Investigating the Effect and Mechanism of 3-Methyladenine Against Diabetic Encephalopathy by Network Pharmacology, Molecular Docking, and Experimental Validation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
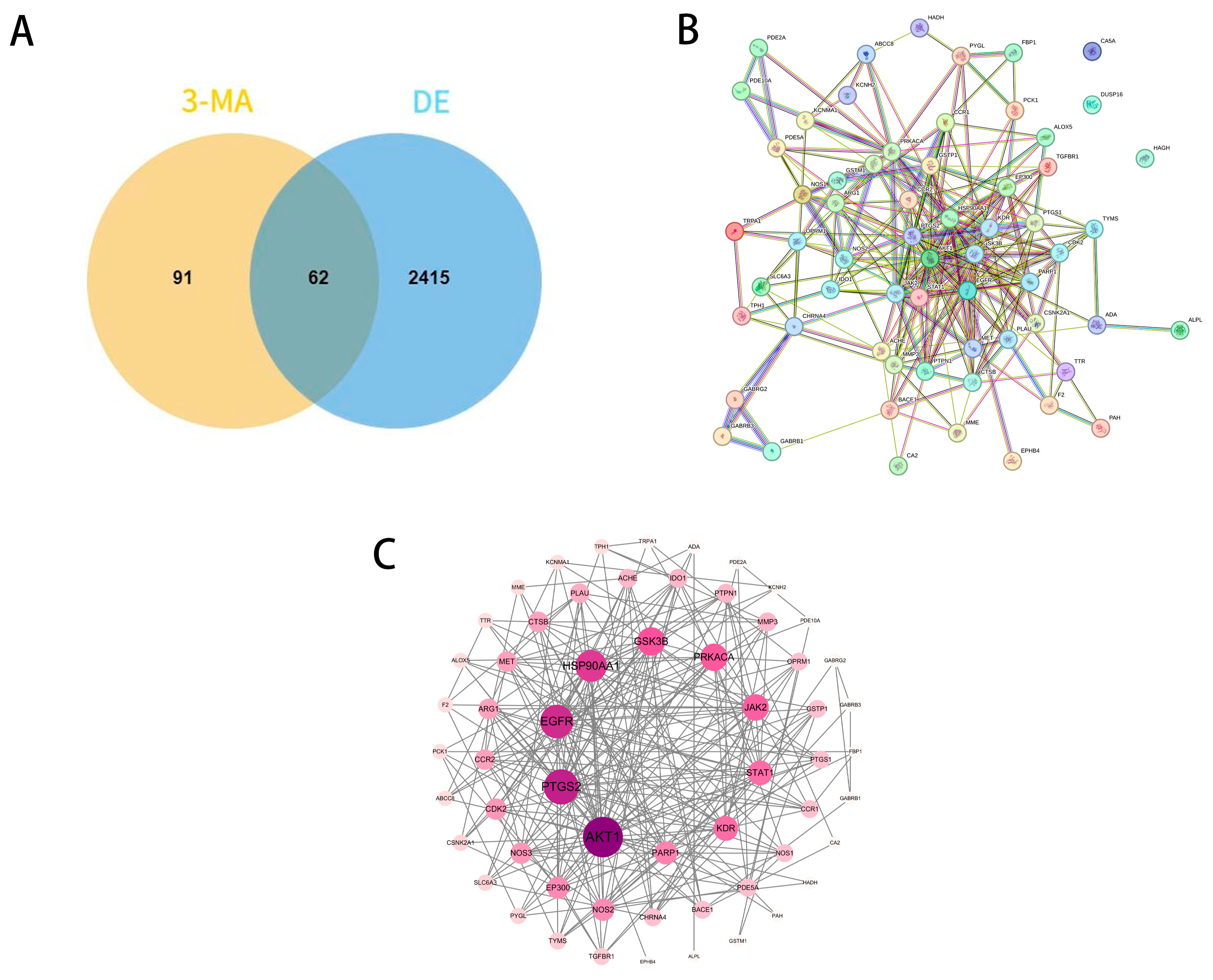
2.1. Collection of 3-MA Targets and DE Targets
2.2. PPI Network Analysis and Core Target Screening
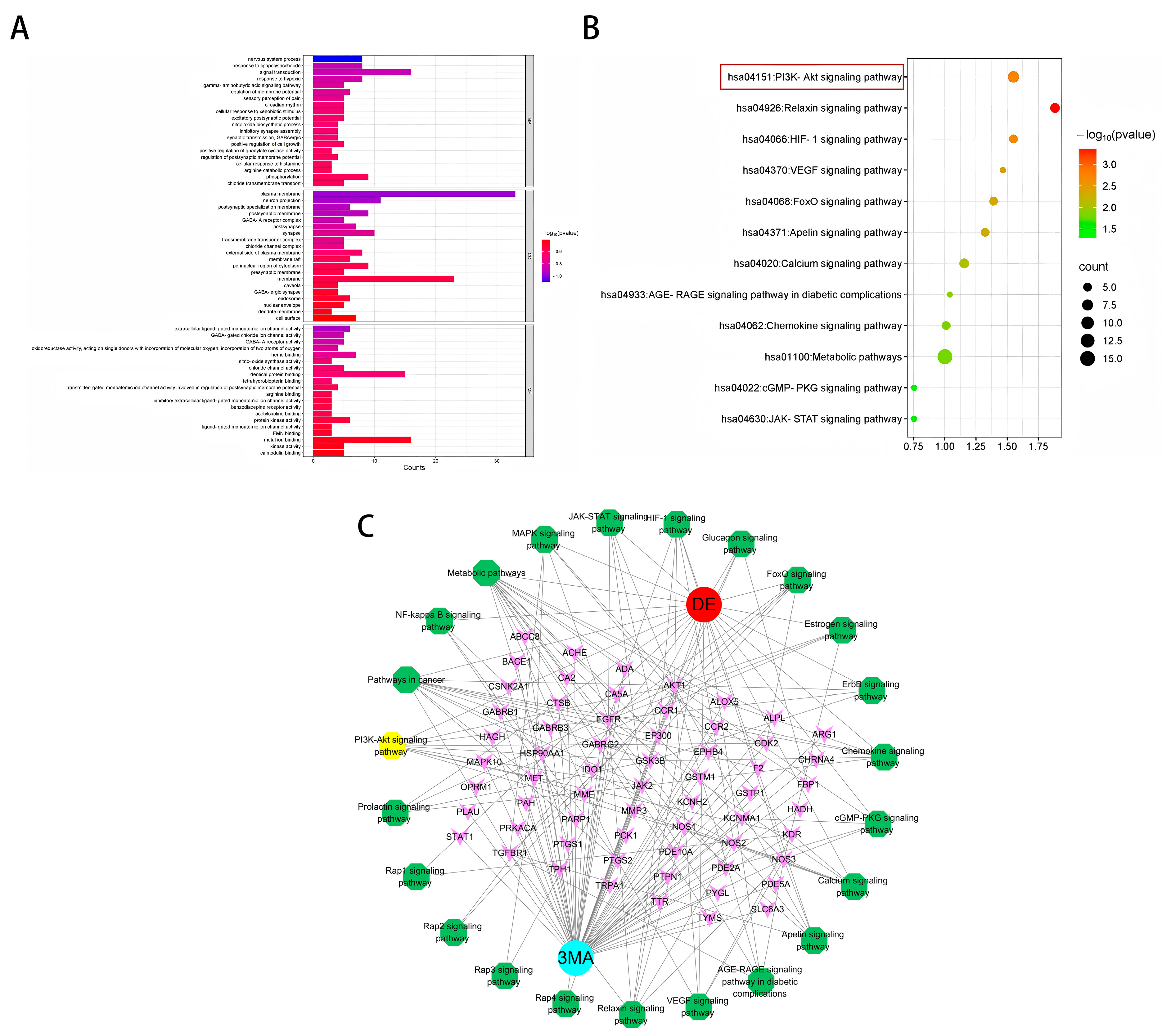
2.3. GO and KEGG Enrichment Analysis
2.4. Construction of the Target–Disease Network
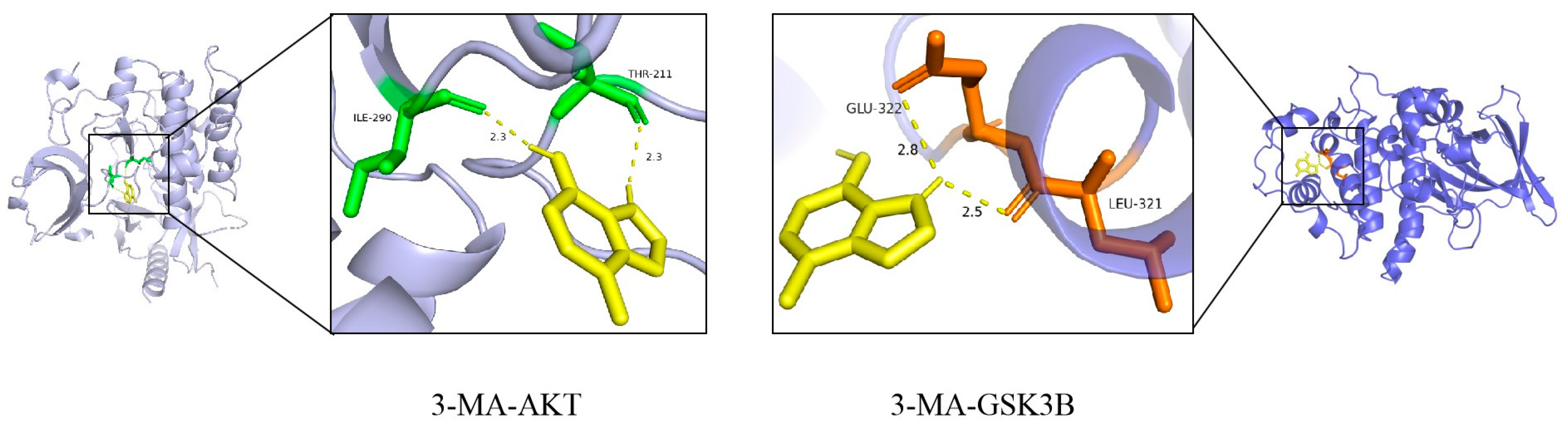
2.5. Predicting Active Compounds of 3-MA Through Molecular Docking
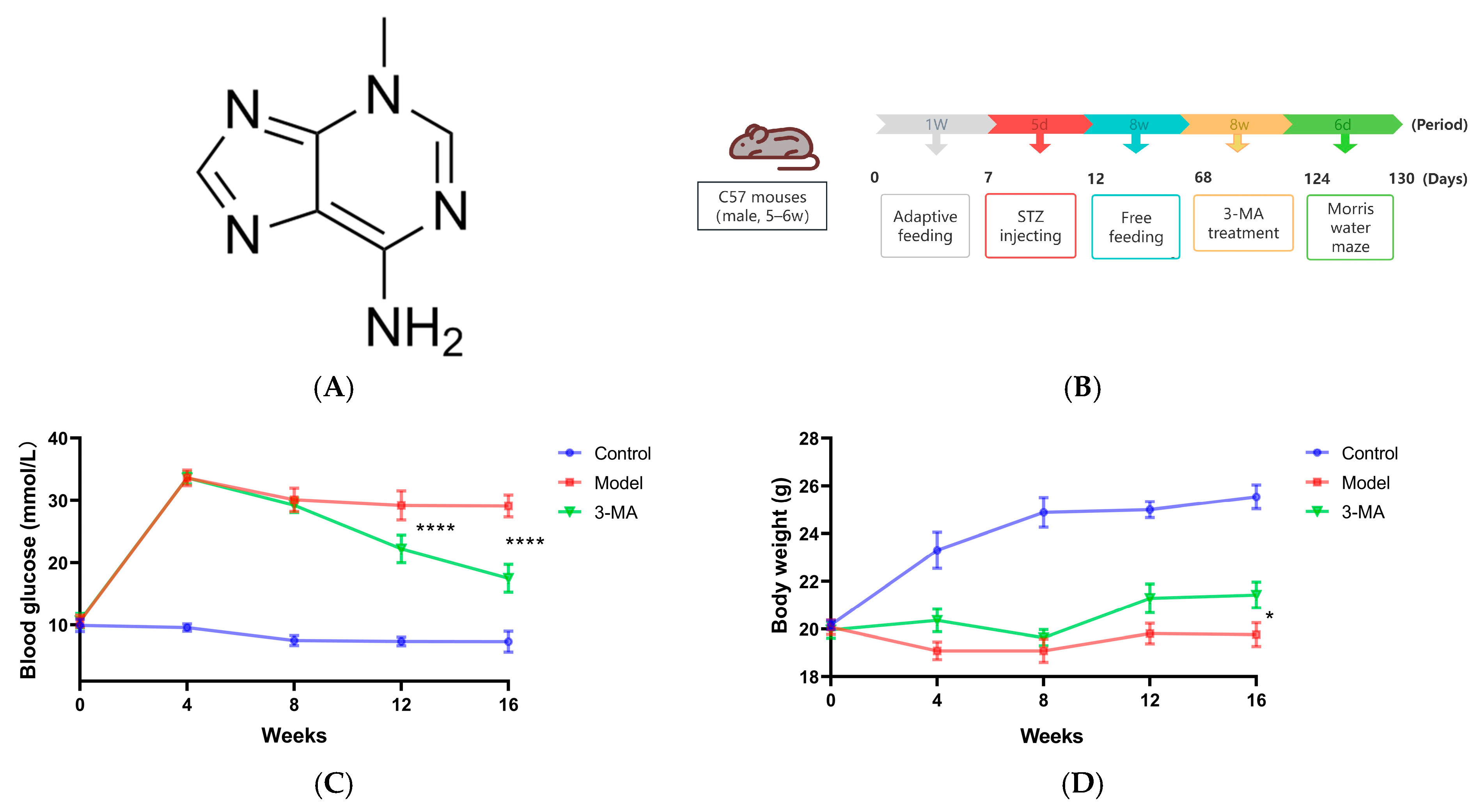
2.6. Effects of 3-MA on RBG and Body Weight
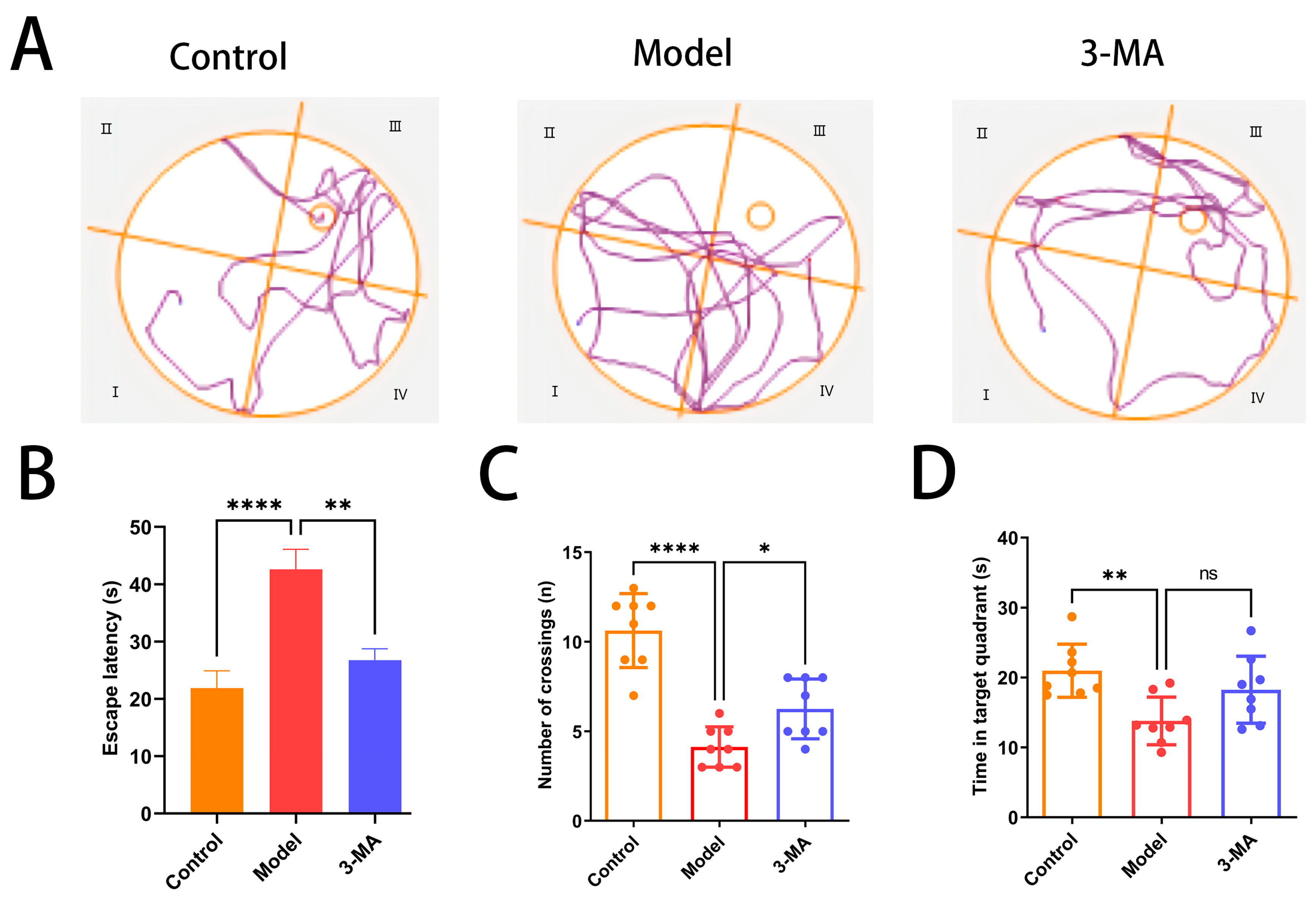
2.7. Effects of 3-MA on Learning and Memory Behavior
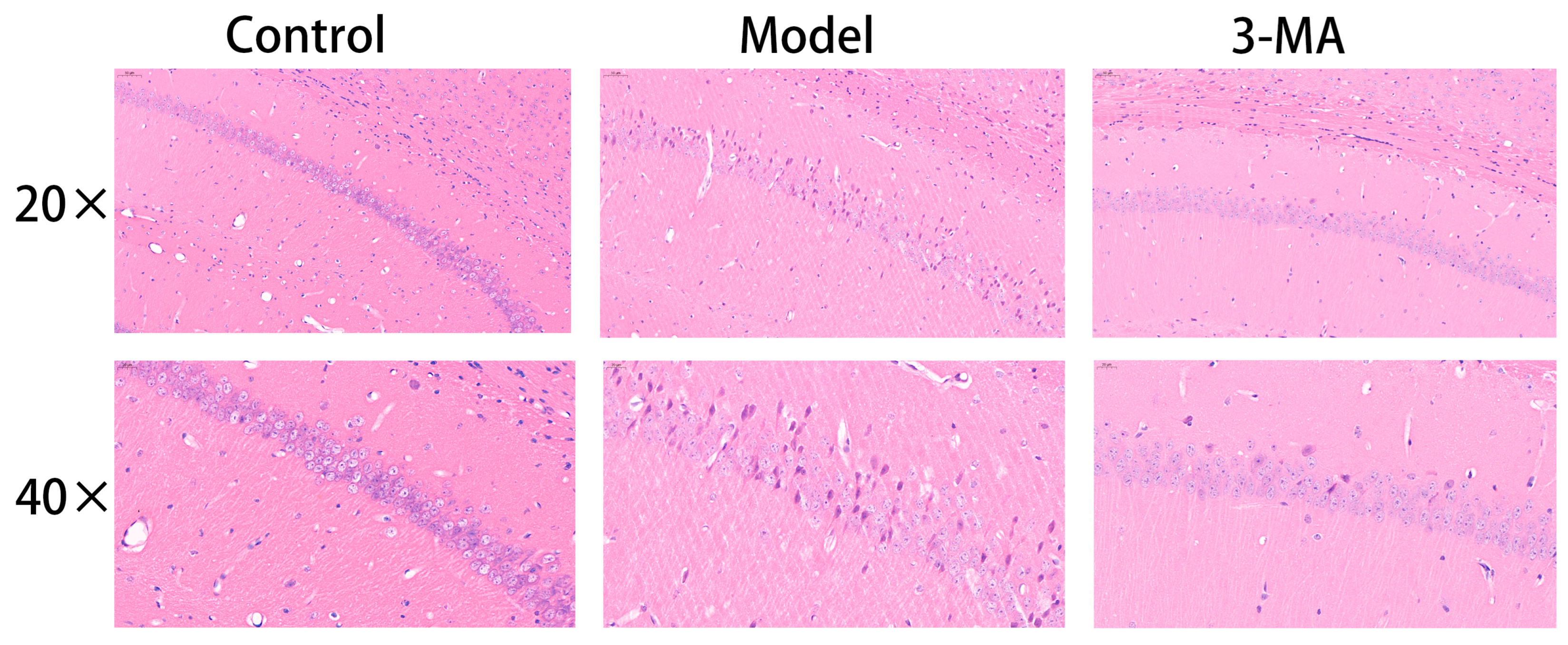
2.8. Effects of 3-MA on the Hippocampus Histopathology
2.9. Effect of 3-MA on the Expressions of APP and Tau Protein in Hippocampus
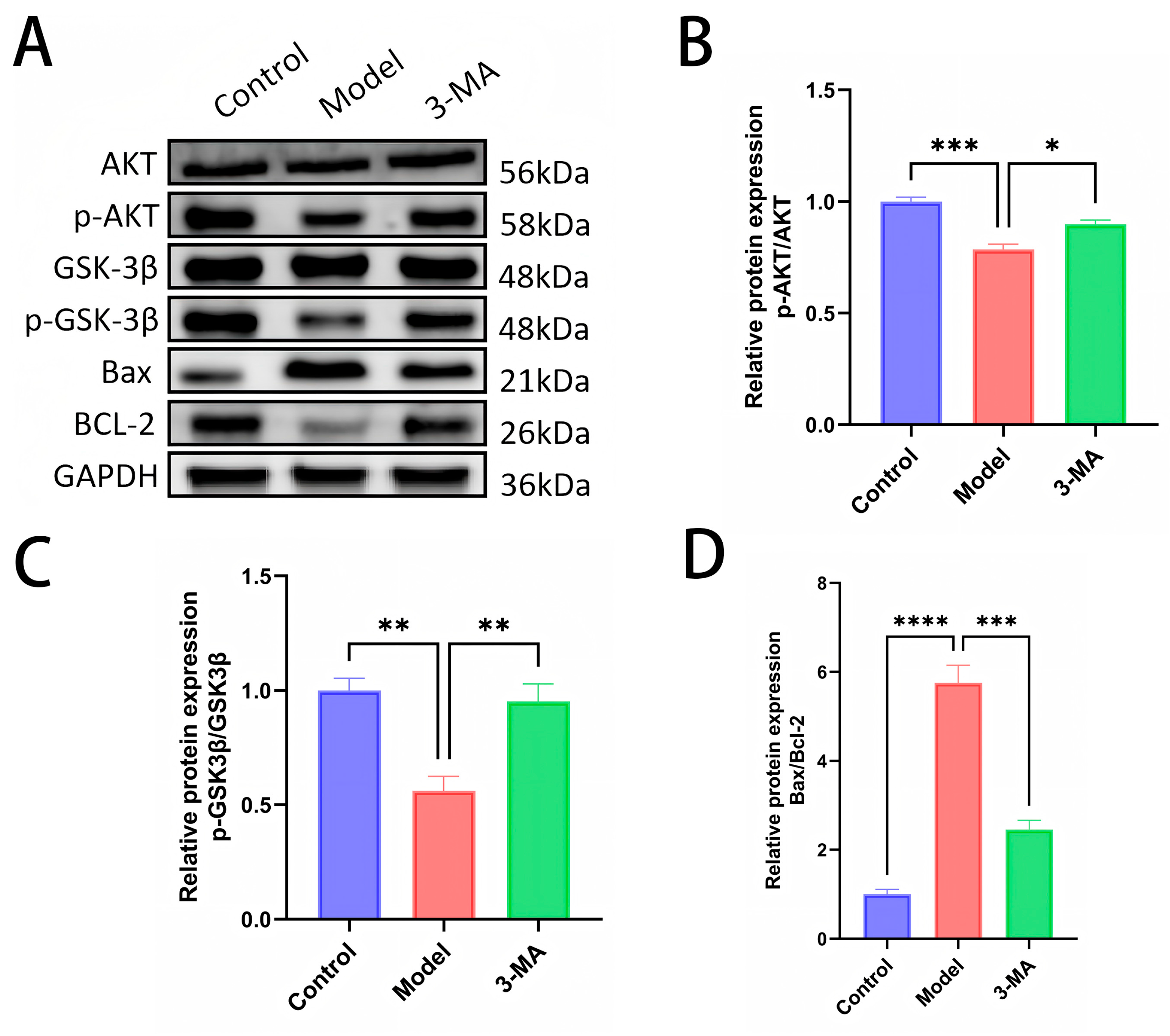
2.10. Western Blot Analysis of Brain Tissues
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. 3-MA Target Prediction
4.3. Search for DE-Related Genes
4.4. Construction of Protein–Protein Interaction (PPI) Network and SeIection of Key Targets
4.5. Cytoscape Gene OntoIogy (GO) and Kyoto EncycIopedia of Gene and Genomes (KEGG) Enrichment AnaIysis
4.6. Constructing the “Drug–Disease–Target–Pathway” Network
4.7. MoIecuIar Docking Verification
4.8. Animals
4.9. Establishment of DE Model and Treatment of 3-MA
4.10. Random Blood Glucose and Body Weight Measurement
4.11. Morris Water Maze
4.12. H/E Staining Analysis
4.13. IHC Analysis
4.14. Western Blot Analysis
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biessels, G.J.; Whitmer, R.A. Cognitive dysfunction in diabetes: How to implement emerging guidelines. Diabetologia 2020, 63, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Li, Y.; He, S.; Huang, N.; Du, M.; Zhai, Q.; Pu, K.; Wu, M.; Yan, C.; Ma, Z.; et al. Reprogramming astrocytic NDRG2/NF-κB/C3 signaling restores the diabetes-associated cognitive dysfunction. EBioMedicine 2023, 93, 104653. [Google Scholar] [CrossRef] [PubMed]
