


Rational Design, Synthesis, and Biological Evaluation of Novel Thiazole/Thiazolidinones Multitarget Anti-Human Immunodeficiency Virus Molecules

 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
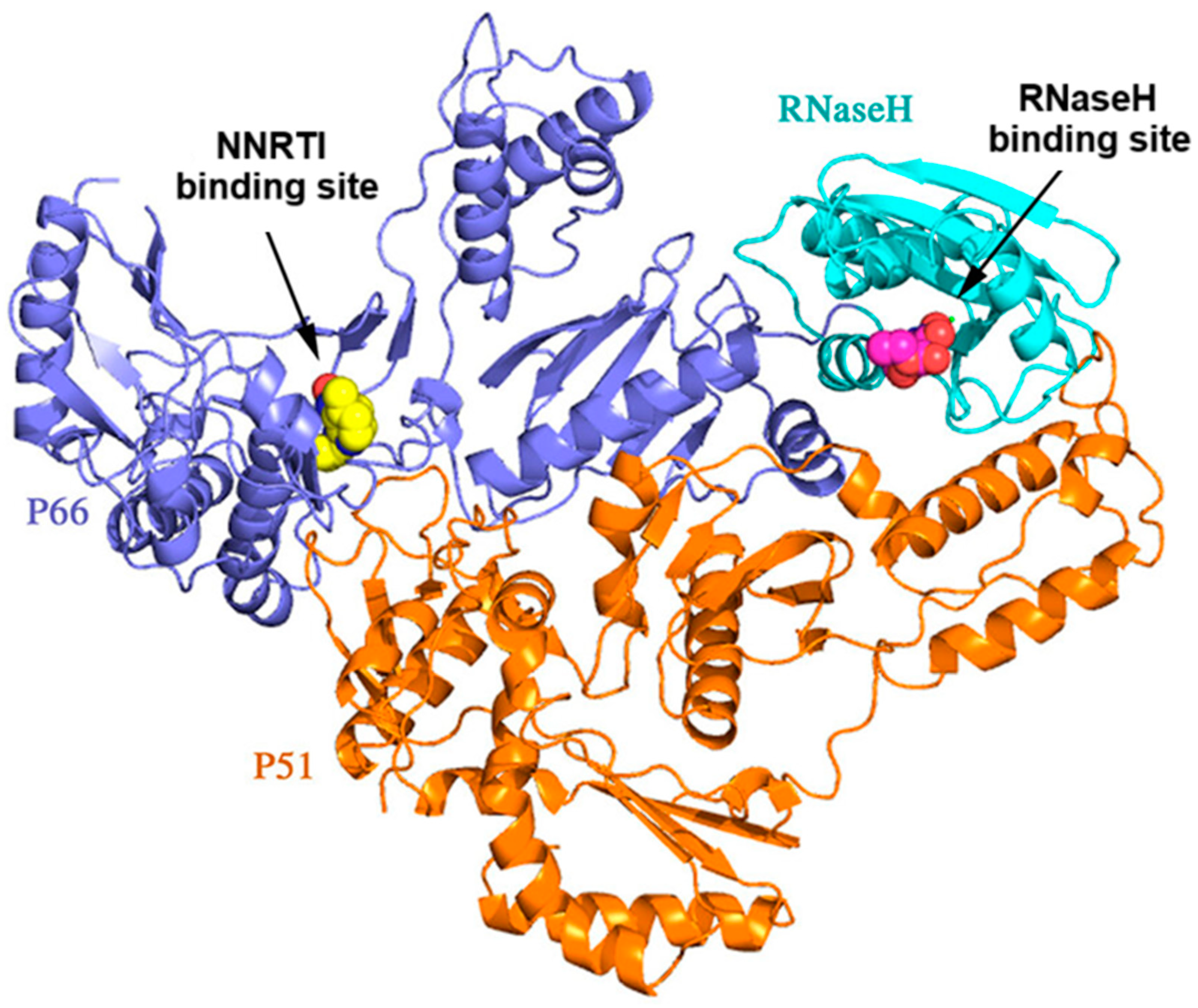
2.1. Rational Design of Compounds
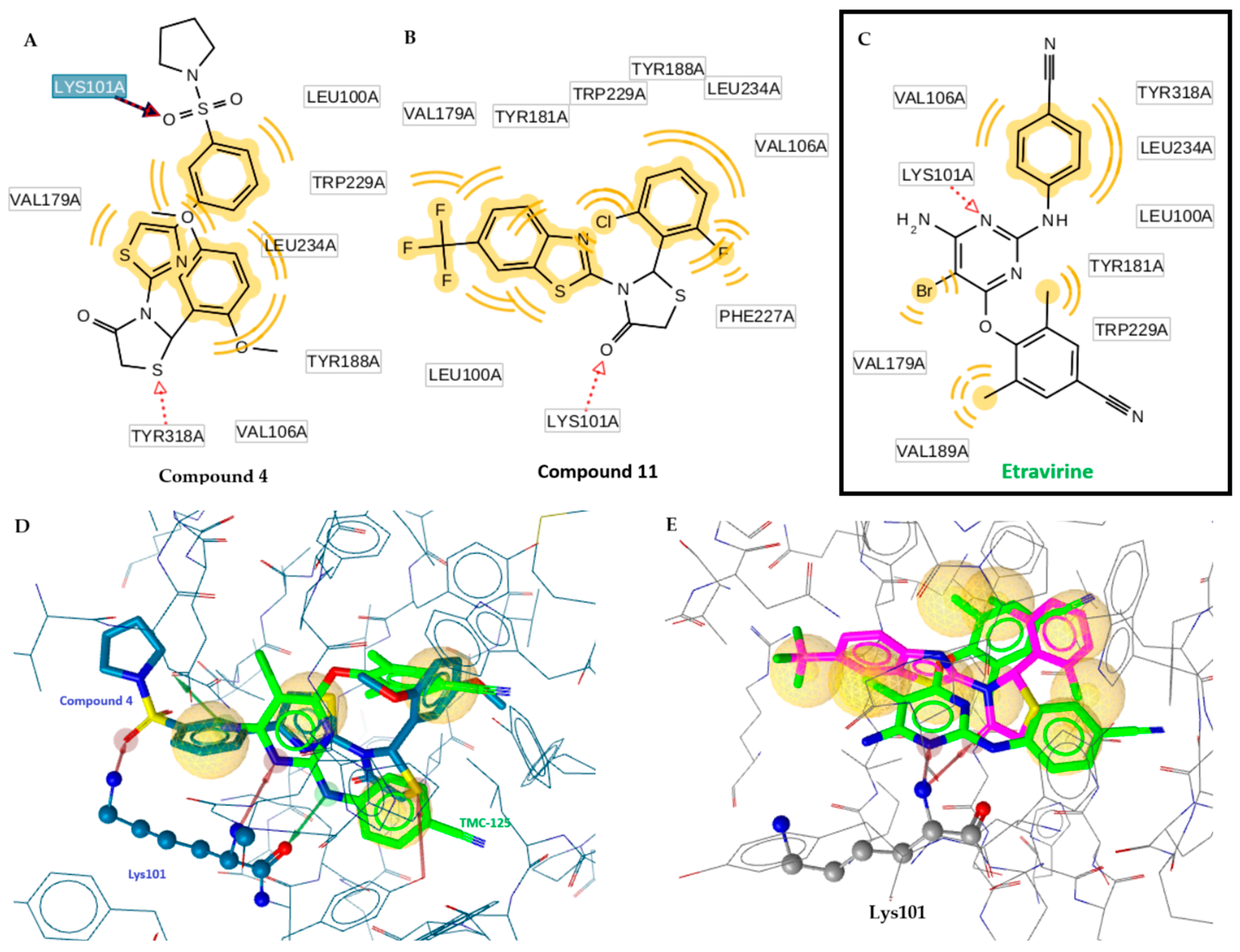
- Remarkably the binding site of NNRTI consists of mainly hydrophobic contacts while only a few hydrogen bonding interactions are needed. The presence of the conventional hydrogen bonding contact between the inhibitor (Figure 2) and the residue Lys101 significantly constitutes to a hydrophilic interaction [19]. This type of H-bond contact may positively contribute to the binding affinity of NNRTI and will serve as an anchor for the inhibitor adopting the most appropriate conformation with the aim of maximizing hydrophobic interaction contacts in the binding site. The O atom of the C=O group of the thiazolidinone moiety can play this role.
- The presence of a thiazole or benzothiazole moiety and aromatic rings facilitate hydrophobic interactions via π-π interactions with the residual amino acids side chains—Tyr181, Tyr188, Trp229, Phe227, Val106, Pro236, Leu100, and Leu234—enhancing the RT inhibition.
- The presence of a benzyl group at position 2 of the thiazolidinone moiety helps the molecule to obtain the “butterfly” configuration required to bind to the allosteric center of NNRTIs and inhibit RT.
