
Cannabinoid-2 Receptor Activation Attenuates Sulfur Mustard Analog 2-Chloroethyl-Ethyl-Sulfide-Induced Acute Lung Injury in Mice
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
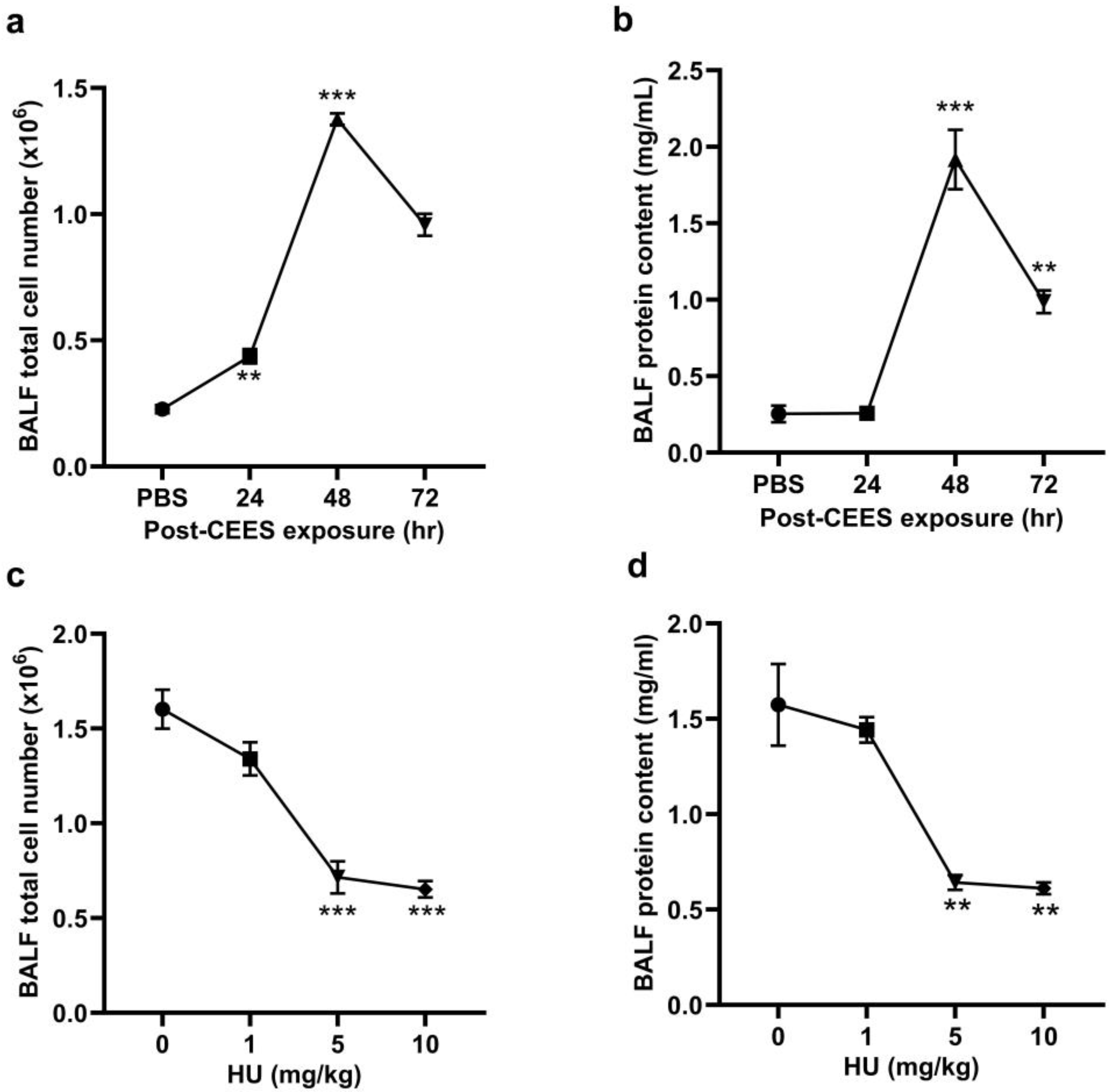
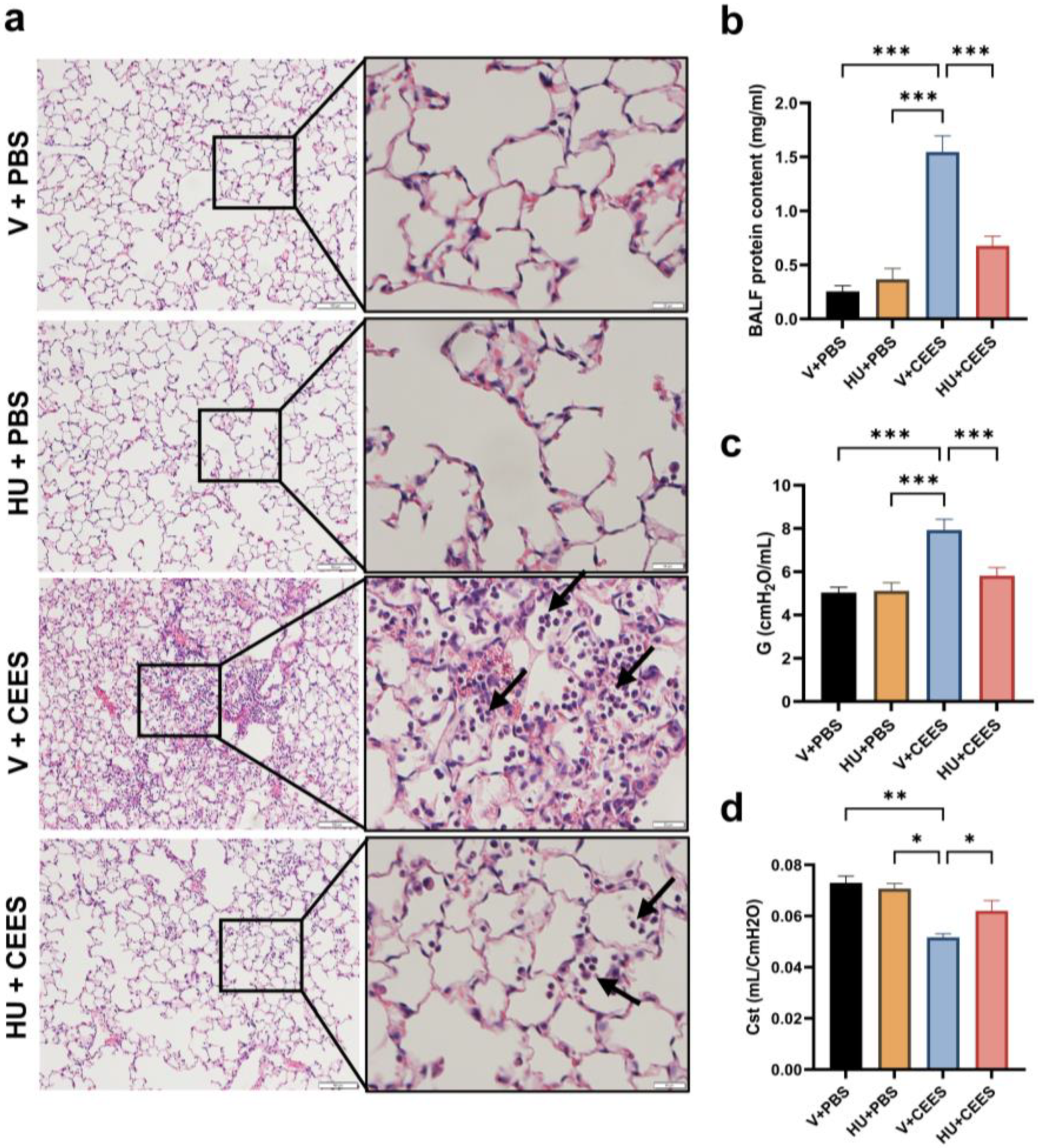
2.1. CB2R Activation Alleviates CEES—Induced Lung Injury and the Decline in Lung Function
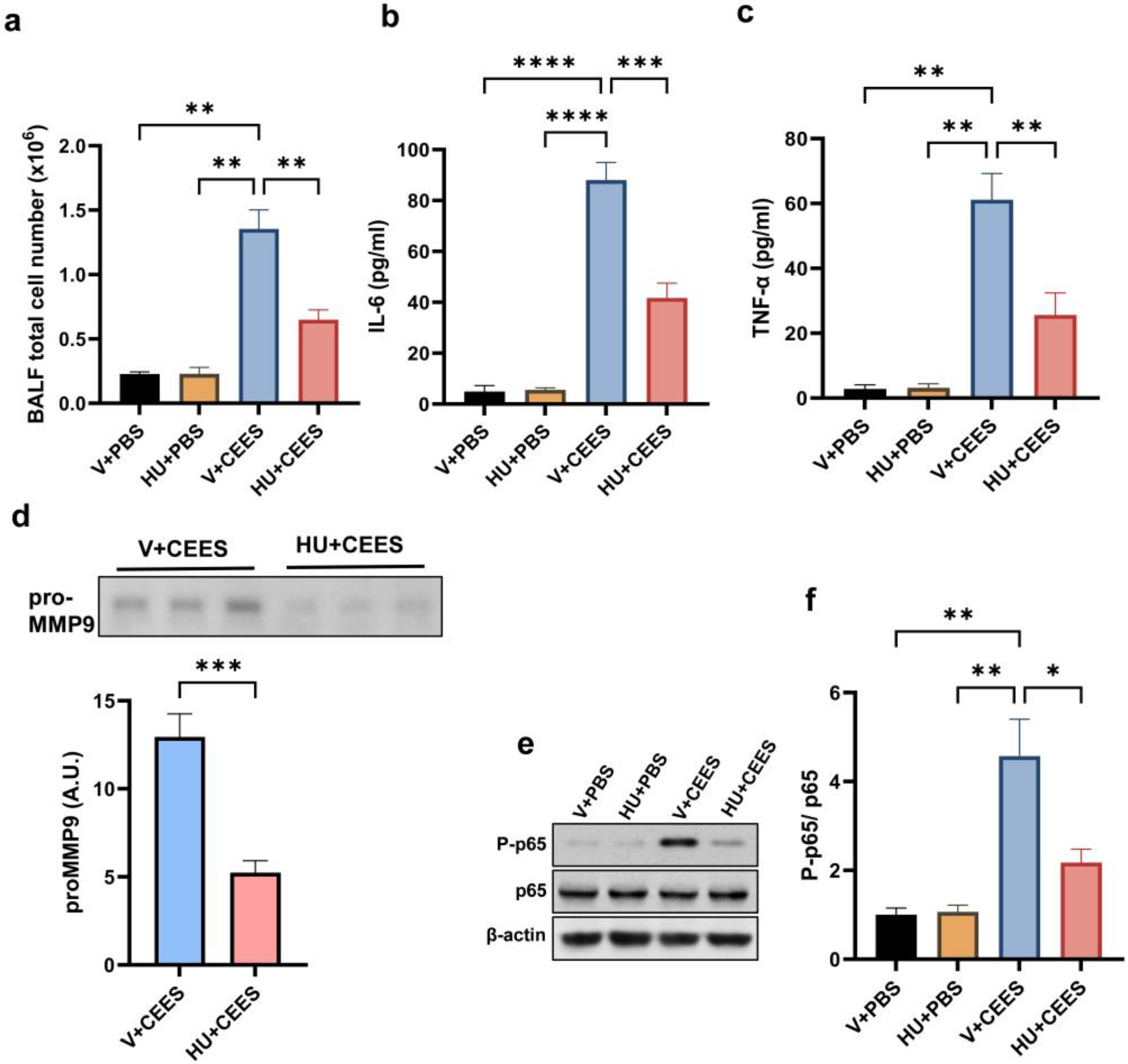
2.2. CB2R Activation Alleviates Lung Inflammation Induced by CEES
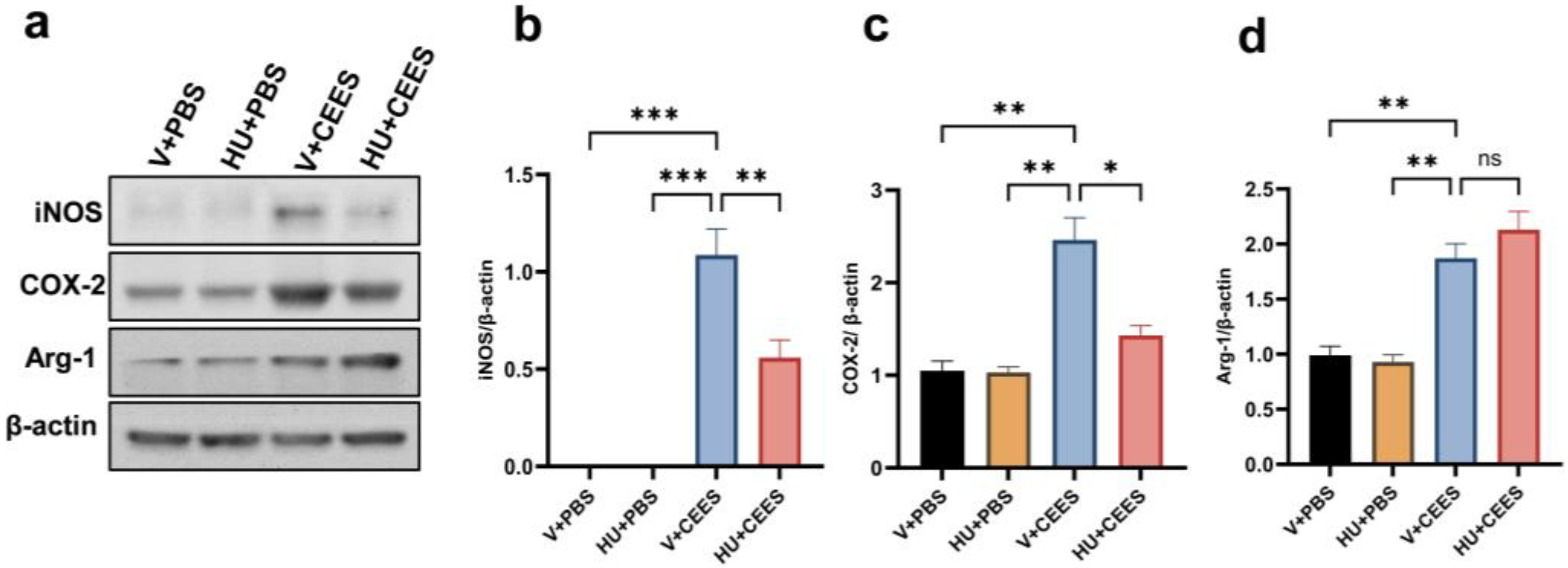
2.3. HU308 Treatment Suppresses CEES-Induced Proinflammatory Macrophage Phenotype
2.4. Pharmacological Activation of CB2R Attenuates the CEES-Induced Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Animal Procedures
4.3. Measurement of Lung Mechanics
4.4. Analysis of Bronchoalveolar Lavage Fluid (BALF)
4.5. Lung Tissue Processing
4.6. Isolation of Mouse Primary Alveolar Macrophages (AMs)
4.7. Immunoblot Analysis
4.8. Detection of Intracellular ROS
4.9. Immunostaining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henretig, F.M.; Kirk, M.A.; McKay, C.A., Jr. Hazardous Chemical Emergencies and Poisonings. N. Engl. J. Med. 2019, 380, 1638–1655. [Google Scholar] [CrossRef] [PubMed]
- Jett, D.A.; Laney, J.W. Civilian research on chemical medical countermeasures. Toxicol. Mech. Methods 2021, 31, 242–243. [Google Scholar] [CrossRef] [PubMed]
- Kehe, K.; Szinicz, L. Medical aspects of sulphur mustard poisoning. Toxicology 2005, 214, 198–209. [Google Scholar] [CrossRef] [PubMed]
- White, C.W.; Rancourt, R.C.; Veress, L.A. Sulfur mustard inhalation: Mechanisms of injury, alteration of coagulation, and fibrinolytic therapy. Ann. N. Y. Acad. Sci. 2016, 1378, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Kempf, C.L.; Song, J.H.; Sammani, S.; Bermudez, T.; Hernon, V.R.; Tang, L.; Cai, H.; Camp, S.M.; Johnson, C.A.; Basiouny, M.S.; et al. TLR4 ligation by eNAMPT, a novel DAMP, is essential to sulfur mustard- induced inflammatory lung injury and fibrosis. Eur. J. Respir. Med. 2024, 6, 389–397. [Google Scholar]
