

Facilitation of Tumor Stroma-Targeted Therapy: Model Difficulty and Co-Culture Organoid Method


Abstract
1. Introduction
2. Challenges in Tumor Stroma Mimicry Modeling
2.1. Intricate Stroma Composition
2.1.1. CAFs
2.1.2. MSC
2.1.3. Other Cells
2.1.4. ECM
2.2. Complex Tumor–Stroma Crosstalk
2.2.1. Crosstalk Approaches
2.2.2. Phenotypes Influenced by Crosstalk
2.2.3. Mutual Reaction
2.2.4. Interaction Within Stroma
2.3. Adaptive Dynamic Remodeling
2.3.1. Multidirectional Remodeling Process
2.3.2. Ambiguous Bilateral Relationship
2.3.3. Cascade Bio-Effect
2.4. Multidimensional Stroma Heterogeneity
2.4.1. Temporal Stroma Heterogeneity
2.4.2. Spatial Stroma Heterogeneity
2.4.3. Heterogeneity Among Different Tumor Types
3. Preclinical Models in Stroma-Targeted Therapy Development
3.1. In Vivo Stromal Models
3.1.1. Spontaneous and Experimental Tumor Animal Models
3.1.2. Genetically Engineered Mice Model (GEMM)
3.1.3. Xenograft Model
3.1.4. Syngeneic Tumor Models
3.2. In Vitro Stromal Models
3.2.1. Two-Dimensional Culture Model
3.2.2. Spheroids, Heterospheroids, and Multicellular Tumor Spheroids (MCTS)
3.2.3. Tumor Organoids
3.2.4. Models Based on Scaffolds
3.2.5. Tumor-on-a-Chip (TOC)
3.3. Ex Vivo Stromal Models
Tumor Slice Culture (TSC)
3.4. In Silico Stromal Models
3.4.1. Public Sequencing Data
3.4.2. Machine Learning (ML)
3.4.3. Computational Models
4. Co-Culture PDTO Application in Stroma Imitation
4.1. Classic Co-Culture
4.1.1. Co-Culture with CAFs
4.1.2. Co-Culture with Other Stromal Cells
4.1.3. Co-Culture with Other TME Components
4.2. Next Generation Co-Culture
4.2.1. Tumor-Organoid-on-a-Chip
4.2.2. Assembloids
4.2.3. Bionic Scaffold
4.2.4. Air–Liquid Interface (ALI)
4.2.5. Patient-Derived Tumor-like Cell Clusters (PTC)
5. Is Co-Cultured PDTO Competent in Stroma-Related Therapy Development?
5.1. Strengths
5.2. Weaknesses
5.3. Opportunity
The Future Trajectory of Co-Cultured PDTO Rests Upon Strategic Combinations
5.4. Treats
6. Conclusions and Discussions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Imparato, G.; Urciuolo, F.; Mazio, C.; Netti, P.A. Capturing the spatial and temporal dynamics of tumor stroma for on-chip optimization of microenvironmental targeting nanomedicine. Lab Chip 2022, 23, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, T.; Xia, R.; Wei, Y.; Wei, X. Targeting the tumor stroma for cancer therapy. Mol. Cancer 2022, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Niu, Y.R.; Wang, Z.H.; Ye, L.L.; Peng, W.B.; Zhou, Q. Cancer-associated fibroblasts: Vital suppressors of the immune response in the tumor microenvironment. Cytokine Growth Factor Rev. 2022, 67, 35–48. [Google Scholar] [CrossRef]
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A. Paget’s “Seed and Soil” Theory of Cancer Metastasis: An Idea Whose Time has Come. Adv. Anat. Pathol. 2019, 26, 69–74. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Ma, H.; Wang, J.; Zhao, X.; Wu, T.; Huang, Z.; Chen, D.; Liu, Y.; Ouyang, G. Periostin Promotes Colorectal Tumorigenesis through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Rep. 2020, 30, 793–806.e6. [Google Scholar] [CrossRef]
- Wang, H.; Xu, H.; Chen, W.; Cheng, M.; Zou, L.; Yang, Q.; Chan, C.B.; Zhu, H.; Chen, C.; Nie, J.; et al. Rab13 Sustains Breast Cancer Stem Cells by Supporting Tumor-Stroma Cross-talk. Cancer Res. 2022, 82, 2124–2140. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef]