- The presence of a dyad of –OH as a substituent of the phenyl group to be involved in the chelation process with two magnesium ions along with an existing hydrophobic phenyl group, which represents one of the most important pharmacophore needed for RNase H binding (Figure 2).
- The presence of a halogen substituent and mainly chlorine in benzothiazole, has been found to increase the inhibitory activity of the compounds [20]. Halogens form electrostatic interactions, which are stronger than H-bonds, and thereby a more stable complex is formed resulting in higher inhibition [21].
2.2. Docking Studies
2.3. Chemistry
2.4. Drug-Likeness Properties
2.5. Biological Evaluation
2.5.1. In Vitro Evaluation of HIV-1 RT RDDP Inhibitory Activity
2.5.2. RT RNase H Polymerase-Independent Cleavage Assay
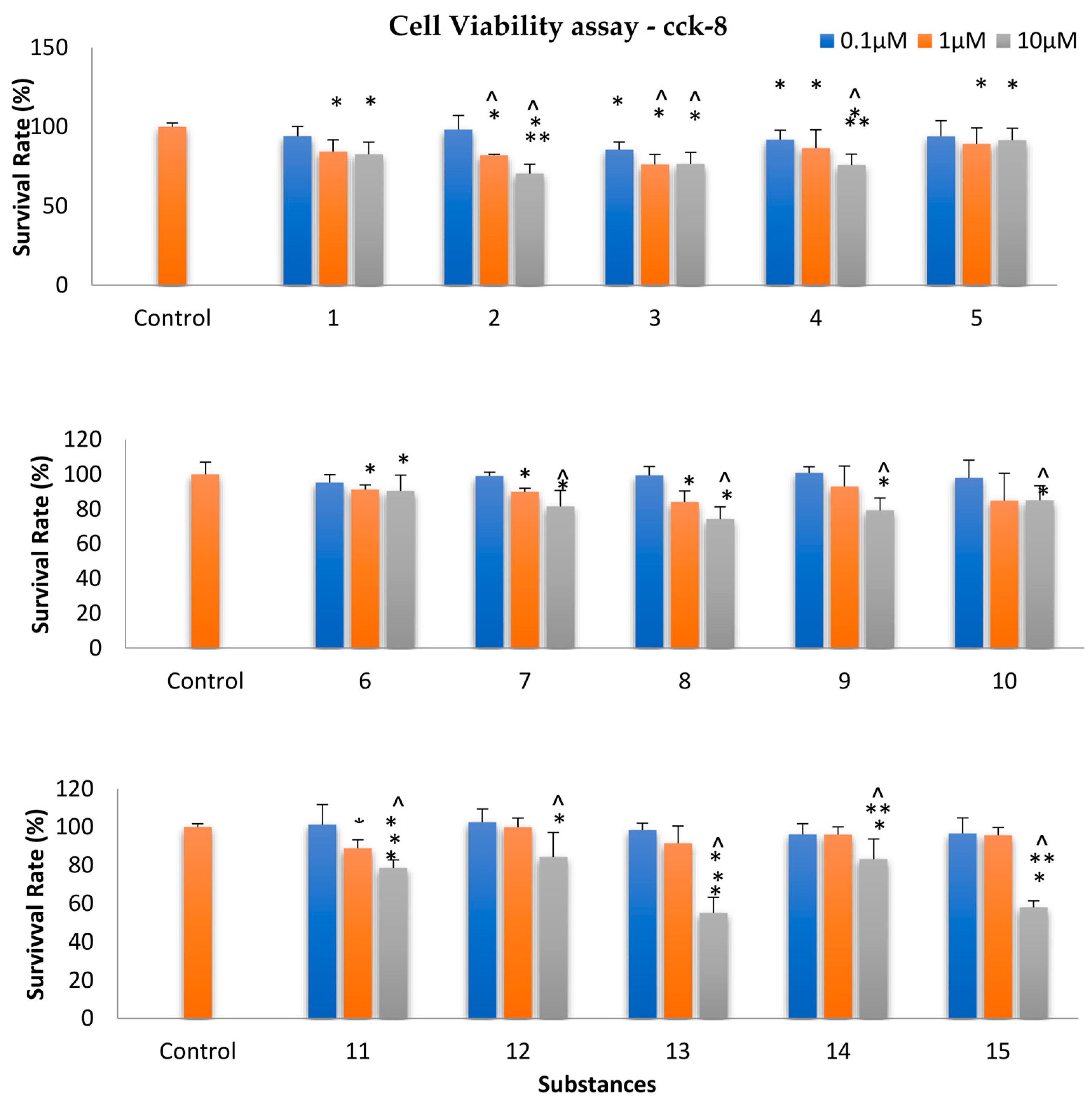
2.6. Assessment of Cellular Viability
3. Materials and Methods
3.1. Docking Studies
3.2. ADMET Properties
3.3. Chemistry
3.3.1. General Procedure for the Synthesis of Thiazolidinones by a Conventional Method
3.3.2. Microwave Irradiation Experiments
3.4. Biological Evaluation
3.4.1. In Vitro Evaluation of HIV-1 RT RDDP Inhibitory Activity
3.4.2. RT RNase H Polymerase-Independent Cleavage Assay
3.5. Evaluation of Cellular Viability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandel, S.E.; Merenstein, D. Human Immunodeficiency Virus (HIV) and Acquired Immunodeficiency Syndrome (AIDS): A Global Health Perspective. Clin. Infect. Dis. 2019, 68, 341–348. [Google Scholar]
- McPhee, S.L.; Papadakis, M.A.; Rabow, M.W. HIV and AIDS. Curr. Med. Diagn. Treat. 2012, 31, 1208–1210. [Google Scholar]
- Tcherepanov, V.; Stranska, R. HIV Reverse Transcriptase and RNase H: Key Enzymes in HIV Replication and Targets for Antiretroviral Therapy. Antivir. Ther. 2021, 26, 15–25. [Google Scholar]
- Esposito, F.; Tramontano, E. Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: Single and dual active site inhibitors. Antivir. Chem. Chemother. 2013, 23, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Schatz, O.; Cromme, F.; Naas, T.; Lindemann, D. Inactivation of the RNase H Domain of HIV-1 Reverse Transcriptase Blocks Viral Infectivity; Gene Regul AIDS. Oncogenesis and AIDS; Papas, T.S., Ed.; Portfolio Publishing Company: Houston, TX, USA, 1990; pp. 293–404. [Google Scholar]
- Roquebert, B.; Leleu, J. The reverse transcriptase of HIV-1: Role of the domain connection and RNase H in anti-retroviral resistors. Virologie 2010, 14, e129–e139. [Google Scholar]