- Veress, L.A.; Hendry-Hofer, T.B.; Loader, J.E.; Rioux, J.S.; Garlick, R.B.; White, C.W. Tissue plasminogen activator prevents mortality from sulfur mustard analog–induced airway obstruction. Am. J. Respir. Cell Mol. Biol. 2013, 48, 439–447. [Google Scholar] [CrossRef]
- Ahmad, S.; Ahmad, A. Emerging targets for treating sulfur mustard–induced injuries. Ann. N. Y. Acad. Sci. 2016, 1374, 123–131. [Google Scholar] [CrossRef]
- Balali—Mood, M.; Hefazi, M. Comparison of early and late toxic effects of sulfur mustard in Iranian veterans. Basic Clin. Pharmacol. Toxicol. 2006, 99, 273–282. [Google Scholar] [CrossRef]
- Ghanei, M.; Adibi, I.; Farhat, F.; Aslani, J. Late respiratory effects of sulfur mustard: How is the early symptoms severity involved? Chronic Respir. Dis. 2008, 5, 95–100. [Google Scholar] [CrossRef]
- Mariappan, N.; Husain, M.; Zafar, I.; Singh, V.; Smithson, K.G.; Crowe, D.R.; Pittet, J.-F.; Ahmad, S.; Ahmad, A. Extracellular nucleic acid scavenging rescues rats from sulfur mustard analog-induced lung injury and mortality. Arch. Toxicol. 2020, 94, 1321–1334. [Google Scholar] [CrossRef]
- Sadeghi, S.; Mosaffa, N.; Hashemi, S.M.; Naghizadeh, M.M.; Ghazanfari, T. The immunomodulatory effects of mesenchymal stem cells on long term pulmonary complications in an animal model exposed to a sulfur mustard analog. Int. Immunopharmacol. 2019, 80, 105879. [Google Scholar] [CrossRef] [PubMed]
