
Uvarinol and Dichamanetin Derived from Uvaria chamae as Potential Dual-Site Inhibitors Against PBP2a in Methicillin Resistant Staphylococcus aureus: An In Silico Study

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
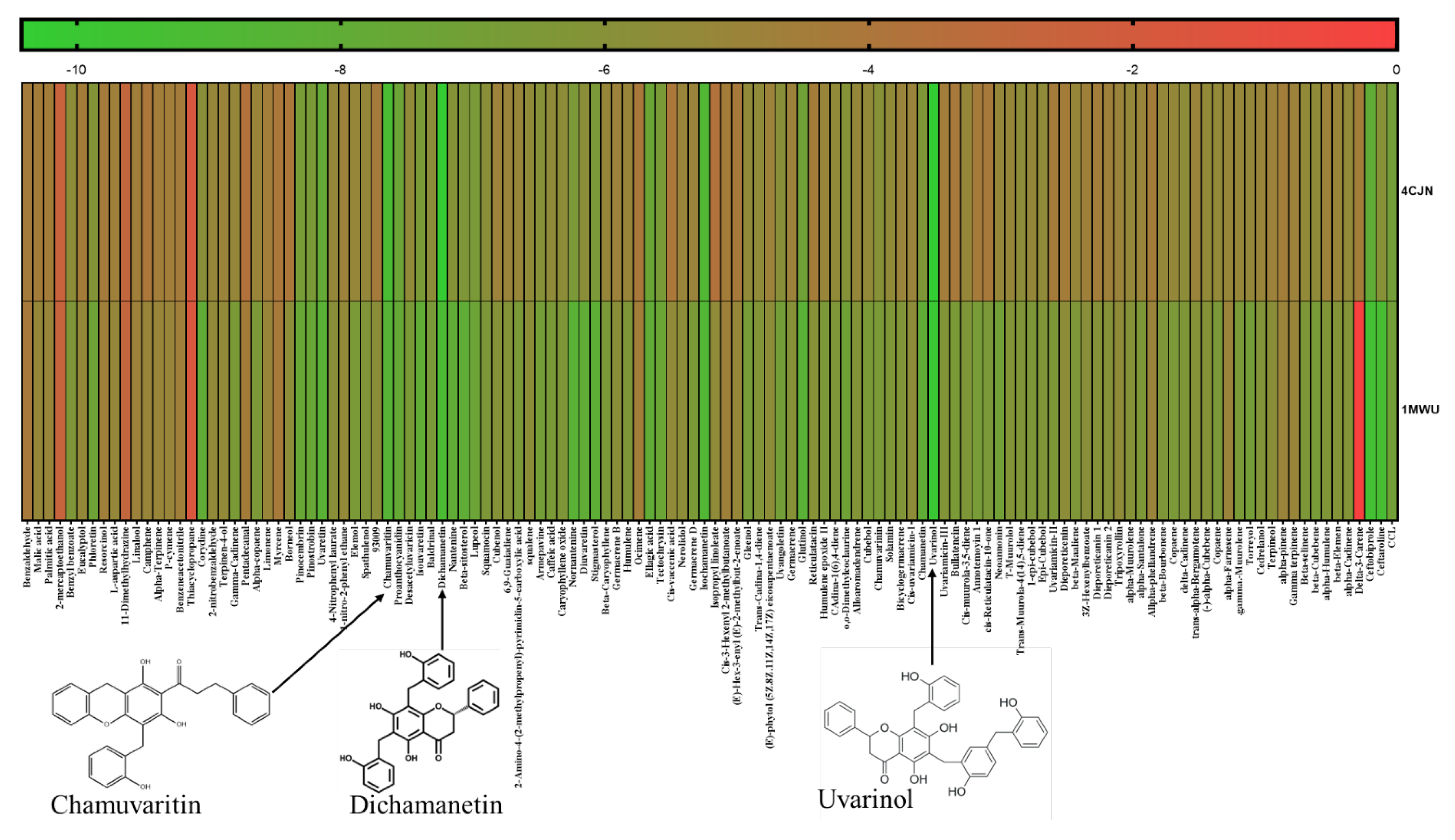
2.1. Molecular Docking
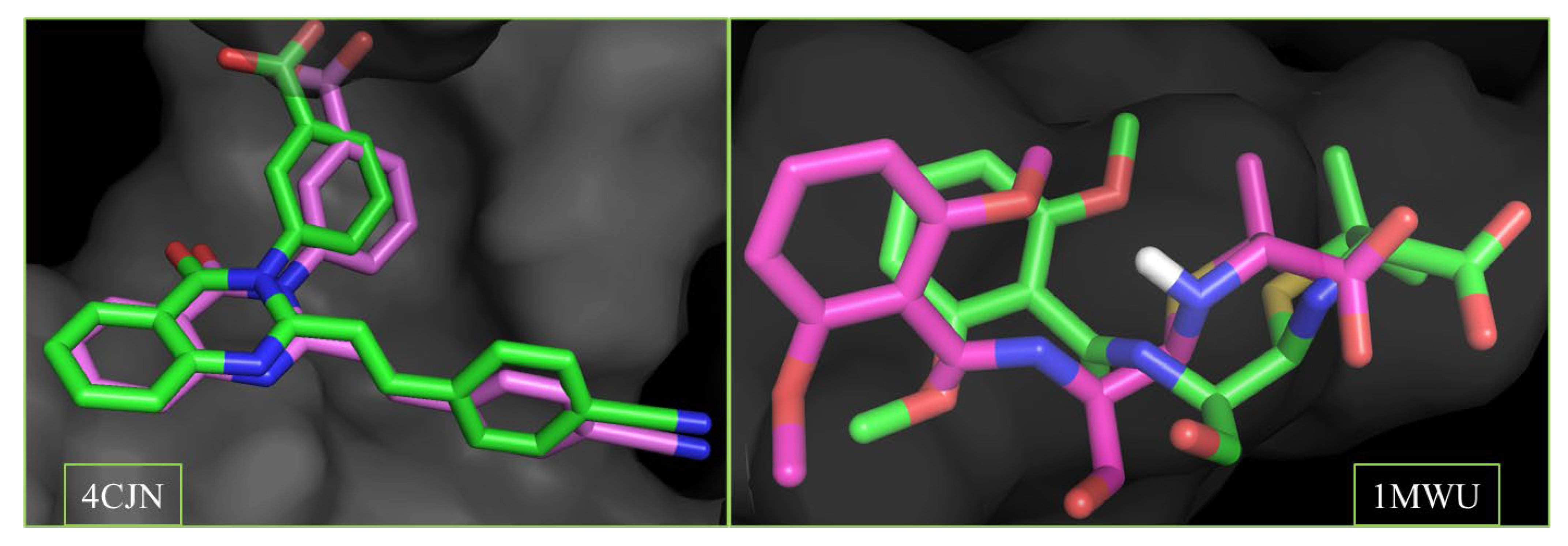
2.2. Validation of Docking Protocol
2.3. Molecular Dynamics (MD) Simulation
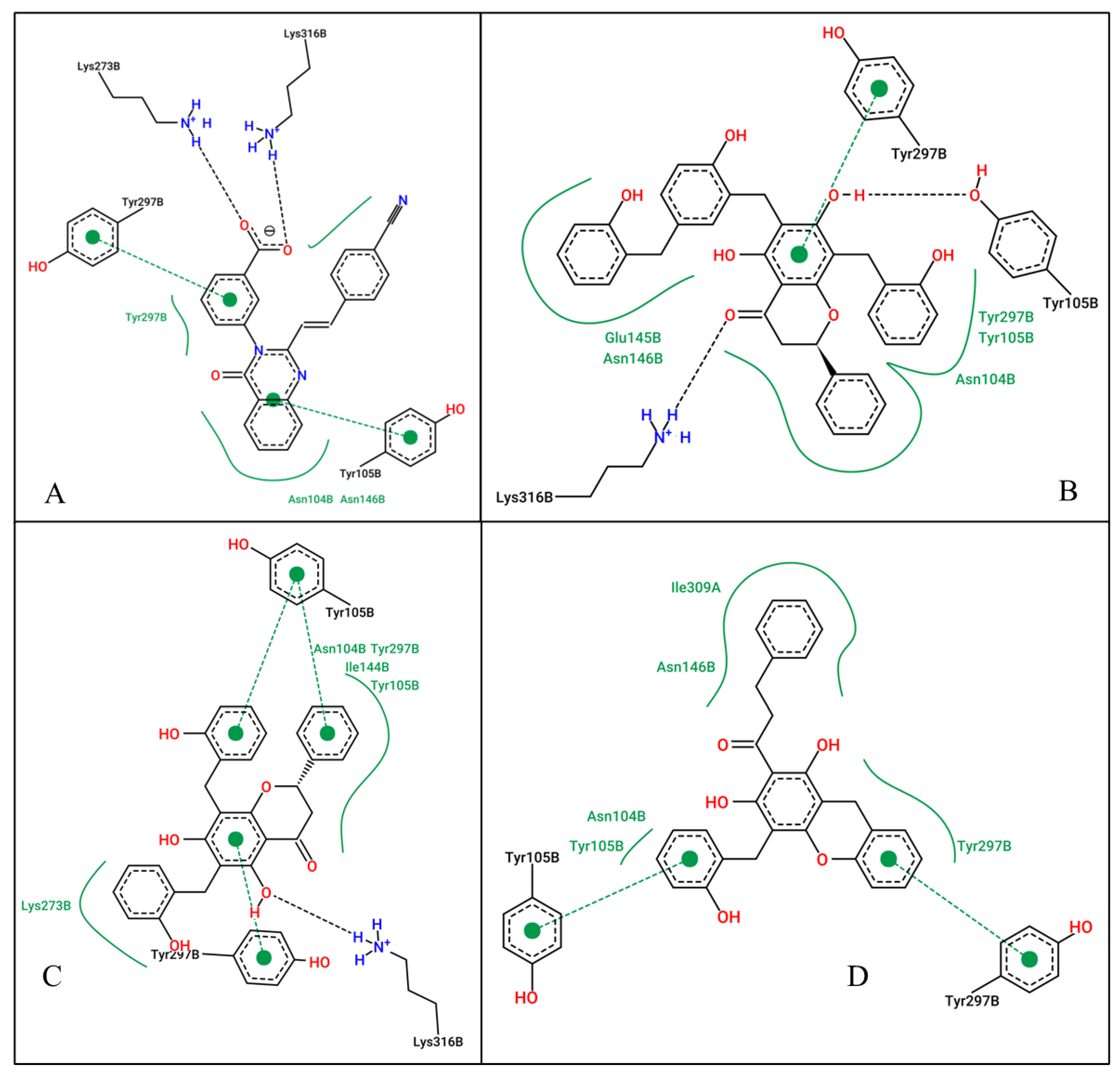
2.4. At the Allosteric Site (4CJN)
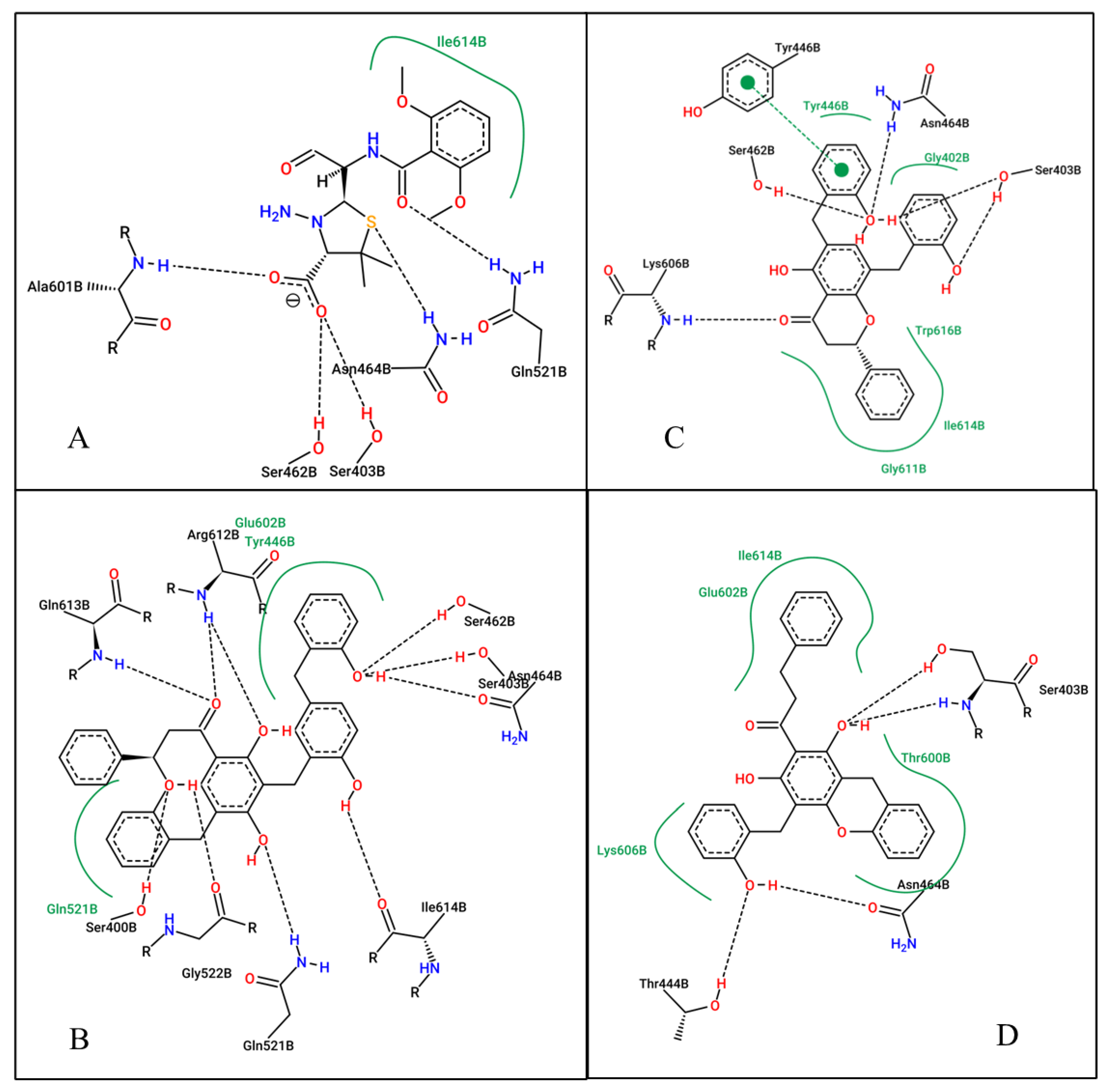
2.5. At the Active Site (1MWU)
2.6. MM/GBSA Binding Free Energy
2.7. Pharmacokinetic Predictions
3. Discussion
4. Materials and Methods
4.1. Virtual Screening and Docking Platform
4.2. Phytochemical Library Generation and Ligand Preparation
4.3. Target Retrieval and Preparation
4.4. Receptor Grid Generation
4.5. Molecular Docking
4.6. Pharmacokinetic Profile
4.7. Molecular Dynamic (MD) Simulation and Trajectory Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Alzahrani, O.R.; Alshabrmi, F.M.; Khalaf, E.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; Soliman, M.E.S.; et al. Non-β-Lactam Allosteric Inhibitors Target Methicillin-Resistant Staphylococcus aureus: An In Silico Drug Discovery Study. Antibiotics 2021, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.K.; Millar, B.C.; Moore, J.E. Antimicrobial Resistance (AMR). Br. J. Biomed. Sci. 2023, 80, 11387. [Google Scholar] [CrossRef]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial Resistance: Prevalence, Economic Burden, Mechanisms of Resistance and Strategies to Overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef]
- Linz, M.S.; Mattappallil, A.; Finkel, D.; Parker, D. Clinical Impact of Staphylococcus aureus Skin and Soft Tissue Infections. Antibiotics 2023, 12, 557. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Ito, T.; Tamai, M.; Nakagawa, S.; Nakamura, Y. The Role of Staphylococcus aureus Quorum Sensing in Cutaneous and Systemic Infections. Inflamm. Regen. 2024, 44, 9. [Google Scholar] [CrossRef]