- Dolatshahi, M.; Sanjari Moghaddam, H.; Saberi, P.; Mohammadi, S.; Aarabi, M.H. Central nervous system microstructural alterations in Type 1 diabetes mellitus: A systematic review of diffusion Tensor imaging studies. Diabetes Res. Clin. Pract. 2023, 205, 110645. [Google Scholar] [CrossRef] [PubMed]
- Haut, F.; Argyrousi, E.K.; Arancio, O. Re-Arranging the Puzzle between the Amyloid-Beta and Tau Pathology: An APP-Centric Approach. Int. J. Mol. Sci. 2023, 25, 259. [Google Scholar] [CrossRef]
- Chen, J.; Fan, A.; Li, S.; Xiao, Y.; Fu, Y.; Chen, J.S.; Zi, D.; Zeng, L.H.; Tan, J. APP mediates tau uptake and its overexpression leads to the exacerbated tau pathology. Cell. Mol. Life Sci. 2023, 80, 123. [Google Scholar] [CrossRef]
- Ehtewish, H.; Arredouani, A.; El-Agnaf, O. Diagnostic, Prognostic, and Mechanistic Biomarkers of Diabetes Mellitus-Associated Cognitive Decline. Int. J. Mol. Sci. 2022, 23, 6144. [Google Scholar] [CrossRef]
- Cheng, L.; Chen, Y.; Guo, D.; Zhong, Y.; Li, W.; Lin, Y.; Miao, Y. mTOR-dependent TFEB activation and TFEB overexpression enhance autophagy-lysosome pathway and ameliorate Alzheimer’s disease-like pathology in diabetic encephalopathy. Cell Commun. Signal. 2023, 21, 91. [Google Scholar] [CrossRef]
- Eid, S.A.; Rumora, A.E.; Beirowski, B.; Bennett, D.L.; Hur, J.; Savelieff, M.G.; Feldman, E.L. New perspectives in diabetic neuropathy. Neuron. 2023, 111, 2623–2641. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, W.; Liu, J.; Xie, M.; Liu, Q.; Li, S. Vascular complications of diabetes: A narrative review. Medicine 2023, 102, e35285. [Google Scholar] [CrossRef]
