
Performance of Imidazoquinoline Glycoconjugate BAIT628 as a TLR7 Agonist Prodrug for Prostate Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
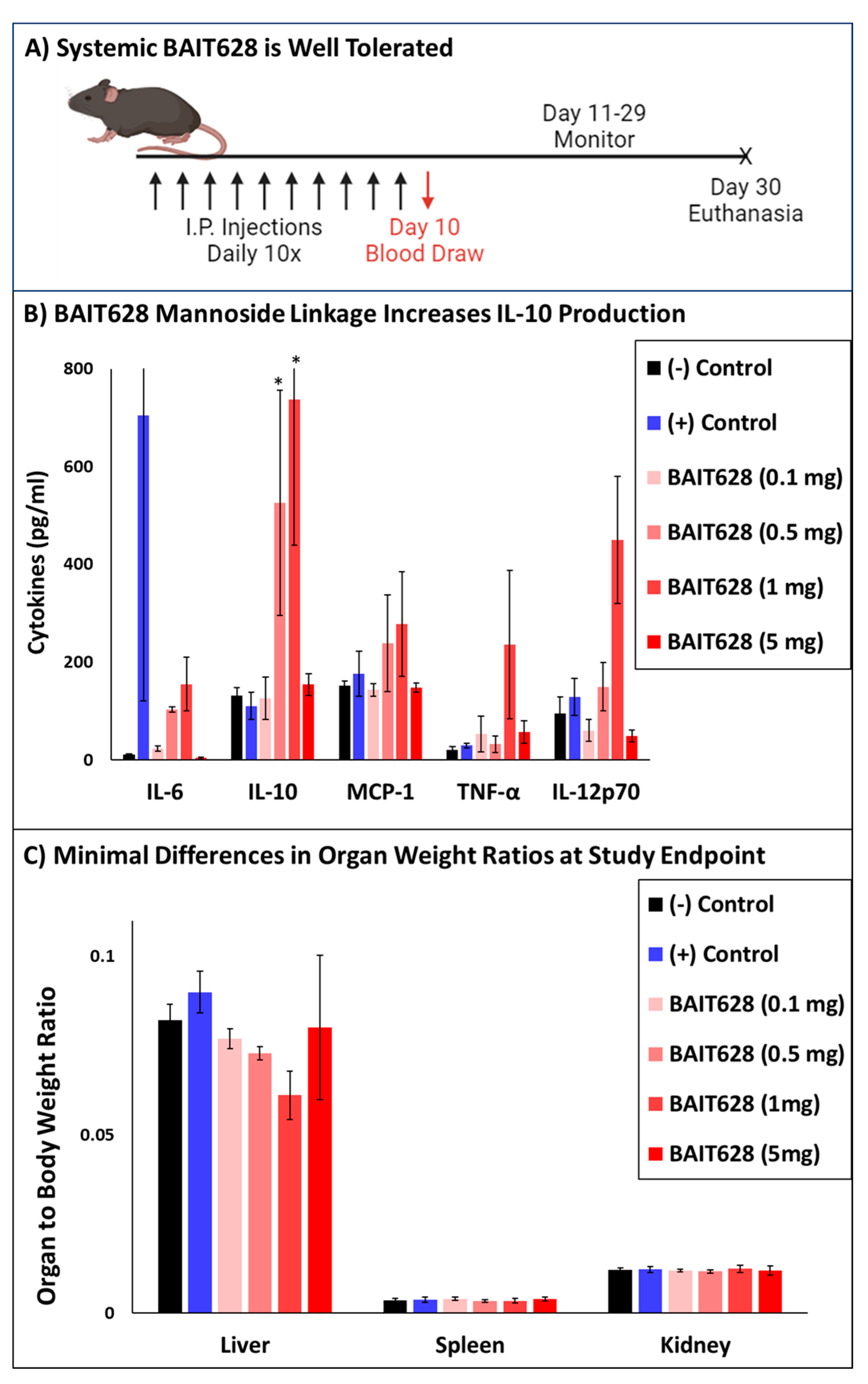
2.1. Systemically Administered BAIT628 Is Better Tolerated than Imiquimod
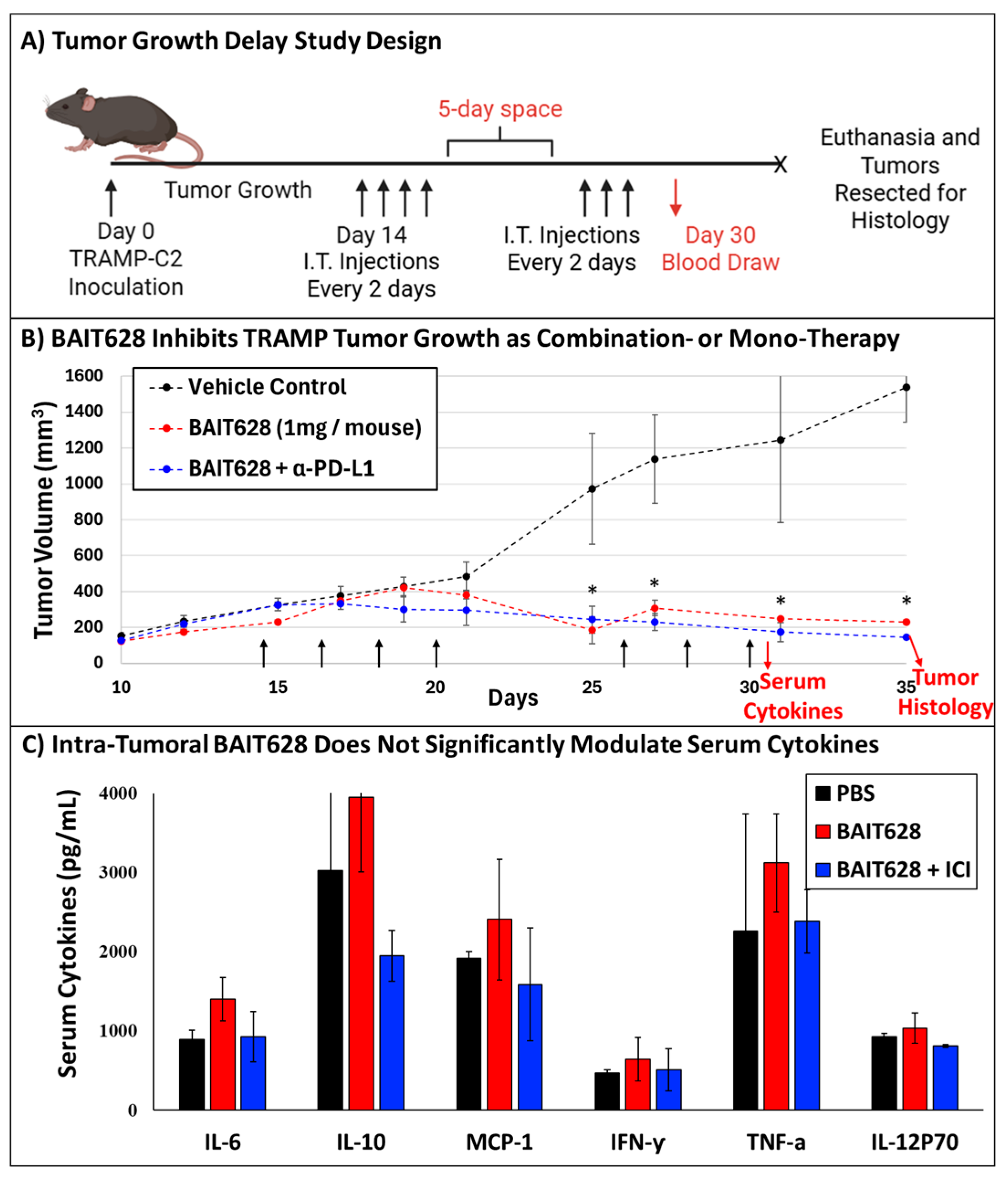
2.2. BAIT628 Significantly Inhibits Tumor Growth and Exhibits Synergy with α-PD-L1 Checkpoint Inhibitor
2.3. Intra-Tumoral BAIT628 Is Well Tolerated and Does Not Modulate Serum Cytokines
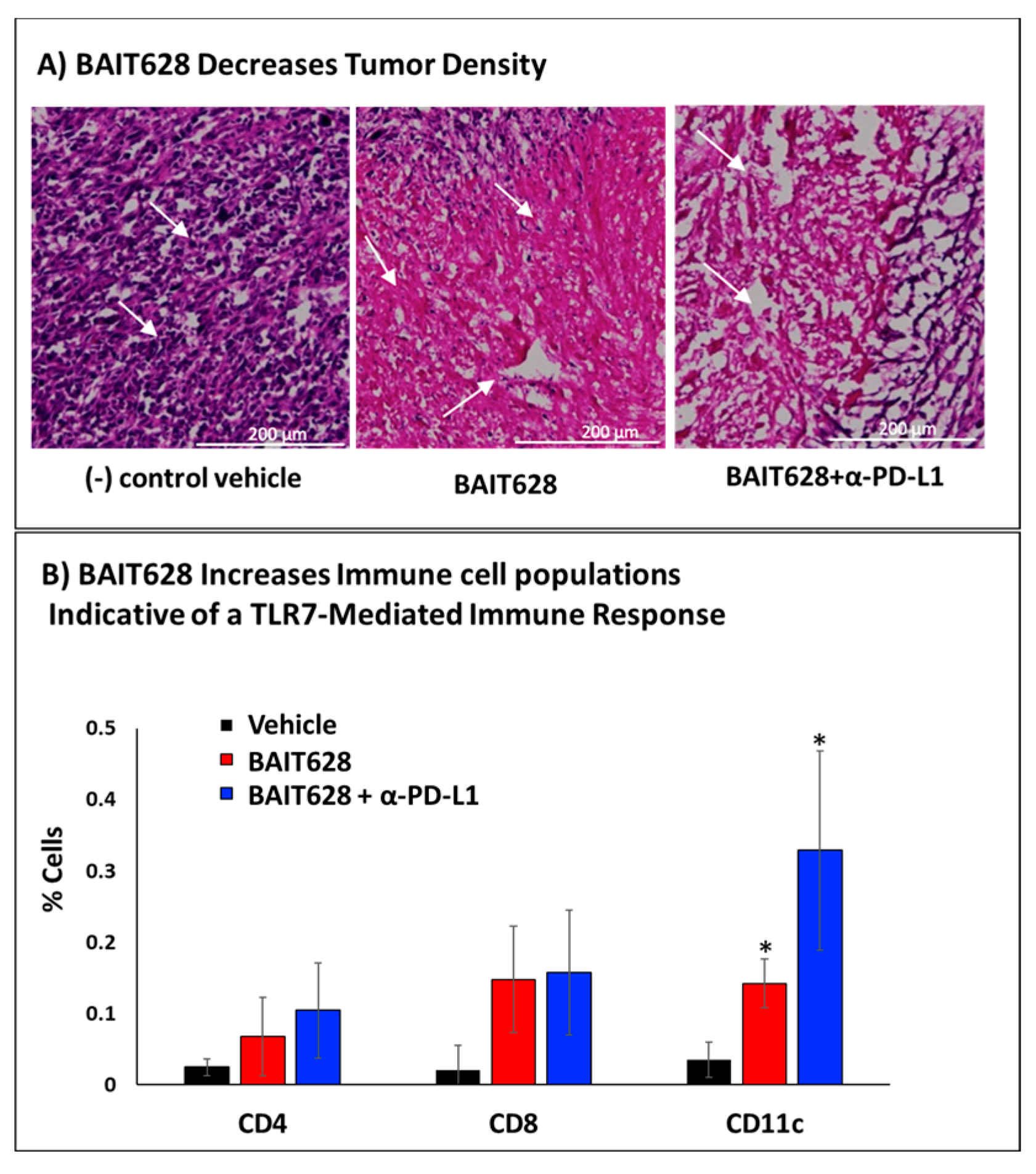
2.4. Histological Examination of Tumor Tissues
2.5. Immunohistochemical Analysis of Tumor-Infiltrating Immune Cells
3. Methods
3.1. Antibodies and Drugs
3.2. Resazurin Assay
3.3. RAW-Blue Cell Culture
3.4. TRAMP-C2 Cell Culture
3.5. Cancer-Mediated Immunogenicity of BAIT628
3.6. Animals
3.7. Safety and Tolerability of Systemic BAIT628
3.8. TRAMP-C2 Tumor Growth and Treatment
3.9. Cytometric Bead Array of Serum Cytokines
3.10. Brightfield Tumor Histology
3.11. Tumor Homogenization for Flow Cytometry
3.12. Tumor Homogenate Immunohistochemistry
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TLR | Toll-like receptor |
| BAIT | Bystander-Assisted ImmunoTherapy |
| MTD | Maximum tolerable dose |
| MDR | Multidrug-resistant |
| TRAMP | Transgenic adenocarcinoma of the mouse prostate |
| DMEM | Dulbecco’s modified Eagle medium |
| PBS | Phosphate-buffered saline |
| ATCC | American Type Culture Collection |
| O.C.T. | Optimal cutting temperature |
| FC | flow cytometry |
| FSC | Forward scatter |
| SSC | Side scatter |
| FSC-A | FSC area |
| FSC-H | FSC height |
| α-man | α-mannosidases |
| IMQ | Imiquimod |
| H&E | Hematoxylin and eosin |
References