- Gnanamani, A.; Hariharan, P.; Paul-Satyaseela, M. Staphylococcus Aureus: Overview of Bacteriology, Clinical Diseases, Epidemiology, Antibiotic Resistance and Therapeutic Approach. In Frontiers in Staphylococcus aureus; InTech: London, UK, 2017. [Google Scholar]
- Vestergaard, M.; Frees, D.; Ingmer, H. Antibiotic Resistance and the MRSA Problem. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Hamed, A.A.; Hassan, H.M.; Belbahri, L.; Rateb, M.E.; Sayed, A.M. Flavonoids as Potential Anti-MRSA Agents through Modulation of PBP2a: A Computational and Experimental Study. Antibiotics 2020, 9, 562. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Cui, L.; Kuroda, M.; Ito, T. The Emergence and Evolution of Methicillin-Resistant Staphylococcus aureus. Trends Microbiol. 2001, 9, 486–493. [Google Scholar] [CrossRef]
- Lim, D.; Strynadka, N.C.J. Structural Basis for the β Lactam Resistance of PBP2a from Methicillin-Resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. [Google Scholar] [CrossRef]
- Lade, H.; Kim, J.-S. Molecular Determinants of β-Lactam Resistance in Methicillin-Resistant Staphylococcus aureus (MRSA): An Updated Review. Antibiotics 2023, 12, 1362. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Bao, Y.; Li, M.; Zhang, Y.; Zhang, F.; Wang, P.; Tao, J.; Tong, H.H.Y.; Guo, J. Unraveling the Mechanism of Ceftaroline-Induced Allosteric Regulation in Penicillin-Binding Protein 2a: Insights for Novel Antibiotic Development against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2023, 67, e0089523. [Google Scholar] [CrossRef] [PubMed]