- Akkoc, Y.; Dalci, K.; Karakas, H.E.; Erbil-Bilir, S.; Yalav, O.; Sakman, G.; Celik, F.; Arikan, S.; Zeybek, U.; Ergin, M.; et al. Tumor-derived CTF1 (cardiotrophin 1) is a critical mediator of stroma-assisted and autophagy-dependent breast cancer cell migration, invasion and metastasis. Autophagy 2023, 19, 306–323. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; Correa de Sampaio, P.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X.; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Maeda, M.; Takeshima, H.; Iida, N.; Hattori, N.; Yamashita, S.; Moro, H.; Yasukawa, Y.; Nishiyama, K.; Hashimoto, T.; Sekine, S.; et al. Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut 2020, 69, 243–251. [Google Scholar] [CrossRef]
- Mo, Y.; Leung, L.L.; Mak, C.S.L.; Wang, X.; Chan, W.S.; Hui, L.M.N.; Tang, H.W.M.; Siu, M.K.Y.; Sharma, R.; Xu, D.; et al. Tumor-secreted exosomal miR-141 activates tumor-stroma interactions and controls premetastatic niche formation in ovarian cancer metastasis. Mol. Cancer 2023, 22, 4. [Google Scholar] [CrossRef]
- Masugi, Y. The Desmoplastic Stroma of Pancreatic Cancer: Multilayered Levels of Heterogeneity, Clinical Significance, and Therapeutic Opportunities. Cancers 2022, 14, 3293. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e422. [Google Scholar] [CrossRef]
- Itoh, G.; Takagane, K.; Fukushi, Y.; Kuriyama, S.; Umakoshi, M.; Goto, A.; Yanagihara, K.; Yashiro, M.; Tanaka, M. Cancer-associated fibroblasts educate normal fibroblasts to facilitate cancer cell spreading and T-cell suppression. Mol. Oncol. 2022, 16, 166–187. [Google Scholar] [CrossRef]
- Uddin, M.N.; Wang, X. Identification of key tumor stroma-associated transcriptional signatures correlated with survival prognosis and tumor progression in breast cancer. Breast Cancer 2022, 29, 541–561. [Google Scholar] [CrossRef]
- Luga, V.; Wrana, J.L. Tumor-stroma interaction: Revealing fibroblast-secreted exosomes as potent regulators of Wnt-planar cell polarity signaling in cancer metastasis. Cancer Res. 2013, 73, 6843–6847. [Google Scholar] [CrossRef]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: Opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Cioni, B.; Zaalberg, A.; van Beijnum, J.R.; Melis, M.H.M.; van Burgsteden, J.; Muraro, M.J.; Hooijberg, E.; Peters, D.; Hofland, I.; Lubeck, Y.; et al. Androgen receptor signalling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat. Commun. 2020, 11, 4498. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Aspizua, S.; González-Masa, A.; Conti, C.J.; García, M.; Chacón-Solano, E.; Larcher, F.; Del Río, M. Humanization of Tumor Stroma by Tissue Engineering as a Tool to Improve Squamous Cell Carcinoma Xenograft. Int. J. Mol. Sci. 2020, 21, 1951. [Google Scholar] [CrossRef]
- Ogawa, Y.; Masugi, Y.; Abe, T.; Yamazaki, K.; Ueno, A.; Fujii-Nishimura, Y.; Hori, S.; Yagi, H.; Abe, Y.; Kitago, M.; et al. Three Distinct Stroma Types in Human Pancreatic Cancer Identified by Image Analysis of Fibroblast Subpopulations and Collagen. Clin. Cancer Res. 2021, 27, 107–119. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, X.; Wang, S.; Trinkle, C. Tumor organoid models in precision medicine and investigating cancer-stromal interactions. Pharmacol. Ther. 2021, 218, 107668. [Google Scholar] [CrossRef]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflamm. 2018, 15, 259. [Google Scholar] [CrossRef]
- Colombo, E.; Cattaneo, M.G. Multicellular 3D Models to Study Tumour-Stroma Interactions. Int. J. Mol. Sci. 2021, 22, 1633. [Google Scholar] [CrossRef]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, 38, e101654. [Google Scholar] [CrossRef]
- Lu, T.; Yang, B.; Wang, R.; Qin, C. Xenotransplantation: Current Status in Preclinical Research. Front. Immunol. 2019, 10, 3060. [Google Scholar] [CrossRef]