- Zhao, L.; Hu, H.; Zhang, L.; Liu, Z.; Huang, Y.; Liu, Q.; Jin, L.; Zhu, M.; Zhang, L. Inflammation in diabetes complications: Molecular mechanisms and therapeutic interventions. MedComm 2024, 5, e516. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, J.; Wang, J.; Zhu, L.; Wang, J.; Zhang, Q. DPP-4 Inhibitors Suppress Tau Phosphorylation and Promote Neuron Autophagy through the AMPK/mTOR Pathway to Ameliorate Cognitive Dysfunction in Diabetic Mellitus. ACS Chem. Neurosci. 2023, 14, 3335–3346. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.J.; Singh, S.; Seksaria, S.; Das Gupta, G.; Singh, A. Inside the diabetic brain: Insulin resistance and molecular mechanism associated with cognitive impairment and its possible therapeutic strategies. Pharmacol. Res. 2022, 182, 106358. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Hu, H.; Guo, R.; Wang, H.; Jiang, H. Mesenchymal Stem Cell Exosomes as a New Strategy for the Treatment of Diabetes Complications. Front. Endocrinol. 2021, 12, 646233. [Google Scholar] [CrossRef] [PubMed]
- Dhas, Y.; Biswas, N.; Divyalakshmi, M.R.; Jones, L.D.; Ashili, S. Repurposing metabolic regulators: Antidiabetic drugs as anticancer agents. Mol. Biomed. 2024, 5, 40. [Google Scholar] [CrossRef]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef]
- Lv, W.; Wang, X.; Xu, Q.; Lu, W. Mechanisms and Characteristics of Sulfonylureas and Glinides. Curr. Top. Med. Chem. 2020, 20, 37–56. [Google Scholar] [CrossRef]
- Fei, Z.; Xu, Y.; Zhang, G.; Liu, Y.; Li, H.; Chen, L. Natural products with potential hypoglycemic activity in T2DM: 2019-2023. Phytochemistry 2024, 223, 114130. [Google Scholar] [CrossRef]
- Zhu, T.; Yao, Y.; Ding, J.; Zhang, C.; Xia, N.; Tao, Y.; Zhang, W.; Qi, H.; Gong, L.; Jiang, P. 3-Methyladenine attenuates neuroinflammation and improves cognitive function in sepsis-associated encephalopathy by inhibiting autophagy. Int. Immunopharmacol. 2024, 139, 112744. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.H.; Nakamoto, Y. Autophagy and Tau Protein. Int. J. Mol. Sci. 2021, 22, 7475. [Google Scholar] [CrossRef]