- Fuda, C.; Suvorov, M.; Vakulenko, S.B.; Mobashery, S. The Basis for Resistance to β-Lactam Antibiotics by Penicillin-Binding Protein 2a of Methicillin-Resistant Staphylococcus aureus. J. Biol. Chem. 2004, 279, 40802–40806. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Kumarasiri, M.; Peng, Z.; Otero, L.H.; Song, W.; Suckow, M.A.; Schroeder, V.A.; Wolter, W.R.; Lastochkin, E.; Antunes, N.T.; et al. Discovery of Antibiotic (E)-3-(3-Carboxyphenyl)-2-(4-Cyanostyryl)Quinazolin-4(3 H)-One. J. Am. Chem. Soc. 2015, 137, 1738–1741. [Google Scholar] [CrossRef]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding Protein 2a of Methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef]
- Lovering, A.L.; Safadi, S.S.; Strynadka, N.C.J. Structural Perspective of Peptidoglycan Biosynthesis and Assembly. Annu. Rev. Biochem. 2012, 81, 451–478. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural Products as a Foundation for Drug Discovery. Curr. Protoc. Pharmacol. 2009, 46, 9–11. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Agbebi, E.A.; Alabi, O.S.; Nkrumah, A.O.; Ogbole, O.O. Evaluation of the Antibacterial and Antifungal Potentials of Peptide-Rich Extracts from Selected Nigerian Plants. Eur. J. Integr. Med. 2022, 54, 102163. [Google Scholar] [CrossRef]
- Clardy, J.; Fischbach, M.A.; Currie, C.R. The Natural History of Antibiotics. Curr. Biol. 2009, 19, R437–R441. [Google Scholar] [CrossRef]
- Al Kazman, B.S.M.; Harnett, J.E.; Hanrahan, J.R. Traditional Uses, Phytochemistry and Pharmacological Activities of Annonacae. Molecules 2022, 27, 3462. [Google Scholar] [CrossRef] [PubMed]
- Abu, T.; Rex-Ogbuku, E.; Idibiye, K. A Review: Secondary Metabolites of Uvaria Chamae p. Beauv. (Annonaceae) and Their Biological Activities. Int. J. Agric. Environ. Food Sci. 2018, 2, 177–185. [Google Scholar] [CrossRef]
- Agbebi, E.A.; Omotuyi, O.I.; Oyinloye, B.E.; Okeke, U.B.; Apanisile, I.; Okor, B.; Adefabijo, D. Ethnomedicine, Phytochemistry, and Pharmacological Activities of Uvaria chamae P. Beauv.: A Comprehensive Review. Naunyn-Schmiedebergs. Arch. Pharmacol. 2024, 397, 5421–5436. [Google Scholar] [CrossRef] [PubMed]