- Yuan, J.; Li, X.; Yu, S. Cancer organoid co-culture model system: Novel approach to guide precision medicine. Front. Immunol. 2022, 13, 1061388. [Google Scholar] [CrossRef]
- Gjorevski, N.; Nikolaev, M.; Brown, T.E.; Mitrofanova, O.; Brandenberg, N.; DelRio, F.W.; Yavitt, F.M.; Liberali, P.; Anseth, K.S.; Lutolf, M.P. Tissue geometry drives deterministic organoid patterning. Science 2022, 375, eaaw9021. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Kramer, M.; Russo, S.; Naik, P.; Arun, G.; Brophy, K.; Andrews, P.; Fan, C.; Perou, C.M.; Preall, J.; et al. Patient-Derived Triple-Negative Breast Cancer Organoids Provide Robust Model Systems That Recapitulate Tumor Intrinsic Characteristics. Cancer Res. 2022, 82, 1174–1192. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.; Liang, X.; Zhang, Y.; Zhao, C.X.; Roberts, M.S.; Wang, H.; Zhang, L.; Crawford, D.H.G. Liver organoid as a 3D in vitro model for drug validation and toxicity assessment. Pharmacol. Res. 2021, 169, 105608. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fu, G.; Zhang, L.; Guan, R.; Tang, P.; Zhang, J.; Rao, X.; Chen, S.; Xu, X.; Zhou, Y.; et al. Assay establishment and validation of a high-throughput organoid-based drug screening platform. Stem Cell Res. Ther. 2022, 13, 219. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef]
- Lewis-Israeli, Y.R.; Wasserman, A.H.; Gabalski, M.A.; Volmert, B.D.; Ming, Y.; Ball, K.A.; Yang, W.; Zou, J.; Ni, G.; Pajares, N.; et al. Self-assembling human heart organoids for the modeling of cardiac development and congenital heart disease. Nat. Commun. 2021, 12, 5142. [Google Scholar] [CrossRef]
- Ebner-Peking, P.; Krisch, L.; Wolf, M.; Hochmann, S.; Hoog, A.; Vári, B.; Muigg, K.; Poupardin, R.; Scharler, C.; Schmidhuber, S.; et al. Self-assembly of differentiated progenitor cells facilitates spheroid human skin organoid formation and planar skin regeneration. Theranostics 2021, 11, 8430–8447. [Google Scholar] [CrossRef]
- Zheng, Y.L.; Hu, N.; Sun, Q.; Wang, C.; Taylor, P.R. Telomere attrition in cancer cells and telomere length in tumor stroma cells predict chromosome instability in esophageal squamous cell carcinoma: A genome-wide analysis. Cancer Res. 2009, 69, 1604–1614. [Google Scholar] [CrossRef]
- Saini, H.; Nikkhah, M. Fabrication Method of a High-Density Co-Culture Tumor-Stroma Platform to Study Cancer Progression. Methods Mol. Biol. 2021, 2258, 241–255. [Google Scholar] [CrossRef]
- Ho, T.; Msallam, R. Tissues and Tumor Microenvironment (TME) in 3D: Models to Shed Light on Immunosuppression in Cancer. Cells 2021, 10, 831. [Google Scholar] [CrossRef]
- Hahn, S.; Oh, B.J.; Kim, H.; Han, I.W.; Shin, S.H.; Kim, G.; Jin, S.M.; Kim, J.H. Anti-cancer effects of metformin in a 3D co-culture model of pancreatic ductal adenocarcinoma. Am. J. Cancer Res. 2023, 13, 1806–1825. [Google Scholar] [PubMed]
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.A.; Ma, B.; Su, J.; et al. Cancer-Associated Fibroblasts Provide a Stromal Niche for Liver Cancer Organoids That Confers Trophic Effects and Therapy Resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, X.; Ding, R.; Yang, L.; Lyu, X.; Zeng, J.; Lei, J.H.; Wang, L.; Bi, J.; Shao, N.; et al. Patient-Derived Organoids Can Guide Personalized-Therapies for Patients with Advanced Breast Cancer. Adv. Sci. 2021, 8, e2101176. [Google Scholar] [CrossRef]
- Kaps, L.; Schuppan, D. Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells 2020, 9, 2027. [Google Scholar] [CrossRef]