- Miller, R.L.; Gerster, J.F.; Owens, M.L.; Slade, H.B.; Tomai, M.A. Review Article Imiquimod applied topically: A novel immune response modifier and new class of drug. Int. J. Immunopharmacol. 1999, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gerster, J.F.; Lindstrom, K.J.; Miller, R.L.; Tomai, M.A.; Birmachu, W.; Bomersine, S.N.; Gibson, S.J.; Imbertson, L.M.; Jacobson, J.R.; Knafla, R.T.; et al. Synthesis and Structure−Activity-Relationships of 1H-Imidazo[4,5-c]quinolines That Induce Interferon Production. J. Med. Chem. 2005, 48, 3481–3491. [Google Scholar] [CrossRef]
- ‘Mac’ Cheever, M.A. Twelve immunotherapy drugs that could cure cancers. Immunol. Rev. 2008, 222, 357–368. [Google Scholar] [CrossRef]
- Bhagchandani, S.; Johnson, J.A.; Irvine, D.J. Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Deliv. Rev. 2021, 175, 113803. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Khong, H.; Dai, Z.; Huang, X.-F.; Wargo, J.A.; Cooper, Z.A.; Vasilakos, J.P.; Hwu, P.; Overwijk, W.W. Effective Innate and Adaptive Antimelanoma Immunity through Localized TLR7/8 Activation. J. Immunol. 2014, 193, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.D.; Antonysamy, M.A.; Tomai, M.A.; Lipson, K.E. Potentiation of the antitumor effects of imidazoquinoline immune response modifiers by cyclophosphamide. Cancer Biol. Ther. 2010, 10, 155–165. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef]
- Kumar, K.; Honda-Okubo, Y.; Sakala, I.G.; Singh, K.N.; Petrovsky, N.; Salunke, D.B. Modulation of the Adjuvant Potential of Imidazoquinoline-Based TLR7/8 Agonists via Alum Adsorption. ACS Med. Chem. Lett. 2024, 15, 1677–1684. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Jiang, M.; Mas-Rosario, J.A.; Fedeli, S.; Cao-Milan, R.; Liu, L.; Winters, K.J.; Hirschbiegel, C.-M.; Nabawy, A.; et al. Polarization of macrophages to an anti-cancer phenotype through in situ uncaging of a TLR 7/8 agonist using bioorthogonal nanozymes. Chem. Sci. 2024, 15, 2486–2494. [Google Scholar] [CrossRef]