- Feng, H.; Cui, Y.; Liu, J.; Liu, M.; Zhou, W.; Yan, Z.; Zhang, H.; Wang, Y.; Wang, X.; Liu, X.; et al. Effects of 3-Methyladenine on Microglia Autophagy and Neuronal Apoptosis After Radiation-Induced Brain Injury. Dose Response 2022, 20, 15593258221100593. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.W.; Yu, W.; Wang, Y.N.; Zhang, X.Y.; Song, S.Q.; Gong, S.Y.; Meng, L.Y.; Gan, C.; Liu, B.J.; Gong, Q. Effects of autophagy inhibitor 3-methyladenine on a diabetic mice model. Int. J. Ophthalmol. 2023, 16, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.T.; Han, S.S.; Xu, X.M.; Sun, H.J.; Zhou, H.; Shang, K.; Liu, Z.H.; Liang, S.J. 3-Methyladenine ameliorates surgery-induced anxiety-like behaviors in aged mice by inhibiting autophagy-induced excessive oxidative stress. Metab. Brain Dis. 2023, 38, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Liu, X.; Yu, J.; Li, K.; Xia, L.; Su, S.; Lin, P.; Zhang, D.; Li, Y. 3-methyladenine ameliorates acute lung injury by inhibiting oxidative damage and apoptosis. Heliyon 2024, 10, e33996. [Google Scholar] [CrossRef]
- Kwon, Y.; Haam, C.E.; Byeon, S.; Choi, S.K.; Lee, Y.H. Effects of 3-methyladenine, an autophagy inhibitor, on the elevated blood pressure and arterial dysfunction of angiotensin II-induced hypertensive mice. Biomed. Pharmacother. 2022, 154, 113588. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, T.; Li, S.; Yang, C.; Zheng, X.; Mao, S.; Liu, N.; Mo, S.; Li, D.; Yang, M.; et al. Inhibition of autophagy attenuates cognitive decline and mitochondrial dysfunction in an Alzheimer’s disease mouse model with chronic cerebral hypoperfusion. Brain Res. 2024, 1850, 149416. [Google Scholar] [CrossRef]
- Luo, A.; Xie, Z.; Wang, Y.; Wang, X.; Li, S.; Yan, J.; Zhan, G.; Zhou, Z.; Zhao, Y.; Li, S. Type 2 diabetes mellitus-associated cognitive dysfunction: Advances in potential mechanisms and therapies. Neurosci. Biobehav. Rev. 2022, 137, 104642. [Google Scholar] [CrossRef]
- Jung, J.Y.; Choi, H.; Son, E.D.; Kim, H.J. 3-Methyladenine Inhibits Procollagen-1 and Fibronectin Expression in Dermal Fibroblasts Independent of Autophagy. Curr. Mol. Med. 2020, 20, 741–750. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Y.; Ren, H.; Ou, L.; Deng, X.; Huang, M.; Wu, X.; Gong, Q. 3-Methyladenine alleviates early renal injury in diabetic mice by inhibiting AKT signaling. Nan Fang Yi Ke Da Xue Xue Bao 2024, 44, 1236–1242. (In Chinese) [Google Scholar] [CrossRef]