- Bokharaei-Salim, F.; Esghaei, M.; Khanaliha, K.; Kalantari, S.; Marjani, A.; Fakhim, A.; Keyvani, H. HIV-1 reverse transcriptase and protease mutations for drug-resistance detection among treatment-experienced and naïve HIV-infected individuals. PLoS ONE 2020, 15, e0229275. [Google Scholar] [CrossRef]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 Reverse Transcriptase Still Remains a New Drug Target: Structure, Function, Classical Inhibitors, and New Inhibitors with Innovative Mechanisms of Actions. Mol. Biol. Int. 2012, 2012, 586401. [Google Scholar] [CrossRef]
- Iyidogan, P.; Anderson, K.S. Current perspectives on HIV-1 antiretroviral drug resistance. Viruses 2014, 6, 4095–4139. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Clark, A.D., Jr.; Lewi, P.J.; Heeres, J.; De Jonge, M.R.; Koymans, L.M.H.; Vinkers, H.M.; De Béthune, M.P.; Boyer, P.L.; Clark, P.; et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (Etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [Google Scholar] [PubMed]
- Corona, A.; Masaoka, T.; Tocco, G.; Tramontano, E.; Le Grice, S.F. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Future Med. Chem. 2013, 5, 2127–2139. [Google Scholar] [CrossRef]
- Klumpp, K.; Hang, J.Q.; Rajendran, S.; Yang, Y.; Derosier, A.; Wong Kai In, P.; Overton, H.; Parkes, K.E.; Cammack, N.; Martin, J.A. Two-metal ion mechanism of RNA cleavage by HIV RNase H and mechanism-based design of selective HIV RNase H inhibitors. Nucleic Acids Res. 2003, 31, 6852–6859. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sarafianos, S.G.; Wang, Z. Cutting into the Substrate Dominance: Pharmacophore and Structure-Based Approaches toward Inhibiting Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H. Acc. Chem. Res. 2020, 53, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Tramontano, E.; Esposito, F.; Badas, R.; Di Santo, R.; Costi, R.; La Colla, P. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]- 2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antivir. Res. 2005, 65, 117–124. [Google Scholar] [CrossRef]
- Petrou, A.; Eleftheriou, P.; Geronikaki, A.; Akrivou, M.G.; Vizirianakis, I. Novel Thiazolidin-4-ones as Potential Non-nucleoside Inhibitors of HIV-1 Reverse Transcriptase. Molecules 2019, 24, 3821. [Google Scholar] [CrossRef]
- Tian, Y.; Zhan, P.; Rai, D.; Zhang, J.; De Clercq, E.; Liu, X. Recent advances in the research of 2,3-diaryl-1,3-thiazolidin-4-one derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Curr. Med. Chem. 2012, 19, 2026–2037. [Google Scholar] [CrossRef]