- Li, H.; Van Herck, S.; Liu, Y.; Hao, Y.; Ding, X.; Nuhn, L.; Zhong, Z.; Combes, F.; Sanders, N.N.; Lienenklaus, S.; et al. Imidazoquinoline-Conjugated Degradable Coacervate Conjugate for Local Cancer Immunotherapy. ACS Biomater. Sci. Eng. 2020, 6, 4993–5000. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Z.; Ma, F.; Deng, B.; Zhao, Y.; Zuo, X.; Wang, H.; Lu, Y. Nano-adjuvant based on lipo-Imiquimod self-assembly for enhanced foot-and-mouth disease virus vaccine immune responses via intradermal immunization. Mater. Today Bio 2025, 31, 101567. [Google Scholar] [CrossRef]
- Czysch, C.; Medina-Montano, C.; Zhong, Z.; Fuchs, A.; Stickdorn, J.; Winterwerber, P.; Schmitt, S.; Deswarte, K.; Raabe, M.; Scherger, M.; et al. Transient Lymph Node Immune Activation by Hydrolysable Polycarbonate Nanogels. Adv. Funct. Mater. 2022, 32, 2203490. [Google Scholar] [CrossRef]
- Fang, S.; Brems, B.M.; Olawode, E.O.; Miller, J.T.; Brooks, T.A.; Tumey, L.N. Design and Characterization of Immune-Stimulating Imidazo[4,5-c]quinoline Antibody-Drug Conjugates. Mol. Pharm. 2022, 19, 3228–3241. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Li, H.; Ge, X.; Liu, Y.; Yin, J.; Li, X.; Liu, Y.; Chen, H.; Huang, L.; Zou, J.; et al. Site-specific nanoswitch circumventing immune resistance via activating TLR and inhibiting PD-L1/PD-1 axis. J. Control. Release 2023, 361, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, B.; Santra, A.; Tah, D.; Goswami, K.; Jana, A.; Girigoswami, A.; Bose, D. Decoding the interaction of an imidazo-pyrimidine derivative with serum proteins: Spectroscopic, computational and structure-activity relationship analysis. Biophys. Chem. 2025, 322, 107435. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Balabanska, R.; Cottreel, E.; Chen, E.; Duan, D.; Jiang, Q.; Patil, A.; Triyatni, M.; Upmanyu, R.; Zhu, Y.; et al. TLR7 agonist RO7020531 versus placebo in healthy volunteers and patients with chronic hepatitis B virus infection: A randomised, observer-blind, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 2023, 23, 496–507. [Google Scholar] [CrossRef]
