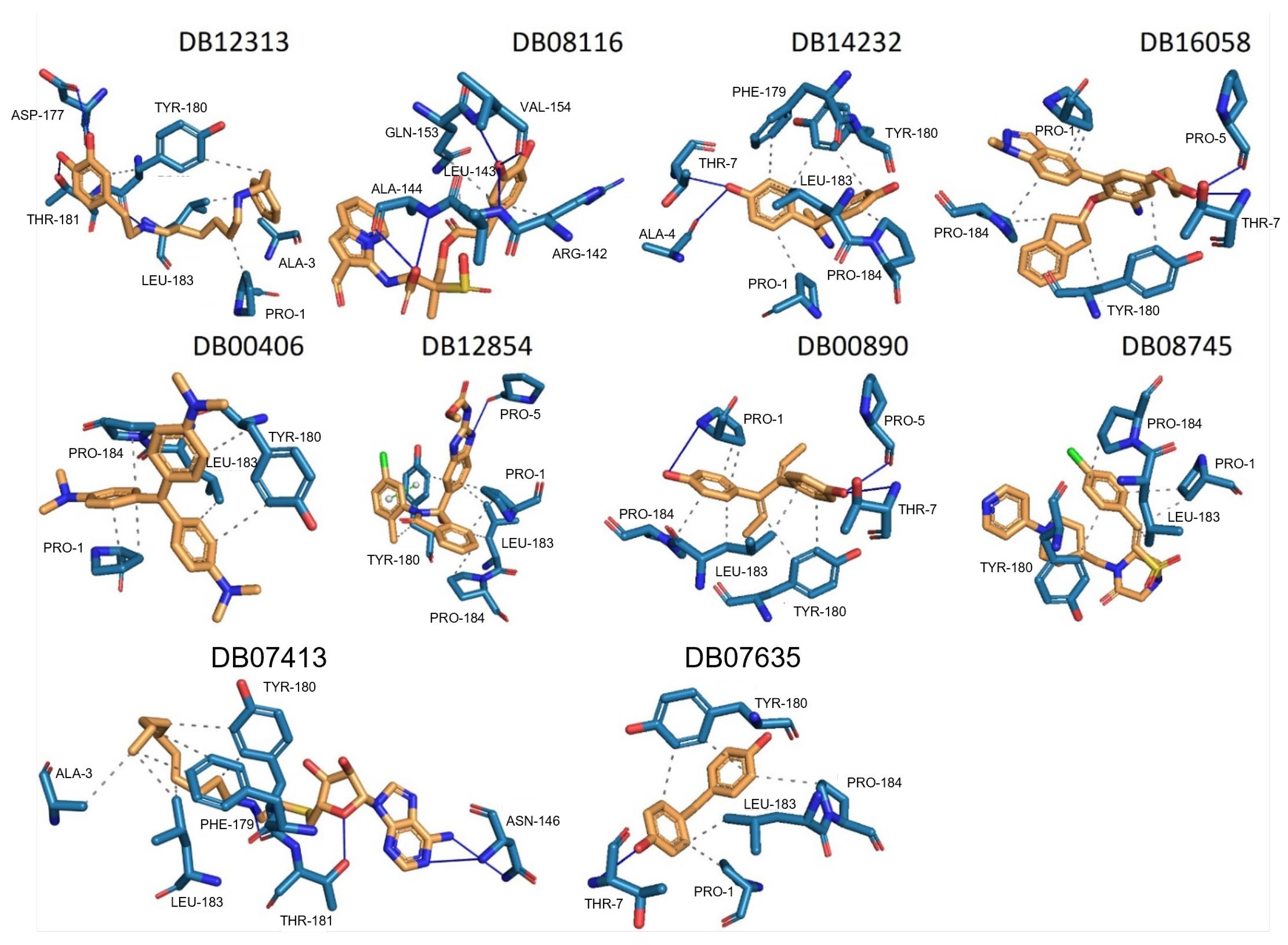
| Compound | Target | Anti-Trypanosoma Effect | Binding Free Energy, Interaction Profile, and Selectivity Index (SI) | Other Functions |
|---|---|---|---|---|
 Dopexamine (DB12313) | Not Available. | Negative | −5.645 kcal/mol. HI. P1, A3, Y180, L183; HB. D177, Y180, T181. SI: −1.367 | Dopexamine has been used in trials studying the diagnostic and treatment of free flap, oral cancer, hypotension, septic shock, and head and neck cancer [35]. |
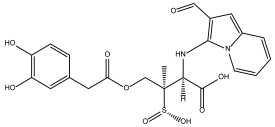 DB08116 | Beta-lactamase SHV-1. | Negative | −6.465 kcal/mol. HI. R142, Q153; HB. L143, A144, V154. SI: −1.242 | Beta-lactamase inhibitor [36]. Offers resistance against a variety of beta-lactam antibiotics. |
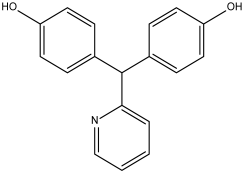 Deacetylbisacodyl (DB14232) | Not Available. | Negative | −6.422 kcal/mol. HI. P1, F179, Y180, L183, P184; HB. A4, T7. SI: −1.086 | Not Available. |
 AK106-001616 (DB16058) | Not Available. | Negative | −7.416 kcal/mol. HI. P1, Y180, P184; HB. P5, T7. SI: −1.083 | AK106-001616 is currently being examined in clinical trial NCT01285752 (An Investigation of AK106-001616 in Individuals Diagnosed with Rheumatoid Arthritis (RA)) [35]. |
 Gentian Violet (DB00406) | DNA. | Positive | −6.561 kcal/mol. HI. P1, Y180, L183, P184. SI: −1.046 | Gentian violet at 250 µg/mL with 2 mg/mL of ascorbic acid and six hours of photoradiation (75 W) sterilized blood samples [37]. Gentian violet has been identified as an acetylcholinesterase inhibitor (AChE) [38]. |
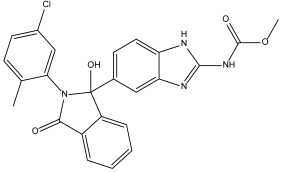 BMS-908662 (DB12854) | Not Available. | Negative | −7.741 kcal/mol. HI. P1, Y180, L183, P184; HB. P5; Ï€-s. Y180. SI: −1.035 | BMS-908662 has been employed in clinical trials investigating its potential for treating melanoma and colorectal cancer [35]. |
 Dienestrol (DB00890) | Not Available. | Positive | −6.444 kcal/mol. HI. P1, Y180, L183, P184; HB. P1, P5, T7. SI: −1.035 | Dienestrol is an estrogenic compound devoid of steroidal properties, utilized for treating atrophic vaginitis and kraurosis vulvae [35]. Some estrogens have been noted to decrease protist parasite infections, including trypanosomes [39,40]. |
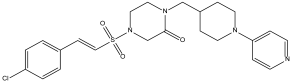 DB08745 | Coagulation factor X. | Negative | −6.521 kcal/mol. HI. P1, Y180, L183, P184. SI: −0.985 | Not available. |