- Maria Rosa Buemi, M.-R.; Rosaria Gitto, R.; Ielo, L.; Pannecouque, C.; Laura De Luca, L. Inhibition of HIV-1 RT activity by a new series of 3-(1,3,4-thiadiazol-2-yl)thiazolidin-4-one derivatives. Bioorg. Med. Chem. 2020, 28, 115431. [Google Scholar]
- Tatar, E.; Küçükgüzel, I.; Ötük, G.; Bİlgin, M.; De Clercq, E.; Andrei, G.; Snoeck, R.; Pannecouque, C.; Kaushik-Basu, N. Synthesis, characterization and biological evaluation of 1,3-thiazolidine-4-ones derived from (2S)-2-benzoylamino-3-methylbutanohydrazide hydrazones. J. Res. Pharm. 2021, 25, 507–518. [Google Scholar] [CrossRef]
- Esnouf, R.; Ren, J.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Tarantino, D.; Nawrozkij, M.B.; Maga, G.; Mai, A. Exploring the role of 2-chloro-6-fluoro substitution in 2-alkylthio-6-benzyl-5-alkylpyrimidin-4(3 H)-ones: Effects in HIV-1-infected cells and in HIV-1 reverse transcriptase enzymes. J. Med. Chem. 2014, 57, 5212–5225. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chemistry 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Fesatidou, M.; Petrou, A.; Geronikaki, A. Design, Synthesis, Biological Evaluation and Molecular Docking Studies of New Thiazolidinone Derivatives as NNRTIs and SARS-CoV-2 Main Protease Inhibitors. Chem. Biodivers. 2024, 21, e202401697. [Google Scholar] [CrossRef]
- Corona, A.; Di Leva, F.S.; Thierry, S. Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by Diketo acid derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- ProteinDataBank (PDB). Available online: https://www.rcsb.org (accessed on 13 October 2023).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Melbourne, Australia. Available online: https://biosig.lab.uq.edu.au/pkcsm/prediction (accessed on 13 October 2023).
- Tratrat, C.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Kostić, M.; Soković, M.; Vizirianakis, I.S.; Theodoroula, N.F.; Haroun, M. Thiazolidin-4-Ones as Potential Antimicrobial Agents: Experimental and In Silico Evaluation. Molecules 2022, 27, 1930. [Google Scholar] [CrossRef] [PubMed]
- Haroun, M.; Tratrat, C.; Petrou, A.; Geronikaki, A.; Ivanov, M.; Ćirić, A.; Soković, M.; Nagaraja, S.; Venugopala, K.N.; Balachandran Nair, A.; et al. Exploration of the Antimicrobial Effects of Benzothiazolylthiazolidin-4-One and In Silico Mechanistic Investigation. Molecules 2021, 26, 4061. [Google Scholar] [CrossRef]