- Hufford, C.D.; Lasswell, W.L.; Hirotsu, K.; Clardy, J. Uvarinol: A Novel Cytotoxic Tribenzylated Flavanone from Uvaria Chamae. J. Org. Chem. 1979, 44, 4709–4710. [Google Scholar] [CrossRef]
- Fall, D.; Gleye, C.; Franck, X.; Laurens, A.; Hocquemiller, R. Cis-Bullatencin, a Linear Acetogenin from Roots of Uvaria Chamae. Nat. Prod. Lett. 2002, 16, 315–321. [Google Scholar] [CrossRef]
- Kaboré, K.; Dibala, C.I.; Sama, H.; Diao, M.; Somda, M.K.; Dicko, M.H. Phenolic Content, Antioxidant Potential, and Antimicrobial Activity of Uvaria chamae (Annonaceae), a Food Plant from Burkina Faso. Biochem. Res. Int. 2024, 2024, 1289859. [Google Scholar] [CrossRef]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial Activity of Flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Nazzaro, F.; Fratianni, F.; Coppola, R. Quorum Sensing and Phytochemicals. Int. J. Mol. Sci. 2013, 14, 12607–12619. [Google Scholar] [CrossRef]
- Ejeje, J.N.; Agbebi, E.A.; Mathenjwa-Goqo, M.S.; Oje, O.A.; Agboinghale, P.E.; Ebe, I.T.; Obafemi, T.O.; Adewole, E.; Omaka, O.N.; Onikanni, S.A.; et al. Computational Investigation of the Therapeutic Potential of Detarium Senegalense in the Management of Erectile Dysfunction. Int. J. Mol. Sci. 2024, 25, 12362. [Google Scholar] [CrossRef]
- Saibu, O.A.; Singh, G.; Omoboyowa, D.A.; Oyejoke, A.K.; Olugbodi, S.A.; Bamisaye, A.; Adeniji, C.B.; Ajayi, T.M.; Akinpelu, Y.I.; Ogunwole, C.A.; et al. Discovery of Putative Natural Compounds Inhibitor of the Germinant Spore Receptor CspC in Clostridioides Difficile Infection: Gaining Insights via In Silico and Bioinformatics Approach. Inform. Med. Unlocked 2023, 42, 101339. [Google Scholar] [CrossRef]