- Hou, Q.; Yuan, J.; Li, S.; Ma, J.; Li, W.; Zhang, B.; Zhao, X.; Zhang, F.; Ma, Y.; Zheng, H.; et al. Autophagic degradation of DHCR7 activates AKT3 and promotes sevoflurane-induced hippocampal neuroinflammation in neonatal mice. Free Radic. Biol. Med. 2024, 222, 304–316. [Google Scholar] [CrossRef]
- Luo, Z.; Wan, Q.; Han, Y.; Li, Z.; Li, B. CAPE-pNO2 ameliorates diabetic brain injury through modulating Alzheimer’s disease key proteins, oxidation, inflammation and autophagy via a Nrf2-dependent pathway. Life Sci. 2021, 287, 119929. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-P.; Zhong, X.-Q.; Luo, Q.; Zhang, Q.-X.; Deng, M.-Z. Autophagic activity of piperine on small intestine in dementia model mice with Parkinson’s disease. Zhongguo Zhong Yao Za Zhi 2020, 45, 5238–5247. (In Chinese) [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, L. Ghrelin inhibits NLRP3 inflammasome activation by upregulating autophagy to improve Alzheimer’s disease. In Vitro Cell. Dev. Biol. Anim. 2023, 59, 665–673. [Google Scholar] [CrossRef]
- Meng, S.; Wang, B.; Li, W. CircAXL Knockdown Alleviates Aβ1-42-Induced Neurotoxicity in Alzheimer’s Disease via Repressing PDE4A by Releasing miR-1306-5p. Neurochem. Res. 2022, 47, 1707–1720. [Google Scholar] [CrossRef]
- Yang, H.; Ping, X.; Cui, Y.; Zheng, S.; Shentu, X. Role of Rapamycin and 3-MA in oxidative damage of HLECs caused by two doses of UVB radiation. Adv. Ophthalmol. Pract. Res. 2022, 3, 15–22. [Google Scholar] [CrossRef]
- Matsuda, S.; Ikeda, Y.; Murakami, M.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y. Roles of PI3K/AKT/GSK3 Pathway Involved in Psychiatric Illnesses. Diseases 2019, 7, 22. [Google Scholar] [CrossRef]