- Chung, S.; Wendeler, M.; Rausch, J.W.; Beilhartz, G.; Gotte, M.; O'Keefe, B.R.; Bermingham, A.; Beutler, J.A.; Liu, S.; Zhuang, X.; et al. Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 2010, 54, 3913–3921. [Google Scholar] [CrossRef]
- Petrou, A.; Zagaliotis, P.; Theodoroula, N.; Mystridis, G.; Vizirianakis, I.S.; Walsh, T.J.; Geronikaki, A. Thiazole/Thiadiazole/Benzothiazole Based Thiazolidin-4-One Derivatives as Potential Inhibitors of Main Protease of SARS-CoV-2. Molecules 2022, 27, 2180. [Google Scholar] [CrossRef]
- Tseligka, E.D.; Rova, A.; Amanatiadou, E.P.; Calabrese, G.; Tsibouklis, J.; Fatouros, D.G.; Vizirianakis, I.S. Pharmacological development of target-specific delocalized lipophilic cation-functionalized carboranes for cancer therapy. Pharm. Res. 2016, 33, 1945–1958. [Google Scholar] [CrossRef] [PubMed]
- Akrivou, M.G.; Demertzidou, V.P.; Theodoroula, N.F.; Chatzopoulou, F.M.; Kyritsis, K.A.; Grigoriadis, N.; Zografos, A.L.; Vizirianakis, I.S. Uncovering the pharmacological response of novel sesquiterpene derivatives that differentially alter gene expression and modulate the cell cycle in cancer cells. Int. J. Oncol. 2018, 53, 2167–2179. [Google Scholar] [CrossRef]

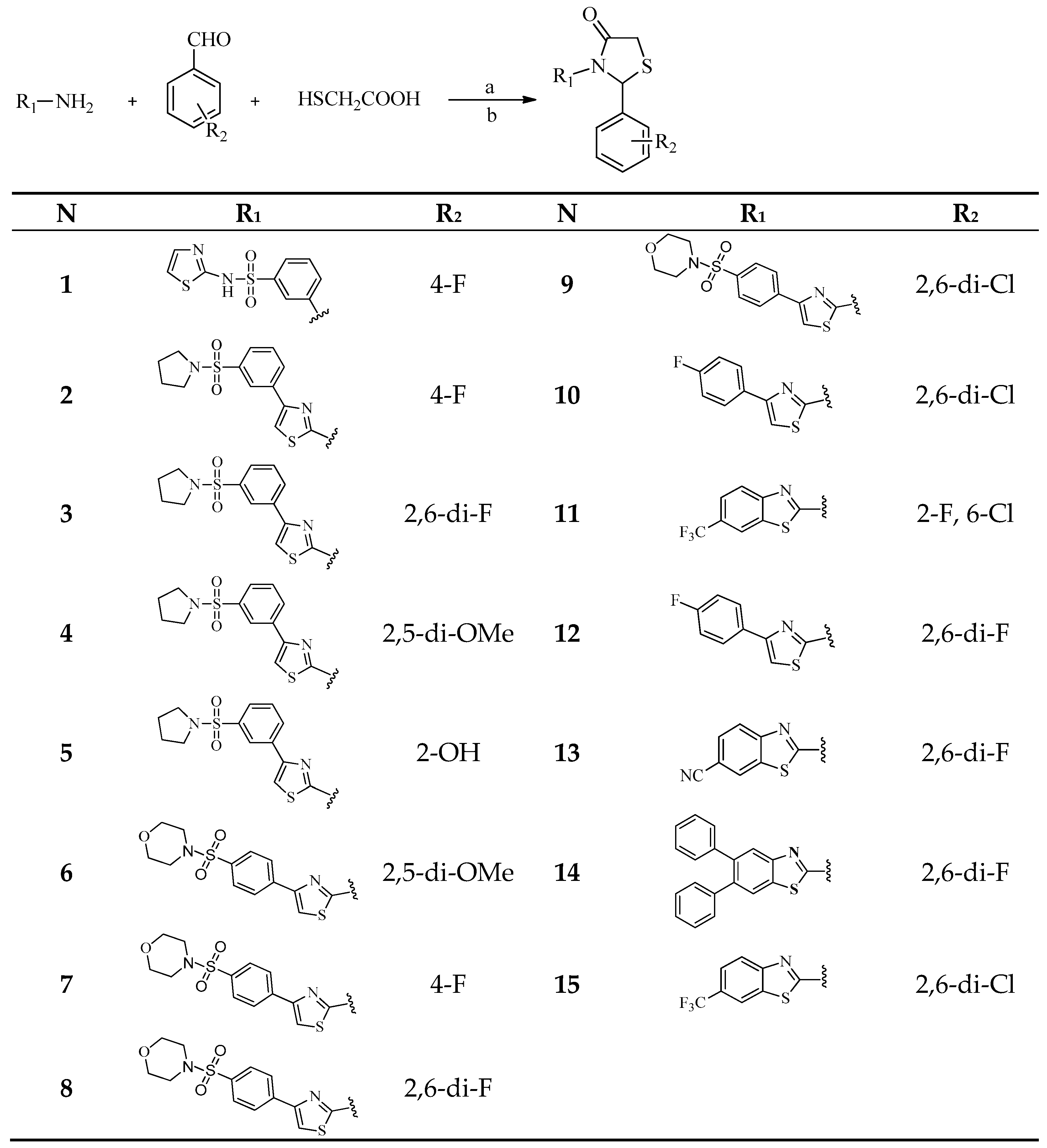


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | HIV-1 Reverse Transcriptase (PDB ID:3MEC) | RT RNase H Active Site (PDB: 6AOC) | ||
|---|---|---|---|---|
| Binding Free Energy (kcal/mol) | Residues Involved in Hydrogen Bonds | Binding Free Energy (kcal/mol) | Residues Involved in Hydrogen Bonds and Interactions with Mg | |
| 1 | −9.25 | Lys101 (O···H, 3.25Å) | −8.85 | Asp498 (O···H, 3.74Å) |
| 2 | −9.56 | Lys101 (O···H, 3.15Å), Lys102 (O···H, 3.57Å), | −7.76 | Asp443 (H···O, 3.25Å) |
| 3 | −9.43 | Lys101 (O···H, 3.55Å), Lys102 (O···H, 3.71Å), | −7.81 | Asp498 (O···H, 2.84Å) |
| 4 | −9.82 | Lys101 (O···H, 3.55Å), Tyr318 (S···H, 2.31Å) | −9.53 | Arg448 (O···H, 2.25Å) Mg2+-O |
| 5 | −8.03 | Lys101 (O···H, 3.47Å) | −9.10 | Mg2+-O |
| 6 | −8.57 | Lys101 (O···H, 3.59Å) | −9.36 | Tyr501(O···H, 3.52Å), Asp443 (O···H, 3.12Å), Ser449(O···H, 3.72Å) |
| 7 | −9.45 | Lys101 (O···H, 2.40Å), Lys103 (O···H, 2.31Å) | −8.62 | Asp498 (O···H, 2.90Å) |
| 8 | −9.31 | Lys101 (O···H, 2.74Å) | −7.73 | Tyr501(O···H, 2.75Å) |
| 9 | −8.50 | Lys101 (O···H, 2.77Å), His235 (O···H, 2.89Å) | −9.03 | Tyr501(O···H, 3.49Å), Asp443 (O···H, 3.71Å) |
| 10 | −8.81 | Lys101 (halogen bond), Thr181 (halogen bond) | −8.46 | Asp498 (O···H, 3.56Å) |