- Hantho, J.D.; Strayer, T.A.; Nielsen, A.E.; Mancini, R.J. An Enzyme-Directed Imidazoquinoline for Cancer Immunotherapy. ChemMedChem 2016, 11, 2496–2500. [Google Scholar] [CrossRef]
- Ryan, A.T.; Pulukuri, A.J.; Davaritouchaee, M.; Abbasi, A.; Hendricksen, A.T.; Opp, L.K.; Burt, A.J.; Nielsen, A.E.; Mancini, R.J. Comparing the immunogenicity of glycosidase-directed resiquimod prodrugs mediated by cancer cell metabolism. Acta Pharmacol. Sin. 2020, 41, 995–1004. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Jaiswal, S.; Velarde de la Cruz, E.E.; Thakare, R.P. Targeting Solute Carrier Transporters (SLCs) as a Therapeutic Target in Different Cancers. Diseases 2024, 12, 63. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Song, M.G.; di Magliano, M.P.; Lyssiotis, C.A.; Kim, S.E. Nutrient transporters: Connecting cancer metabolism to therapeutic opportunities. Oncogene 2023, 42, 711–724. [Google Scholar] [CrossRef]
- Domenichini, A.; Adamska, A.; Falasca, M. ABC transporters as cancer drivers: Potential functions in cancer development. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z.; Zhao, Z.; Pang, J.; Chen, J. Recent Progress in the Development of Glucose Transporter (GLUT) Inhibitors. J. Med. Chem. 2025, 68, 1033–1050. [Google Scholar] [CrossRef]
- Pandey, A.; Alcaraz, M.; Saggese, P.; Soto, A.; Gomez, E.; Jaldu, S.; Yanagawa, J.; Scafoglio, C. Exploring the Role of SGLT2 Inhibitors in Cancer: Mechanisms of Action and Therapeutic Opportunities. Cancers 2025, 17, 466. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, G.; Hediger, M.A. Transporter-Mediated Drug Delivery. Molecules 2023, 28, 1151. [Google Scholar] [CrossRef]
- Legigan, T.; Clarhaut, J.; Tranoy-Opalinski, I.; Monvoisin, A.; Renoux, B.; Thomas, M.; Le Pape, A.; Lerondel, S.; Papot, S. The First Generation of β-Galactosidase-Responsive Prodrugs Designed for the Selective Treatment of Solid Tumors in Prodrug Monotherapy. Angew. Chem. Int. Ed. 2012, 51, 11606–11610. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef]