- Macalalad, M.A.B.; Gonzales, A.A. In Silico Screening and Identification of Antidiabetic Inhibitors Sourced from Phytochemicals of Philippine Plants against Four Protein Targets of Diabetes (PTP1B, DPP-4, SGLT-2, and FBPase). Molecules 2023, 28, 5301. [Google Scholar] [CrossRef] [PubMed]
- Rani, N.; Vijayakumar, S.; Thanga Velan, L.P.; Arunachalam, A. Quercetin 3-O-Rutinoside Mediated Inhibition of PBP2a: Computational and Experimental Evidence to Its Anti-MRSA Activity. Mol. BioSyst. 2014, 10, 3229–3237. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.-A.W.; Dokla, E.M.E.; Serya, R.A.T.; Abouzid, K.A.M. Penicillin Binding Protein 2a: An Overview and a Medicinal Chemistry Perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Azmal, M.; Prima, F.S.; Zaman, B.; Hossain, M.M.; Mishu, M.A.; Ghosh, A. Retention of Methicillin Susceptibility in Staphylococcus aureus Using Natural Adjuvant as an Allosteric Modifier of Penicillin-Binding Protein 2a. Comput. Biol. Med. 2024, 181, 109070. [Google Scholar] [CrossRef]
- Ambade, S.S.; Gupta, V.K.; Bhole, R.P.; Khedekar, P.B.; Chikhale, R.V. A Review on Five and Six-Membered Heterocyclic Compounds Targeting the Penicillin-Binding Protein 2 (PBP2A) of Methicillin-Resistant Staphylococcus aureus (MRSA). Molecules 2023, 28, 7008. [Google Scholar] [CrossRef]
- Mora-Ochomogo, M.; Lohans, C.T. β-Lactam Antibiotic Targets and Resistance Mechanisms: From Covalent Inhibitors to Substrates. RSC Med. Chem. 2021, 12, 1623–1639. [Google Scholar] [CrossRef]
- Urgaonkar, S.; La Pierre, H.S.; Meir, I.; Lund, H.; RayChaudhuri, D.; Shaw, J.T. Synthesis of Antimicrobial Natural Products Targeting FtsZ: (±)-Dichamanetin and (±)-2‘ ‘‘-Hydroxy-5‘ ‘-Benzylisouvarinol-B. Org. Lett. 2005, 7, 5609–5612. [Google Scholar] [CrossRef]
- Pereira, F.; Madureira, A.M.; Sancha, S.; Mulhovo, S.; Luo, X.; Duarte, A.; Ferreira, M.-J.U. Cleistochlamys Kirkii Chemical Constituents: Antibacterial Activity and Synergistic Effects against Resistant Staphylococcus aureus strains. J. Ethnopharmacol. 2016, 178, 180–187. [Google Scholar] [CrossRef]