| 11 | −9.13 | Lys101 (O···H, 2.54Å) | −7.70 | Asn265 (O···H, 3.57Å) |
| 12 | −8.58 | Lys101 (O···H, 3.76Å) | −8.65 | Tyr501(O···H, 3.30Å), Asp443 (O···H, 3.89Å) |
| 13 | −9.52 | Lys101 (O···H, 2.33Å) | −8.59 | Asn265 (Ν···H, 3.52Å) |
| 14 | −7.93 | Lys101 (O···H, 2.82Å) | −7.94 | Asn265 (S···H, 3.15Å) |
| 15 | −8.51 | Lys101 (O···H, 3.54Å) | −8.19 | Gly444 (O···H, 3.64Å) |
| TMC-125 | −8.24 | Lys101 (N···H, 3.22Å) | - | - |
| N-hydroxythieno- pyrimidine-2,4-dione | - | - | −10.79 | Asp498 (N···H, 2.54Å) Mg2+-N |
| Property | Model Name | Predicted Value | Unit | |||
|---|---|---|---|---|---|---|
| Etravirine | 4 | 6 | 11 | |||
| Absorption | Water solubility | −3.718 | −5.545 | −5.281 | −6.527 | Numeric (log mol/L) |
| Absorption | Caco2 permeability | 0.694 | 0.73 | 0.588 | 1.085 | Numeric (log Papp in 10−6 cm/s) |
| Absorption | Intestinal absorption (human) | 86.939 | 99.051 | 100 | 90.963 | Numeric (% Absorbed) |
| Absorption | Skin permeability | −2.737 | −2.838 | −2.827 | −2.704 | Numeric (log Kp) |
| Absorption | P-Glycoprotein substrate | Yes | No | No | No | Categorical (Yes/No) |
| Absorption | P-Glycoprotein I inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Absorption | P-Glycoprotein II inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Distribution | VDss (human) | 0.051 | 0.127 | −0.09 | 0.172 | Numeric (log L/kg) |
| Distribution | Fraction unbound (human) | 0 | 0.046 | 0.062 | 0.038 | Numeric (Fu) |
| Distribution | BBB permeability | −0.13 | −1.545 | −1.739 | 0.246 | Numeric (log BB) |
| Distribution | CNS permeability | −1.981 | −2.936 | −3.117 | −1.451 | Numeric (log PS) |
| Metabolism | CYP2D6 substrate | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP3A4 substrate | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Metabolism | CYP2D6 inhibitor | No | No | No | No | Categorical (Yes/No) |
| Metabolism | CYP3A4 inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Metabolism | CYP1A2 inhibitor | Yes | No | No | Yes | Categorical (Yes/No) |
| Metabolism | CYP2C19 inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Metabolism | CYP2C9 inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Excretion | Total Clearance | −0.425 | 0.467 | 0.819 | 0.135 | Numeric (log ml/min/kg) |
| Excretion | Renal OCT2 substrate | No | Yes | Yes | Yes | Categorical (Yes/No) |
| Toxicity | AMES toxicity | No | No | No | No | Categorical (Yes/No) |
| Toxicity | Max. Tolerated dose (human) | 0.467 | −0.211 | −0.205 | −0.075 | Numeric (log mg/kg/day) |