- Sandberg, T.P.; Stuart, M.; Oosting, J.; Tollenaar, R.; Sier, C.F.M.; Mesker, W.E. Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer 2019, 19, 284. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, X.; An, R.; Bai, J.; Dong, J.; Cai, H.; Zhu, H.; Zhong, W.; Chen, W.; Liu, A.; et al. Peritumoral tertiary lymphoid structure and tumor stroma percentage predict the prognosis of patients with non-metastatic colorectal cancer. Front. Immunol. 2022, 13, 962056. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, J.; Koh, V.; So, J.B.Y.; Yong, W.P.; Zavros, Y. A Preclinical Human-Derived Autologous Gastric Cancer Organoid/Immune Cell Co-Culture Model to Predict the Efficacy of Targeted Therapies. J. Vis. Exp. 2021, 173, e61443. [Google Scholar] [CrossRef]
- Magré, L.; Verstegen, M.M.A.; Buschow, S.; van der Laan, L.J.W.; Peppelenbosch, M.; Desai, J. Emerging organoid-immune co-culture models for cancer research: From oncoimmunology to personalized immunotherapies. J. Immunother. Cancer 2023, 11, e006290. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e1916. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e1512. [Google Scholar] [CrossRef]
- Rodrigues, J.; Heinrich, M.A.; Teixeira, L.M.; Prakash, J. 3D In Vitro Model (R)evolution: Unveiling Tumor-Stroma Interactions. Trends Cancer 2021, 7, 249–264. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef]
- Hu, D.; Li, Z.; Zheng, B.; Lin, X.; Pan, Y.; Gong, P.; Zhuo, W.; Hu, Y.; Chen, C.; Chen, L.; et al. Cancer-associated fibroblasts in breast cancer: Challenges and opportunities. Cancer Commun. 2022, 42, 401–434. [Google Scholar] [CrossRef]
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordoñez, A.; Reina-Campos, M.; Kuo, H.H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The lactate-NAD+ axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. 2022, 39, 110792. [Google Scholar] [CrossRef]
- Arebro, J.; Towle, R.; Lee, C.M.; Bennewith, K.L.; Garnis, C. Extracellular vesicles promote activation of pro-inflammatory cancer-associated fibroblasts in oral cancer. Front. Cell Dev. Biol. 2023, 11, 1240159. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Wang, Z. CAF heterogeneity and dynamics. Nat. Cell Biol. 2022, 24, 1686. [Google Scholar] [CrossRef]
- Luo, H.; Xia, X.; Huang, L.B.; An, H.; Cao, M.; Kim, G.D.; Chen, H.N.; Zhang, W.H.; Shu, Y.; Kong, X.; et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 2022, 13, 6619. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.C.; Chung, J.Y.; Xue, V.W.; Xiao, J.; Meng, X.M.; Huang, X.R.; Zhou, S.; Chan, A.S.; Tsang, A.C.; Cheng, A.S.; et al. Smad3 Promotes Cancer-Associated Fibroblasts Generation via Macrophage-Myofibroblast Transition. Adv. Sci. 2022, 9, e2101235. [Google Scholar] [CrossRef]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef]
- Park, S.J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K.; et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 173, 3844–3854. [Google Scholar] [CrossRef]
- Chomarat, P.; Banchereau, J.; Davoust, J.; Palucka, A.K. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 2000, 1, 510–514. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Hamberger, F.; Ravichandra, A.; Miller, M.; Nair, A.; Affo, S.; Filliol, A.; Chin, L.; Savage, T.M.; Yin, D.; et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Investig. 2021, 131, e146987. [Google Scholar] [CrossRef]
- Hughes, R.M.; Simons, B.W.; Khan, H.; Miller, R.; Kugler, V.; Torquato, S.; Theodros, D.; Haffner, M.C.; Lotan, T.; Huang, J.; et al. Asporin Restricts Mesenchymal Stromal Cell Differentiation, Alters the Tumor Microenvironment, and Drives Metastatic Progression. Cancer Res. 2019, 79, 3636–3650. [Google Scholar] [CrossRef]