- Pulukuri, A.J.; Burt, A.J.; Opp, L.K.; McDowell, C.M.; Davaritouchaee, M.; Nielsen, A.E.; Mancini, R.J. Acquired Drug Resistance Enhances Imidazoquinoline Efflux by P-Glycoprotein. Pharmaceuticals 2021, 14, 1292. [Google Scholar] [CrossRef]
- Foster, B.A.; Gingrich, J.R.; Kwon, E.D.; Madias, C.; Greenberg, N.M. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997, 57, 3325–3330. [Google Scholar]
- Burt, A.J.; Hantho, J.D.; Nielsen, A.E.; Mancini, R.J. An Enzyme-Directed Imidazoquinoline Activated by Drug Resistance. Biochemistry 2018, 57, 2184–2188. [Google Scholar] [CrossRef]
- He, L.; Fan, C.; Kapoor, A.; Ingram, A.J.; Rybak, A.P.; Austin, R.C.; Dickhout, J.; Cutz, J.-C.; Scholey, J.; Tang, D. α-Mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Nat. Commun. 2011, 2, 307. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.-W.; Davidson, S.; Bhat, G. Markers of malignant prostate cancer cells: Golgi localization of α-mannosidase 1A at GM130-GRASP65 site and appearance of high mannose N-glycans on cell surface. Biochem. Biophys. Res. Commun. 2020, 527, 406–410. [Google Scholar] [CrossRef]
- Gonzalez, P.S.; O’Prey, J.; Cardaci, S.; Barthet, V.J.A.; Sakamaki, J.; Beaumatin, F.; Roseweir, A.; Gay, D.M.; Mackay, G.; Malviya, G.; et al. Mannose impairs tumour growth and enhances chemotherapy. Nature 2018, 563, 719–723. [Google Scholar] [CrossRef]
- Nan, F.; Sun, Y.; Liang, H.; Zhou, J.; Ma, X.; Zhang, D. Mannose: A Sweet Option in the Treatment of Cancer and Inflammation. Front. Pharmacol. 2022, 13, 877543. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, L.; McColl, A.; Graham, G.; Cavanagh, J. The Systemic Response to Topical Aldara Treatment is Mediated Through Direct TLR7 Stimulation as Imiquimod Enters the Circulation. Sci. Rep. 2017, 7, 16570. [Google Scholar] [CrossRef] [PubMed]