- Rancourt, R.C.; Ahmad, A.; Veress, L.A.; Rioux, J.S.; Garlick, R.B.; White, C.W. Antifibrinolytic mechanisms in acute airway injury after sulfur mustard analog inhalation. Am. J. Respir. Cell Mol. Biol. 2014, 51, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Venosa, A.; Malaviya, R.; Choi, H.; Gow, A.J.; Laskin, J.D.; Laskin, D.L. Characterization of distinct macrophage subpopulations during nitrogen mustard-induced lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 54, 436–446. [Google Scholar] [CrossRef]
- Venosa, A.; Gow, J.G.; Hall, L.; Malaviya, R.; Gow, A.J.; Laskin, J.D.; Laskin, D.L. Regulation of nitrogen mustard-induced lung macrophage activation by valproic acid, a histone deacetylase inhibitor. Toxicol. Sci. 2017, 157, 222–234. [Google Scholar] [CrossRef]
- Gould, N.S.; White, C.W.; Day, B.J. A role for mitochondrial oxidative stress in sulfur mustard analog 2-chloroethyl ethyl sulfide-induced lung cell injury and antioxidant protection. J. Pharmacol. Exp. Ther. 2009, 328, 732–739. [Google Scholar] [CrossRef]
- Malaviya, R.; Bellomo, A.; Abramova, E.; Croutch, C.R.; Roseman, J.; Tuttle, R.; Peters, E.; Casillas, R.P.; Sunil, V.R.; Laskin, J.D.; et al. Pulmonary injury and oxidative stress in rats induced by inhaled sulfur mustard is ameliorated by anti-tumor necrosis factor-α antibody. Toxicol. Appl. Pharmacol. 2021, 428, 115677. [Google Scholar] [CrossRef]
- Marzo, V.D. Erratum: Corrigendum: New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.-R.; LaViolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [PubMed]
- Rakotoarivelo, V.; Mayer, T.Z.; Simard, M.; Flamand, N.; Di Marzo, V. The impact of the CB2 Cannabinoid receptor in inflammatory diseases: An update. Molecules 2024, 29, 3381. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Zhao, H.; Zheng, Q.; Xiao, L.; Zhao, M. CB2 receptor activation ameliorates the proinflammatory activity in acute lung injury induced by paraquat. BioMed Res. Int. 2014, 2014, 971750. [Google Scholar] [CrossRef]