- Canzonetta, C.; Pelosi, A.; Di Matteo, S.; Veneziani, I.; Tumino, N.; Vacca, P.; Munari, E.; Pezzullo, M.; Theuer, C.; De Vito, R.; et al. Identification of neuroblastoma cell lines with uncommon TAZ+/mesenchymal stromal cell phenotype with strong suppressive activity on natural killer cells. J. Immunother. Cancer 2021, 9, e001313. [Google Scholar] [CrossRef]
- Kishi, S.; Fujiwara-Tani, R.; Honoki, K.; Sasaki, R.; Mori, S.; Ohmori, H.; Sasaki, T.; Miyagawa, Y.; Kawahara, I.; Kido, A.; et al. Oxidized high mobility group B-1 enhances metastability of colorectal cancer via modification of mesenchymal stem/stromal cells. Cancer Sci. 2022, 113, 2904–2915. [Google Scholar] [CrossRef]
- Tu, Z.; Karnoub, A.E. Mesenchymal stem/stromal cells in breast cancer development and management. Semin. Cancer Biol. 2022, 86, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Timaner, M.; Tsai, K.K.; Shaked, Y. The multifaceted role of mesenchymal stem cells in cancer. Semin. Cancer Biol. 2020, 60, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Luo, M.; Wei, X. Mesenchymal stem/stromal cells in cancer therapy. J. Hematol. Oncol. 2021, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Atiya, H.I.; Frisbie, L.; Goldfeld, E.; Orellana, T.; Donnellan, N.; Modugno, F.; Calderon, M.; Watkins, S.; Zhang, R.; Elishaev, E.; et al. Endometriosis-Associated Mesenchymal Stem Cells Support Ovarian Clear Cell Carcinoma through Iron Regulation. Cancer Res. 2022, 82, 4680–4693. [Google Scholar] [CrossRef]
- Zarubova, J.; Hasani-Sadrabadi, M.M.; Norris, S.C.P.; Majedi, F.S.; Xiao, C.; Kasko, A.M.; Li, S. Cell-Taxi: Mesenchymal Cells Carry and Transport Clusters of Cancer Cells. Small 2022, 18, e2203515. [Google Scholar] [CrossRef]
- Pasanen, I.; Pietilä, M.; Lehtonen, S.; Lehtilahti, E.; Hakkarainen, T.; Blanco Sequeiros, R.; Lehenkari, P.; Kuvaja, P. Mesenchymal stromal cells from female donors enhance breast cancer cell proliferation in vitro. Oncology 2015, 88, 214–225. [Google Scholar] [CrossRef]
- Yu, S.; Zhou, Y.; Niu, L.; Qiao, Y.; Yan, Y. Mesenchymal stem cell-derived exosome mir-342-3p inhibits metastasis and chemo-resistance of breast cancer through regulating ID4. Genes Genom. 2022, 44, 539–550. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef]
- Olejarz, W.; Kubiak-Tomaszewska, G.; Chrzanowska, A.; Lorenc, T. Exosomes in Angiogenesis and Anti-angiogenic Therapy in Cancers. Int. J. Mol. Sci. 2020, 21, 5840. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.V.; Luo, T.; Hass, R. Spontaneous Fusion of MSC with Breast Cancer Cells Can Generate Tumor Dormancy. Int. J. Mol. Sci. 2021, 22, 5930. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. MSC stimulate ovarian tumor growth during intercellular communication but reduce tumorigenicity after fusion with ovarian cancer cells. Cell Commun. Signal. 2018, 16, 67. [Google Scholar] [CrossRef] [PubMed]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in cancer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250, 555–572. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.K.; Englisch, A.; Brenner, B.; Smith, T.; Hoyos, V.; Suzuki, M.; Brenner, M.K. Mesenchymal stromal cell delivery of oncolytic immunotherapy improves CAR-T cell antitumor activity. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Zhang, B.; Wu, C.; Yu, F.; Han, B.; Li, B.; Li, L. Therapeutic roles of mesenchymal stem cell-derived extracellular vesicles in cancer. J. Hematol. Oncol. 2021, 14, 136. [Google Scholar] [CrossRef]