- Kumar, M.; Bansal, N. Implications of Phosphoinositide 3-Kinase-Akt (PI3K-Akt) Pathway in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 354–385. [Google Scholar] [CrossRef]
- Lei, L.; Luo, Y.; Kang, D.; Yang, F.; Meng, D.; Wang, J.Z.; Liu, R.; Wang, X.; Li, H.L. Gypenoside IX restores Akt/GSK-3β pathway and alleviates Alzheimer’s disease-like neuropathology and cognitive deficits. Aging 2023, 15, 14172–14191. [Google Scholar] [CrossRef]
- Sharma, M.; Dey, C.S. Role of Akt isoforms in neuronal insulin signaling and resistance. Cell. Mol. Life Sci. 2021, 78, 7873–7898. [Google Scholar] [CrossRef]
- Moreno-Jiménez, E.P.; Flor-García, M.; Hernández-Vivanco, A.; Terreros-Roncal, J.; Rodríguez-Moreno, C.B.; Toni, N.; Méndez, P.; Llorens-Martín, M. GSK-3β orchestrates the inhibitory innervation of adult-born dentate granule cells in vivo. Cell. Mol. Life Sci. 2023, 80, 225. [Google Scholar] [CrossRef]
- Pan, J.; Yao, Q.; Wang, Y.; Chang, S.; Li, C.; Wu, Y.; Shen, J.; Yang, R. The role of PI3K signaling pathway in Alzheimer’s disease. Front. Aging Neurosci. 2024, 16, 1459025. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shin, W.; Kim, K.; Lee, S.; Lee, E.J.; Kim, J.; Kweon, H.; Lee, E.; Park, H.; Kang, M.; et al. NGL-3 in the regulation of brain development, Akt/GSK3b signaling, long-term depression, and locomotive and cognitive behaviors. PLoS Biol. 2019, 17, e2005326. [Google Scholar] [CrossRef] [PubMed]
- Li, L.F.; Gao, Y.; Xu, Y.; Su, D.J.; Yang, Q.; Liu, A.; Wang, S.Y.; Tang, X.L.; Zhao, J.; Luo, L.; et al. Praeruptorin C alleviates cognitive impairment in type 2 diabetic mice through restoring PI3K/AKT/GSK3β pathway. Phytother. Res. 2023, 37, 4838–4850. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s Disease: Mechanisms and Nutritional Aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, S.; Li, J.; Jiang, T.; Feng, L.; Pei, J.; Wang, G.; Ouyang, L.; Liu, B. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm. Sin. B 2021, 11, 3015–3034. [Google Scholar] [CrossRef]