- Kido, L.A.; De Almeida Lamas, C.; Maróstica, M.R.; Cagnon, V.H.A. Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model: A good alternative to study PCa progression and chemoprevention approaches. Life Sci. 2019, 217, 141–147. [Google Scholar] [CrossRef]
- Hurwitz, A.A.; Foster, B.A.; Allison, J.P.; Greenberg, N.M.; Kwon, E.D. The TRAMP Mouse as a Model for Prostate Cancer. Curr. Protoc. Immunol. 2001, 45, 20.5.1–20.5.23. [Google Scholar] [CrossRef]
- Han, J.-H.; Lee, J.; Jeon, S.-J.; Choi, E.-S.; Cho, S.-D.; Kim, B.-Y.; Kim, D.-J.; Park, J.-H.; Park, J.-H. In vitro and in vivo growth inhibition of prostate cancer by the small molecule Imiquimod. Int. J. Oncol. 2013, 42, 2087–2093. [Google Scholar] [CrossRef]
- Karwacki, J.; Kiełbik, A.; Szlasa, W.; Sauer, N.; Kowalczyk, K.; Krajewski, W.; Saczko, J.; Kulbacka, J.; Szydełko, T.; Małkiewicz, B. Boosting the Immune Response—Combining Local and Immune Therapy for Prostate Cancer Treatment. Cells 2022, 11, 2793. [Google Scholar] [CrossRef]
- Demaria, O.; Pagni, P.P.; Traub, S.; de Gassart, A.; Branzk, N.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Flavell, R.A.; Alexopoulou, L. TLR8 deficiency leads to autoimmunity in mice. J. Clin. Investig. 2010, 120, 3651–3662. [Google Scholar] [CrossRef]
- Desnues, B.; Macedo, A.B.; Roussel-Queval, A.; Bonnardel, J.; Henri, S.; Demaria, O.; Alexopoulou, L. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc. Natl. Acad. Sci. USA 2014, 111, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, J.R.; Barrios, R.J.; Kattan, M.W.; Nahm, H.S.; Finegold, M.J.; Greenberg, N.M. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997, 57, 4687–4691. [Google Scholar] [PubMed]
- Jeet, V.; Ow, K.; Doherty, E.; Curley, B.; Russell, P.J.; Khatri, A. Broadening of transgenic adenocarcinoma of the mouse prostate (TRAMP) model to represent late stage androgen depletion independent cancer. Prostate 2008, 68, 548–562. [Google Scholar] [CrossRef]