- Cook, L.M.; Frieling, J.S.; Nerlakanti, N.; McGuire, J.J.; Stewart, P.A.; Burger, K.L.; Cleveland, J.L.; Lynch, C.C. Betaglycan drives the mesenchymal stromal cell osteogenic program and prostate cancer-induced osteogenesis. Oncogene 2019, 38, 6959–6969. [Google Scholar] [CrossRef]
- Wang, S.; Su, X.; Xu, M.; Xiao, X.; Li, X.; Li, H.; Keating, A.; Zhao, R.C. Exosomes secreted by mesenchymal stromal/stem cell-derived adipocytes promote breast cancer cell growth via activation of Hippo signaling pathway. Stem Cell Res. Ther. 2019, 10, 117. [Google Scholar] [CrossRef]
- Cogliati, B.; Yashaswini, C.N.; Wang, S.; Sia, D.; Friedman, S.L. Friend or foe? The elusive role of hepatic stellate cells in liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 647–661. [Google Scholar] [CrossRef]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef]
- Nan, L.; Qin, T.; Xiao, Y.; Qian, W.; Li, J.; Wang, Z.; Ma, J.; Ma, Q.; Wu, Z. Pancreatic Stellate Cells Facilitate Perineural Invasion of Pancreatic Cancer via HGF/c-Met Pathway. Cell Transplant. 2019, 28, 1289–1298. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhou, J.; Li, L.; Liao, S.; He, J.; Zhou, S.; Zhou, Y. Pericytes in the tumor microenvironment. Cancer Lett. 2023, 556, 216074. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.M.; Jiang, X.; Zhao, X.; Meng, Q.; Wu, S.; Chen, Y.; Kong, X.; Qiu, X.; Su, L.; Huang, C.; et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat. Commun. 2021, 12, 6011. [Google Scholar] [CrossRef] [PubMed]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yao, J.; Yan, M.; Xie, Y.; Liu, P.; Mao, Y.; Li, X. The role of matrix stiffness in cancer stromal cell fate and targeting therapeutic strategies. Acta Biomater. 2022, 150, 34–47. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, H.; Wang, J.; Liu, Y.; Luo, T.; Hua, H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J. Hematol. Oncol. 2022, 15, 34. [Google Scholar] [CrossRef]
- Wu, B.; Liu, D.A.; Guan, L.; Myint, P.K.; Chin, L.; Dang, H.; Xu, Y.; Ren, J.; Li, T.; Yu, Z.; et al. Stiff matrix induces exosome secretion to promote tumour growth. Nat. Cell Biol. 2023, 25, 415–424. [Google Scholar] [CrossRef]
- Patwardhan, S.; Mahadik, P.; Shetty, O.; Sen, S. ECM stiffness-tuned exosomes drive breast cancer motility through thrombospondin-1. Biomaterials 2021, 279, 121185. [Google Scholar] [CrossRef]
- Wei, X.; Lou, H.; Zhou, D.; Jia, Y.; Li, H.; Huang, Q.; Ma, J.; Yang, Z.; Sun, C.; Meng, Y.; et al. TAGLN mediated stiffness-regulated ovarian cancer progression via RhoA/ROCK pathway. J. Exp. Clin. Cancer Res. 2021, 40, 292. [Google Scholar] [CrossRef]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef]
- Cottler, P.S.; Kang, H.; Nash, V.; Salopek, L.; Bruce, A.C.; Spiller, K.L.; Campbell, C.A. Immunomodulation of Acellular Dermal Matrix Through Interleukin 4 Enhances Vascular Infiltration. Ann. Plast. Surg. 2022, 88, S466–S472. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Chen, H.; Lee, B.H.; Lee, J.Y.H.; Yip, Y.S.; Tan, N.S.; Tan, L.P. Epithelial-mesenchymal transition of cancer cells using bioengineered hybrid scaffold composed of hydrogel/3D-fibrous framework. Sci. Rep. 2019, 9, 8997. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, P.; Briatico-Vangosa, F.; Bianchi, E.; Pellegata, A.F.; Hartung de Hartungen, A.; Corti, P.; Dubini, G. Bioprinting of Matrigel Scaffolds for Cancer Research. Polymers 2021, 13, 2026. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.X.; Zhou, B.; Cheng, Y.G.; Xu, J.W.; Wang, L.; Zhang, G.Y.; Hu, S.Y. Crosstalk between stromal cells and cancer cells in pancreatic cancer: New insights into stromal biology. Cancer Lett. 2017, 392, 83–93. [Google Scholar] [CrossRef]