| Toxicity | hERG I inhibitor | No | No | No | No | Categorical (Yes/No) |
| Toxicity | hERG II inhibitor | Yes | Yes | Yes | Yes | Categorical (Yes/No) |
| Toxicity | Oral Rat Acute Toxicity (LD50) | 2.699 | 2.871 | 1.528 | 2.985 | Numeric (mol/kg) |
| Toxicity | Hepatotoxicity | Yes | Yes | Yes | No | Categorical (Yes/No) |
| Toxicity | Skin Sensitization | No | No | No | No | Categorical (Yes/No) |
| Entry | Inhibition at 4 μM, (%) | IC50 (μM) | Entry | Inhibition at 4 μM, (%) | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 68.30 | 2.23 ± 0.15 | 9 | 2.48 | >4 |
| 2 | 8.36 | >4 | 10 | 1.59 | >4 |
| 3 | 24.27 | >4 | 11 | 92.37 | 0.32 ± 0.03 |
| 4 | 86.59 | 0.58 ± 0.07 | 12 | 5.86 | >4 |
| 5 | 45.20 | >4 | 13 | 21.37 | >4 |
| 6 | 81.28 | 0.72 ± 0.06 | 14 | 7.90 | >4 |
| 7 | 11.98 | >4 | 15 | 8.52 | >4 |
| 8 | 32.96 | >4 | Nevirapine | 93.84 | 0.31 ± 0.06 |
| Entry | IC50 (μM) | Entry | IC50 (μM) |
|---|---|---|---|
| 1 | 14.30 ± 1.3 | 9 | 9.01 ± 0.1 |
| 2 | >50 | 10 | >50 |
| 3 | >50 | 11 | 25.37 ± 0.4 |
| 4 | 1.47 ± 0.2 | 12 | >50 |
| 5 | 18.01 ± 2.1 | 13 | 16.40 ± 1.7 |
| 6 | 2.50 ± 0.3 | 14 | >50 |
| 7 | >50 | 15 | >50 |
| 8 | >50 | RDS1759 [23] | 7.3 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tratrat, C.; Petrou, A.; Fesatidou, M.; Haroun, M.; Chohan, M.; Geronikaki, A. Rational Design, Synthesis, and Biological Evaluation of Novel Thiazole/Thiazolidinones Multitarget Anti-Human Immunodeficiency Virus Molecules. Pharmaceuticals 2025, 18, 298. https://doi.org/10.3390/ph18030298
Tratrat C, Petrou A, Fesatidou M, Haroun M, Chohan M, Geronikaki A. Rational Design, Synthesis, and Biological Evaluation of Novel Thiazole/Thiazolidinones Multitarget Anti-Human Immunodeficiency Virus Molecules. Pharmaceuticals. 2025; 18(3):298. https://doi.org/10.3390/ph18030298
Chicago/Turabian StyleTratrat, Christophe, Anthi Petrou, Maria Fesatidou, Micheline Haroun, Mohamad Chohan, and Athina Geronikaki. 2025. "Rational Design, Synthesis, and Biological Evaluation of Novel Thiazole/Thiazolidinones Multitarget Anti-Human Immunodeficiency Virus Molecules" Pharmaceuticals 18, no. 3: 298. https://doi.org/10.3390/ph18030298
APA StyleTratrat, C., Petrou, A., Fesatidou, M., Haroun, M., Chohan, M., & Geronikaki, A. (2025). Rational Design, Synthesis, and Biological Evaluation of Novel Thiazole/Thiazolidinones Multitarget Anti-Human Immunodeficiency Virus Molecules. Pharmaceuticals, 18(3), 298. https://doi.org/10.3390/ph18030298