- Nana, O.; Guidana, A.; Momeni, J. Optimization of Ultrasonic-Assisted Extraction of Total Flavonoids from Uvaria Chamae Root Bark by Response Surface Methodology and Its Antioxidant Activity. Eur. Mod. Stud. J. 2024, 8, 186–202. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for Ligand-Receptor Docking. Curr. Protoc. Bioinforma. 2008, 24, 8–14. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Compound | Docking Score | Inhibition Constant (Ki) | Key Interaction |
|---|---|---|---|---|
| 4CJN | CCL (QNZ) | −9.16 | 192.16 nM | H-bond: LYS273, LYS316 H-phobic: ASN104, ASN146, TYR297 Pi-pi: TYR105, TYR297 |
| Dichamanetin | −10.32 | 27.07 nM | H-bond: LYS316 H-phobic: ASN104, TYR105, ILE144, LYS273, TYR297 Pi-pi: TYR105, TYR297 | |
| Uvarinol | −10.42 | 22.89 nM | H-bond: LYS316, TYR105 H-phobic: ASN104, TYR105, GLU154, ASN146, TYR297 Pi-pi: TYR297 | |
| Chamuvaritin | −9.17 | 189.39 nM | H-phobic: ASN104, TYR105, ASN146, TYR297, ILE309 Pi-pi: TYR105, TYR297 | |
| 1MWU | CCL (7EP) | −8.49 | 601.23 nM | H-bond: SER403, SER462, ASN464, GLN 521, ALA601 H-phobic: ILE614 |
| Dichamanetin | −12.01 | 1.57 nM | H-bond: SER403, TYR 446, SER462, ASN464, LYS606 H-phobic: GLY402, TYR446, GLY611, ILE614, TRP616 | |
| Uvarinol | −14.94 | 0.01 nM | H-bond: SER400, SER403, SER462, ASN464, GLN 521, GLY522, ARG612, GLN613, ILE614 H-phobic: TRY446, GLN521, GLU602 | |
| Chamuvaritin | −11.25 | 5.66 nM | H-bond: SER403, THR444, ASN464 H-phobic: THR600, GLU602, LYS606, ILE614 |
| Receptor | Ligand | P_RMSF | PL_RMSD | rGyr | MolSA | SASA | PSA |
|---|---|---|---|---|---|---|---|
| 4CJN | Quinazolinone | 2.182 ± 0.043 | 4.083 ± 0.024 | 4.478 ± 0.002 | 362.8 ± 0.09 | 291.2 ± 1.92 | 179.8 ± 0.11 |