- Fu, Q.; Zheng, Y.; Dong, X.; Wang, L.; Jiang, C.G. Activation of cannabinoid receptor type 2 by JWH133 alleviates bleomycin-induced pulmonary fibrosis in mice. Oncotarget 2017, 8, 103486–103498. [Google Scholar] [CrossRef]
- Costola-De-Souza, C.; Ribeiro, A.; Ferraz-De-Paula, V.; Calefi, A.S.; Aloia, T.P.A.; Gimenes-Júnior, J.A.; De Almeida, V.I.; Pinheiro, M.L.; Palermo-Neto, J. Monoacylglycerol lipase (MAGL) inhibition attenuates acute lung injury in mice. PLoS ONE 2013, 8, e77706. [Google Scholar] [CrossRef]
- Nagre, N.; Nicholson, G.; Cong, X.; Lockett, J.; Pearson, A.C.; Chan, V.; Kim, W.-K.; Vinod, K.Y.; Catravas, J.D. Activation of cannabinoid-2 receptor protects against pseudomonas aeruginosa induced acute lung injury and inflammation. Respir. Res. 2022, 23, 326. [Google Scholar] [CrossRef]
- Benedetti, A.; Comporti, M.; Esterbauer, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta 1980, 620, 281–296. [Google Scholar] [CrossRef]
- Donvito, G.; Nass, S.R.; Wilkerson, J.L.; Curry, Z.A.; Schurman, L.D.; Kinsey, S.G.; Lichtman, A.H. The endogenous cannabinoid system: A budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacology 2018, 43, 52–79. [Google Scholar] [CrossRef]
- Cabral, G.A.; Griffin-Thomas, L. Emerging role of the cannabinoid receptor CB2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [Google Scholar] [CrossRef] [PubMed]
- Soethoudt, M.; Grether, U.; Fingerle, J.; Grim, T.W.; Fezza, F.; De Petrocellis, L.; Ullmer, C.; Rothenhäusler, B.; Perret, C.; Van Gils, N.; et al. Cannabinoid CB2 receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 2017, 8, 13958. [Google Scholar] [CrossRef] [PubMed]