 DB07413 | Hydroxymycolate synthase MmaA4. | Negative | −5.476 kcal/mol. HI. A3, F179, Y180, L183; HB. N146, Y180, T181. SI: −1.035 | S-Adenosylmethionine-dependent methyltransferases (AdoMet-MTs) inhibitor [41]. |
 DB07635 | Lanosterol 14-alpha demethylase. | Positive | −6.201 kcal/mol. HI. P1, Y180, L183, P184; HB. T7. SI: −0.977 | This compound inhibits 14 alpha-demethylase (CYP51), disrupting the production of sterols and impeding the growth of M. tuberculosis within a mouse macrophage model [42]. |
| ID | T. cruzi a IC50 (μM ± SD) NINOA | T. cruzi a IC50 (μM ± SD) A1 | J774.2 b CC50 (μM ± SD) | c SI NINOA | c SI A1 |
|---|---|---|---|---|---|
| DB07635 | >200 | >200 | >200 | ND | ND |
| DB00890 | 70.4094 ± 0.7681 | 37.2594 ± 0.0174 | >200 | 2.84 | 5.36 |
| Nfx | 7.09 ± 0.12 | 19.30 ± 0.08 | 164.20 ± 0.25 | 23.15 | 8.50 |
| Bzn | 30.3 ± 0.03 | 39.08 ± 0.07 | 133.90 ± 0.06 | 4.41 | 3.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaona-López, C.; Méndez-Álvarez, D.; Gonzalez-Gonzalez, A.; Avalos-Navarro, G.; Paz-González, A.D.; Moreno-Rodríguez, A.; Nogueda-Torres, B.; Rivera, G. In Silico Investigation of TATA-Binding Protein as a Therapeutic Target for Chagas Disease: Insights into FDA Drug Repositioning. Pharmaceuticals 2025, 18, 845. https://doi.org/10.3390/ph18060845
Gaona-López C, Méndez-Álvarez D, Gonzalez-Gonzalez A, Avalos-Navarro G, Paz-González AD, Moreno-Rodríguez A, Nogueda-Torres B, Rivera G. In Silico Investigation of TATA-Binding Protein as a Therapeutic Target for Chagas Disease: Insights into FDA Drug Repositioning. Pharmaceuticals. 2025; 18(6):845. https://doi.org/10.3390/ph18060845
Chicago/Turabian StyleGaona-López, Carlos, Domingo Méndez-Álvarez, Alonzo Gonzalez-Gonzalez, Guadalupe Avalos-Navarro, Alma D. Paz-González, Adriana Moreno-Rodríguez, Benjamín Nogueda-Torres, and Gildardo Rivera. 2025. "In Silico Investigation of TATA-Binding Protein as a Therapeutic Target for Chagas Disease: Insights into FDA Drug Repositioning" Pharmaceuticals 18, no. 6: 845. https://doi.org/10.3390/ph18060845
APA StyleGaona-López, C., Méndez-Álvarez, D., Gonzalez-Gonzalez, A., Avalos-Navarro, G., Paz-González, A. D., Moreno-Rodríguez, A., Nogueda-Torres, B., & Rivera, G. (2025). In Silico Investigation of TATA-Binding Protein as a Therapeutic Target for Chagas Disease: Insights into FDA Drug Repositioning. Pharmaceuticals, 18(6), 845. https://doi.org/10.3390/ph18060845