- Wu, Y.; Ye, L.; Yuan, Y.; Jiang, T.; Guo, X.; Wang, Z.; Xu, K.; Xu, Z.; Liu, Y.; Zhong, X.; et al. Autophagy Activation is Associated with Neuroprotection in Diabetes-associated Cognitive Decline. Aging Dis. 2019, 10, 1233–1245. [Google Scholar] [CrossRef]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef]
- Guo, D.; Xie, J.; Zhao, J.; Huang, T.; Guo, X.; Song, J. Resveratrol protects early brain injury after subarachnoid hemorrhage by activating autophagy and inhibiting apoptosis mediated by the Akt/mTOR pathway. Neuroreport 2018, 29, 368–379. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2025 update. Nucleic Acids Res. 2025, 53, D1516–D1525. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef]
- Gu, S.; Lai, L.H. Associating 197 Chinese herbal medicine with drug targets and diseases using the similarity ensemble approach. Acta Pharmacol. Sin. 2020, 41, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Du, H.; Zhang, S.; Yuan, K.; Yang, Z.; Wu, M. Integrated Metabolomics and Network Pharmacology Study on the Mechanism of Rehmanniae radix Extract for Treating Thrombosis. Drug Des. Dev. Ther. 2024, 18, 4859–4875. [Google Scholar] [CrossRef] [PubMed]
- Velankar, S.; Burley, S.K.; Kurisu, G.; Hoch, J.C.; Markley, J.L. The Protein Data Bank Archive. Methods Mol. Biol. 2021, 2305, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Rosignoli, S.; Paiardini, A. Boosting the Full Potential of PyMOL with Structural Biology Plugins. Biomolecules 2022, 12, 1764. [Google Scholar] [CrossRef]
- Akinlade, O.M.; Owoyele, B.V.; Soladoye, A.O. Streptozotocin-induced type 1 and 2 diabetes in rodents: A model for studying diabetic cardiac autonomic neuropathy. Afr. Health Sci. 2021, 21, 719–727. [Google Scholar] [CrossRef]
- Othman, M.Z.; Hassan, Z.; Che Has, A.T. Morris water maze: A versatile and pertinent tool for assessing spatial learning and memory. Exp. Anim. 2022, 71, 264–280. [Google Scholar] [CrossRef]
- Du, R.; Pei, H.; He, Z.; Wang, J.; Zhou, X.; Li, W.; Zhu, D.; Zhang, C. Astragalin improves cognitive disorder in Alzheimer’s disease: Based on network pharmacology and molecular docking simulation. CNS Neurosci. Ther. 2024, 30, e14799. [Google Scholar] [CrossRef]