| Dichamanetin | 1.674 ± 0.027 | 4.752 ± 0.018 | 4.312 ± 0.002 | 400.9 ± 0.12 | 224.2 ± 0.94 | 124.1 ± 0.14 | |
| Uvarinol | 1.956 ± 0.040 | 3.386 ± 0.016 | 5.452 ± 0.002 | 496.6 ± 0.13 | 444.4 ± 1.172 | 164.8 ± 0.18 | |
| Ceftaroline | 1.810 ± 0.033 | 2.625 ± 0.015 | 6.147 ± 0.011 | 533.3 ± 0.26 | 355.9 ± 1.602 | 371.1 ± 0.25 | |
| 1MWU | 7EP (CCL) | 1.809 ± 0.030 | 4.448 ± 0.027 | 3.859 ± 0.002 | 347.1 ± 0.11 | 447.8 ± 7.417 | 181.8 ± 0.20 |
| Dichamanetin | 2.139 ± 0.039 | 3.036 ± 0.021 | 4.303 ± 0.002 | 402.1 ± 0.15 | 213.9 ± 7.967 | 142.5 ± 0.32 | |
| Uvarinol | 2.052 ± 0.039 | 2.955 ± 0.022 | 5.294 ± 0.006 | 499.6 ± 0.13 | 294.9 ± 8.826 | 170.1 ± 0.21 | |
| Ceftaroline | 2.030 ± 0.040 | 3.550 ± 0.023 | 6.450 ± 0.004 | 541.6 ± 0.13 | 195.0 ± 1.134 | 337.6 ± 0.32 |
| Receptor | Ligand | MM/GBSA |
|---|---|---|
| 4CJN | Quinazolinone | −37.25 ± 0.76 |
| Dichamanetin | −43.48 ± 0.50 | |
| Uvarinol | −44.30 ± 0.42 | |
| Ceftaroline | −65.82 ± 0.81 | |
| 1MWU | 7EP (CCL) | −48.31 ± 0.87 |
| Dichamanetin | −54.01 ± 1.35 | |
| Uvarinol | −70.82 ± 0.87 | |
| Ceftaroline | −103.2 ± 0.67 |
| Compound | MW | #H-Bond Acceptors | #H-Bond Donors | TPSA | Consensus Log P | #Lipinski Violation | Synthetic Accessibility |
|---|---|---|---|---|---|---|---|
| Quinazolinone | 393.39 | 5 | 1 | 95.98 | 3.61 | 0 | 3.10 |
| 7EP | 382.43 | 7 | 3 | 139.26 | 0.05 | 0 | 4.01 |
| Ceftaroline | 684.68 | 12 | 4 | 340.13 | −1.16 | 2 | 5.75 |
| Dichamanetin | 468.50 | 6 | 4 | 107.22 | 4.44 | 0 | 4.15 |
| Uvarinol | 574.62 | 7 | 5 | 127.45 | 5.63 | 1 | 4.70 |
| GI Absorption | BBB Permeant | P-Glycoprotein Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor | Bioavailability Score | |
|---|---|---|---|---|---|---|---|---|---|
| Quinazolinone | High | No | No | No | No | Yes | No | No | 0.56 |
| 7EP | High | No | Yes | No | No | No | No | No | 0.55 |
| Ceftaroline | Low | No | Yes | No | No | No | No | No | 0.11 |