- Sunil, V.R.; Vayas, K.N.; Abramova, E.V.; Rancourt, R.; Cervelli, J.A.; Malaviya, R.; Goedken, M.; Venosa, A.; Gow, A.J.; Laskin, J.D.; et al. Lung injury, oxidative stress and fibrosis in mice following exposure to nitrogen mustard. Toxicol. Appl. Pharmacol. 2020, 387, 114798. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Hernandez, A.; Mendoza, R.P.; Nguyen, K.; Harder, A.; Evans, C.M.; Bauer, A.K.; Tewari-Singh, N.; Brown, J.M. Mast Cells Promote Nitrogen Mustard-Mediated Toxicity in the Lung Associated with Proinflammatory Cytokine and Bioactive Lipid Mediator Production. Toxicol. Sci. 2021, 184, 127–141. [Google Scholar] [CrossRef]
- Smith, L.C.; Venosa, A.; Gow, A.J.; Laskin, J.D.; Laskin, D.L. Transcriptional profiling of lung macrophages during pulmonary injury induced by nitrogen mustard. Ann. N. Y. Acad. Sci. 2020, 1480, 146–154. [Google Scholar] [CrossRef]
- Chen, X.; Tang, J.; Shuai, W.; Meng, J.; Feng, J.; Han, Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef]
- Braga, T.T.; Agudelo, J.S.; Camara, N.O. Macrophages during the fibrotic process: M2 as friend and foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef]
- McClintock, S.D.; Hoesel, L.M.; Das, S.K.; Till, G.O.; Neff, T.; Kunkel, R.G.; Smith, M.G.; Ward, P.A. Attenuation of half sulfur mustard gas-induced acute lung injury in rats. Appl. Toxicol. 2006, 26, 126–131. [Google Scholar] [CrossRef]
- Day, B.J. Oxidative Stress: An intersection Between radiation and sulfur mustard lung injury. Disaster Med. Public Health Prep. 2024, 18, e86. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Pan, H.; Patel, V.; Mukhopadhyay, B.; Bátkai, S.; Gao, B.; Haskó, G.; Pacher, P. Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free. Radic. Biol. Med. 2010, 48, 457–467. [Google Scholar] [CrossRef]