- Bonorden, M.J.L.; Grossmann, M.E.; Ewing, S.A.; Rogozina, O.P.; Ray, A.; Nkhata, K.J.; Liao, D.J.; Grande, J.P.; Cleary, M.P. Growth and Progression of TRAMP Prostate Tumors in Relationship to Diet and Obesity. Prostate Cancer 2012, 2012, 543970. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, K.A.; Norgard, M.A.; Levasseur, P.R.; Olson, B.; Burfeind, K.G.; Buenafe, A.C.; Zhu, X.; Jeng, S.; McWeeney, S.K.; Marks, D.L. Persistent Toll-like receptor 7 stimulation induces behavioral and molecular innate immune tolerance. Brain Behav. Immun. 2019, 82, 338–353. [Google Scholar] [CrossRef]
- Drobits, B.; Holcmann, M.; Amberg, N.; Swiecki, M.; Grundtner, R.; Hammer, M.; Colonna, M.; Sibilia, M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Investig. 2012, 122, 575–585. [Google Scholar] [CrossRef]
- Valdman, A.; Jaraj, S.J.; Compérat, E.; Charlotte, F.; Roupret, M.; Pisa, P.; Egevad, L. Distribution of Foxp3-, CD4- and CD8-positive lymphocytic cells in benign and malignant prostate tissue. APMIS 2010, 118, 360–365. [Google Scholar] [CrossRef]
- Godino, J.; Schuhmacher, A.J. Determination and Isolation of Immune Populations from Brain Tumor Microenvironments. Methods Mol. Biol. 2019, 1884, 177–188. [Google Scholar] [CrossRef]
- Ingelshed, K.; Melssen, M.M.; Spiegelberg, D. Protocol for in vivo immune cell analysis in subcutaneous murine tumor models using advanced flow cytometry. STAR Protoc. 2025, 6, 103505. [Google Scholar] [CrossRef]
- Balbaied, T.; Moore, E. Resazurin-Based Assay for Quantifying Living Cells during Alkaline Phosphatase (ALP) Release. Appl. Sci. 2020, 10, 3840. [Google Scholar] [CrossRef]
- Richardson, C.A. The power of automated behavioural homecage technologies in characterizing disease progression in laboratory mice: A review. Appl. Anim. Behav. Sci. 2015, 163, 19–27. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef] [PubMed]
- BD Biosciences Mouse Inflammation CBA. Available online: https://www.bdj.co.jp/pdf/64-01-8100055-552364-A.pdf (accessed on 7 March 2025).