- Kemi, N.; Eskuri, M.; Herva, A.; Leppänen, J.; Huhta, H.; Helminen, O.; Saarnio, J.; Karttunen, T.J.; Kauppila, J.H. Tumour-stroma ratio and prognosis in gastric adenocarcinoma. Br. J. Cancer 2018, 119, 435–439. [Google Scholar] [CrossRef]
- Yang, J.; Ye, H.; Fan, X.; Li, Y.; Wu, X.; Zhao, M.; Hu, Q.; Ye, Y.; Wu, L.; Li, Z.; et al. Artificial intelligence for quantifying immune infiltrates interacting with stroma in colorectal cancer. J. Transl. Med. 2022, 20, 451. [Google Scholar] [CrossRef]
- Ma, Z.; Li, X.; Mao, Y.; Wei, C.; Huang, Z.; Li, G.; Yin, J.; Liang, X.; Liu, Z. Interferon-dependent SLC14A1+ cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell 2022, 40, 1550–1565.e1557. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Uchihara, T.; Miyake, K.; Yonemura, A.; Komohara, Y.; Itoyama, R.; Koiwa, M.; Yasuda, T.; Arima, K.; Harada, K.; Eto, K.; et al. Extracellular Vesicles from Cancer-Associated Fibroblasts Containing Annexin A6 Induces FAK-YAP Activation by Stabilizing β1 Integrin, Enhancing Drug Resistance. Cancer Res. 2020, 80, 3222–3235. [Google Scholar] [CrossRef]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef]
- Becker, L.M.; O’Connell, J.T.; Vo, A.P.; Cain, M.P.; Tampe, D.; Bizarro, L.; Sugimoto, H.; McGow, A.K.; Asara, J.M.; Lovisa, S.; et al. Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020, 31, 107701. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e110. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Chen, D.; Cai, J.; Yuan, Z.; Huang, B.; Li, Y.; Wang, H.; Luo, Q.; Kuang, Y.; Liang, W.; et al. Enhancing cancer-associated fibroblast fatty acid catabolism within a metabolically challenging tumor microenvironment drives colon cancer peritoneal metastasis. Mol. Oncol. 2021, 15, 1391–1411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Qi, R.; Bai, Y.; Li, K.; Liu, N.; Xu, Y.; Dal, E.; Wang, Y.; Lin, R.; Wang, H.; Liu, Z.; et al. Cancer-associated fibroblasts suppress ferroptosis and induce gemcitabine resistance in pancreatic cancer cells by secreting exosome-derived ACSL4-targeting miRNAs. Drug Resist. Updates 2023, 68, 100960. [Google Scholar] [CrossRef]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- You, J.; Li, M.; Cao, L.M.; Gu, Q.H.; Deng, P.B.; Tan, Y.; Hu, C.P. Snail1-dependent cancer-associated fibroblasts induce epithelial-mesenchymal transition in lung cancer cells via exosomes. QJM Mon. J. Assoc. Physicians 2019, 112, 581–590. [Google Scholar] [CrossRef]
- Shi, L.; Zhu, W.; Huang, Y.; Zhuo, L.; Wang, S.; Chen, S.; Zhang, B.; Ke, B. Cancer-associated fibroblast-derived exosomal microRNA-20a suppresses the PTEN/PI3K-AKT pathway to promote the progression and chemoresistance of non-small cell lung cancer. Clin. Transl. Med. 2022, 12, e989. [Google Scholar] [CrossRef]
- Chen, B.; Sang, Y.; Song, X.; Zhang, D.; Wang, L.; Zhao, W.; Liang, Y.; Zhang, N.; Yang, Q. Exosomal miR-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting USP28. Theranostics 2021, 11, 3932–3947. [Google Scholar] [CrossRef]
- Li, Z.; Low, V.; Luga, V.; Sun, J.; Earlie, E.; Parang, B.; Shobana Ganesh, K.; Cho, S.; Endress, J.; Schild, T.; et al. Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression. Nat. Commun. 2022, 13, 6239. [Google Scholar] [CrossRef]