- Huang, W.; Xiong, Y.; Chen, Y.; Cheng, Y.; Wang, R. NOX2 is involved in CB2-mediated protection against lung ischemia-reperfusion injury in mice. Int. J. Clin. Exp. Pathol. 2020, 13, 277–285. [Google Scholar] [PubMed] [PubMed Central]
- Zheng, R.; Po, I.; Mishin, V.; Black, A.T.; Heck, D.E.; Laskin, D.L.; Sinko, P.J.; Gerecke, D.R.; Gordon, M.K.; Laskin, J.D. The generation of 4-hydroxynonenal, an electrophilic lipid peroxidation end product, in rabbit cornea organ cultures treated with UVB light and nitrogen mustard. Toxicol. Appl. Pharmacol. 2013, 272, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Nagre, N.; Cong, X.; Terrazas, C.; Pepper, I.; Schreiber, J.M.; Fu, H.; Sill, J.M.; Christman, J.W.; Satoskar, A.R.; Zhao, X. inhibition of macrophage complement receptor CRIg by TRIM72 polarizes innate immunity of the lung. Am. J. Respir. Cell Mol. Biol. 2018, 58, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Devos, F.C.; Maaske, A.; Robichaud, A.; Pollaris, L.; Seys, S.; Lopez, C.A.; Verbeken, E.; Tenbusch, M.; Lories, R.; Nemery, B.; et al. Forced expiration measurements in mouse models of obstructive and restrictive lung diseases. Respir. Res. 2017, 18, 123. [Google Scholar] [CrossRef]
- Salazar, E.; Knowles, J.H.; Schwingshackl, A.; Lopez, B.; Teng, B.; Lesage, F.; Belperio, J.; Olcese, R.; Waters, C.M.; Robichaud, A.; et al. An analysis of pressure-volume characteristics of the lungs. J. Appl. Physiol. 1964, 19, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Bormann, T.; Maus, R.; Stolper, J.; Tarrés, M.T.; Brandenberger, C.; Wedekind, D.; Jonigk, D.; Welte, T.; Gauldie, J.; Kolb, M.; et al. Role of matrix metalloprotease-2 and MMP-9 in experimental lung fibrosis in mice. Respir. Res. 2022, 23, 180. [Google Scholar] [CrossRef]