- Day, C.E. (Ed.) Histopathology: Methods and Protocols; Humana: New York, NY, USA, 2014; Volume 1180. [Google Scholar] [CrossRef]
- Almeida, A.; Fein, M.; Egeblad, M. Multi-color Flow Cytometry for Comprehensive Analysis of the Tumor Immune Infiltrate in a Murine Model of Breast Cancer. BIO-Protoc. 2021, 11, e4012. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Chang, H.; Radbruch, A.; Akdis, M.; Andrä, I.; Annunziato, F.; Bacher, P.; Barnaba, V.; Battistini, L.; Bauer, W.M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies*. Eur. J. Immunol. 2017, 47, 1584–1797. [Google Scholar] [CrossRef]
- Haroon, M.; Sultana, S.; Najibi, S.A.; Wang, E.T.; Michaelson, A.; Al Muied, P.S.M.; Nielsen, A.E.; Mancini, R.J. Efflux-Enhanced Imidazoquinolines To Exploit Chemoresistance. ACS Omega 2025, 10, 12319–12333. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najibi, S.A.; Pranto, S.M.A.M.; Haroon, M.; Nielsen, A.E.; Mancini, R.J. Performance of Imidazoquinoline Glycoconjugate BAIT628 as a TLR7 Agonist Prodrug for Prostate Cancer. Pharmaceuticals 2025, 18, 804. https://doi.org/10.3390/ph18060804
Najibi SA, Pranto SMAM, Haroon M, Nielsen AE, Mancini RJ. Performance of Imidazoquinoline Glycoconjugate BAIT628 as a TLR7 Agonist Prodrug for Prostate Cancer. Pharmaceuticals. 2025; 18(6):804. https://doi.org/10.3390/ph18060804
Chicago/Turabian StyleNajibi, Seyedeh A., S. M. Al Muied Pranto, Muhammad Haroon, Amy E. Nielsen, and Rock J. Mancini. 2025. "Performance of Imidazoquinoline Glycoconjugate BAIT628 as a TLR7 Agonist Prodrug for Prostate Cancer" Pharmaceuticals 18, no. 6: 804. https://doi.org/10.3390/ph18060804
APA StyleNajibi, S. A., Pranto, S. M. A. M., Haroon, M., Nielsen, A. E., & Mancini, R. J. (2025). Performance of Imidazoquinoline Glycoconjugate BAIT628 as a TLR7 Agonist Prodrug for Prostate Cancer. Pharmaceuticals, 18(6), 804. https://doi.org/10.3390/ph18060804