- Ma, X.; Nan, Y.; Huang, C.; Li, X.; Yang, Y.; Jiang, W.; Ye, M.; Liu, Q.; Niu, Y.; Yuan, L. Expression of αA-crystallin (CRYAA) in vivo and in vitro models of age-related cataract and the effect of its silencing on HLEB3 cells. Aging 2023, 15, 4498–4509. [Google Scholar] [CrossRef]
- Caruso, G.; Godos, J.; Privitera, A.; Lanza, G.; Castellano, S.; Chillemi, A.; Bruni, O.; Ferri, R.; Caraci, F.; Grosso, G. Phenolic Acids and Prevention of Cognitive Decline: Polyphenols with a Neuroprotective Role in Cognitive Disorders and Alzheimer’s Disease. Nutrients 2022, 14, 819. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, J.; Song, J.; Fan, Z.; Zhang, R.; Wang, Q.; Yi, K.; Gong, Q.; Liu, B. Investigating the Effect and Mechanism of 3-Methyladenine Against Diabetic Encephalopathy by Network Pharmacology, Molecular Docking, and Experimental Validation. Pharmaceuticals 2025, 18, 605. https://doi.org/10.3390/ph18050605
Chu J, Song J, Fan Z, Zhang R, Wang Q, Yi K, Gong Q, Liu B. Investigating the Effect and Mechanism of 3-Methyladenine Against Diabetic Encephalopathy by Network Pharmacology, Molecular Docking, and Experimental Validation. Pharmaceuticals. 2025; 18(5):605. https://doi.org/10.3390/ph18050605
Chicago/Turabian StyleChu, Jiaxin, Jianqiang Song, Zhuolin Fan, Ruijun Zhang, Qiwei Wang, Kexin Yi, Quan Gong, and Benju Liu. 2025. "Investigating the Effect and Mechanism of 3-Methyladenine Against Diabetic Encephalopathy by Network Pharmacology, Molecular Docking, and Experimental Validation" Pharmaceuticals 18, no. 5: 605. https://doi.org/10.3390/ph18050605
APA StyleChu, J., Song, J., Fan, Z., Zhang, R., Wang, Q., Yi, K., Gong, Q., & Liu, B. (2025). Investigating the Effect and Mechanism of 3-Methyladenine Against Diabetic Encephalopathy by Network Pharmacology, Molecular Docking, and Experimental Validation. Pharmaceuticals, 18(5), 605. https://doi.org/10.3390/ph18050605