- Cui, H.; Jiang, D.; Banerjee, S.; Xie, N.; Kulkarni, T.; Liu, R.-M.; Duncan, S.R.; Liu, G. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight 2020, 5, e134539. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.; Chen, J.; Sang, T.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. Overexpression of NAG-1/GDF15 prevents hepatic steatosis through inhibiting oxidative stress-mediated dsDNA release and AIM2 inflammasome activation. Redox Biol. 2022, 52, 102322. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicholson, G.; Richards, N.; Lockett, J.; Ly, M.B.; Nair, R.V.; Kim, W.-K.; Vinod, K.Y.; Nagre, N. Cannabinoid-2 Receptor Activation Attenuates Sulfur Mustard Analog 2-Chloroethyl-Ethyl-Sulfide-Induced Acute Lung Injury in Mice. Pharmaceuticals 2025, 18, 236. https://doi.org/10.3390/ph18020236
Nicholson G, Richards N, Lockett J, Ly MB, Nair RV, Kim W-K, Vinod KY, Nagre N. Cannabinoid-2 Receptor Activation Attenuates Sulfur Mustard Analog 2-Chloroethyl-Ethyl-Sulfide-Induced Acute Lung Injury in Mice. Pharmaceuticals. 2025; 18(2):236. https://doi.org/10.3390/ph18020236
Chicago/Turabian StyleNicholson, Gregory, Nicholas Richards, Janette Lockett, My Boi Ly, Raj V. Nair, Woong-Ki Kim, K. Yaragudri Vinod, and Nagaraja Nagre. 2025. "Cannabinoid-2 Receptor Activation Attenuates Sulfur Mustard Analog 2-Chloroethyl-Ethyl-Sulfide-Induced Acute Lung Injury in Mice" Pharmaceuticals 18, no. 2: 236. https://doi.org/10.3390/ph18020236
APA StyleNicholson, G., Richards, N., Lockett, J., Ly, M. B., Nair, R. V., Kim, W.-K., Vinod, K. Y., & Nagre, N. (2025). Cannabinoid-2 Receptor Activation Attenuates Sulfur Mustard Analog 2-Chloroethyl-Ethyl-Sulfide-Induced Acute Lung Injury in Mice. Pharmaceuticals, 18(2), 236. https://doi.org/10.3390/ph18020236