| Dichamanetin | High | No | No | No | Yes | Yes | No | No | 0.55 |
| Uvarinol | Low | No | No | No | No | No | No | No | 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agbebi, E.A.; Adeyemi, S.O.; Adewale, A.I.; Ajofoyinbo, O.S.; Olugbogi, E.A.; Oyinloye, O.M.; Anuoluwa, I.A.; Agbebi, T.O.; Ajiboye, B.O.; Oyinloye, B.E. Uvarinol and Dichamanetin Derived from Uvaria chamae as Potential Dual-Site Inhibitors Against PBP2a in Methicillin Resistant Staphylococcus aureus: An In Silico Study. Pharmaceuticals 2025, 18, 529. https://doi.org/10.3390/ph18040529
Agbebi EA, Adeyemi SO, Adewale AI, Ajofoyinbo OS, Olugbogi EA, Oyinloye OM, Anuoluwa IA, Agbebi TO, Ajiboye BO, Oyinloye BE. Uvarinol and Dichamanetin Derived from Uvaria chamae as Potential Dual-Site Inhibitors Against PBP2a in Methicillin Resistant Staphylococcus aureus: An In Silico Study. Pharmaceuticals. 2025; 18(4):529. https://doi.org/10.3390/ph18040529
Chicago/Turabian StyleAgbebi, Emmanuel Ayodeji, Shalom Oluwafunke Adeyemi, Adetola Ibukunoluwa Adewale, Omolara Seun Ajofoyinbo, Ezekiel Abiola Olugbogi, Oluwatoyin Mary Oyinloye, Iyadunni Adesola Anuoluwa, Timothy Oluwaseyi Agbebi, Basiru Olaitan Ajiboye, and Babatunji Emmanuel Oyinloye. 2025. "Uvarinol and Dichamanetin Derived from Uvaria chamae as Potential Dual-Site Inhibitors Against PBP2a in Methicillin Resistant Staphylococcus aureus: An In Silico Study" Pharmaceuticals 18, no. 4: 529. https://doi.org/10.3390/ph18040529
APA StyleAgbebi, E. A., Adeyemi, S. O., Adewale, A. I., Ajofoyinbo, O. S., Olugbogi, E. A., Oyinloye, O. M., Anuoluwa, I. A., Agbebi, T. O., Ajiboye, B. O., & Oyinloye, B. E. (2025). Uvarinol and Dichamanetin Derived from Uvaria chamae as Potential Dual-Site Inhibitors Against PBP2a in Methicillin Resistant Staphylococcus aureus: An In Silico Study. Pharmaceuticals, 18(4), 529. https://doi.org/10.3390/ph18040529