- Zhao, X.; He, Y.; Chen, H. Autophagic tumor stroma: Mechanisms and roles in tumor growth and progression. Int. J. Cancer 2013, 132, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Tu, B.; Li, H.; Pang, H.; Zhang, N.; Fan, M.; Bai, J.; Wang, W.; Shu, Z.; DuFort, C.C.; et al. Cancer-associated fibroblasts employ NUFIP1-dependent autophagy to secrete nucleosides and support pancreatic tumor growth. Nat. Cancer 2022, 3, 945–960. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xu, G.; Wang, Y.; Xu, Z.; Liu, X.; Xu, X.; Ren, G.; Tian, K. Oxidative stress induced autophagy in cancer associated fibroblast enhances proliferation and metabolism of colorectal cancer cells. Cell Cycle 2017, 16, 73–81. [Google Scholar] [CrossRef]
- Pan, Z.; Xu, T.; Bao, L.; Hu, X.; Jin, T.; Chen, J.; Chen, J.; Qian, Y.; Lu, X.; Li, L.; et al. CREB3L1 promotes tumor growth and metastasis of anaplastic thyroid carcinoma by remodeling the tumor microenvironment. Mol. Cancer 2022, 21, 190. [Google Scholar] [CrossRef]
- Li, X.; Yong, T.; Wei, Z.; Bie, N.; Zhang, X.; Zhan, G.; Li, J.; Qin, J.; Yu, J.; Zhang, B.; et al. Reversing insufficient photothermal therapy-induced tumor relapse and metastasis by regulating cancer-associated fibroblasts. Nat. Commun. 2022, 13, 2794. [Google Scholar] [CrossRef]
- Chen, S.; Morine, Y.; Tokuda, K.; Yamada, S.; Saito, Y.; Nishi, M.; Ikemoto, T.; Shimada, M. Cancer-associated fibroblast-induced M2-polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor-1 pathway. Int. J. Oncol. 2021, 59, 59. [Google Scholar] [CrossRef]
- Timperi, E.; Gueguen, P.; Molgora, M.; Magagna, I.; Kieffer, Y.; Lopez-Lastra, S.; Sirven, P.; Baudrin, L.G.; Baulande, S.; Nicolas, A.; et al. Lipid-Associated Macrophages Are Induced by Cancer-Associated Fibroblasts and Mediate Immune Suppression in Breast Cancer. Cancer Res. 2022, 82, 3291–3306. [Google Scholar] [CrossRef]
- Zeng, W.; Xiong, L.; Wu, W.; Li, S.; Liu, J.; Yang, L.; Lao, L.; Huang, P.; Zhang, M.; Chen, H.; et al. CCL18 signaling from tumor-associated macrophages activates fibroblasts to adopt a chemoresistance-inducing phenotype. Oncogene 2023, 42, 224–237. [Google Scholar] [CrossRef]
- Hong, C.L.; Yu, I.S.; Pai, C.H.; Chen, J.S.; Hsieh, M.S.; Wu, H.L.; Lin, S.W.; Huang, H.P. CD248 Regulates Wnt Signaling in Pericytes to Promote Angiogenesis and Tumor Growth in Lung Cancer. Cancer Res. 2022, 82, 3734–3750. [Google Scholar] [CrossRef]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Chen, Y.; Liu, J.T.; Bao, H.; Wang, W.B.; Qi, Y.X.; Lv, F. Extracellular matrix stiffness controls VEGF(165) secretion and neuroblastoma angiogenesis via the YAP/RUNX2/SRSF1 axis. Angiogenesis 2022, 25, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Grant, E.; Bucklain, F.A.; Ginn, L.; Laity, P.; Ciani, B.; Bryant, H.E. Progesterone receptor expression contributes to gemcitabine resistance at higher ECM stiffness in breast cancer cell lines. PLoS ONE 2022, 17, e0268300. [Google Scholar] [CrossRef]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. J. Lab. Clin. Med. 2019, 209, 55–67. [Google Scholar] [CrossRef]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Huang, D.Q.; Nguyen, M.H. Global burden of hepatitis B virus: Current status, missed opportunities and a call for action. Nature reviews. Gastroenterol. Hepatol. 2023, 20, 524–537. [Google Scholar] [CrossRef]
- Ono, H.; Murase, Y.; Yamashita, H.; Kato, T.; Asano, D.; Ishikawa, Y.; Watanabe, S.; Ueda, H.; Akahoshi, K.; Ogawa, K.; et al. RRM1 is mediated by histone acetylation through gemcitabine resistance and contributes to invasiveness and ECM remodeling in pancreatic cancer. Int. J. Oncol. 2023, 62, 51. [Google Scholar] [CrossRef]
- Sada, M.; Ohuchida, K.; Horioka, K.; Okumura, T.; Moriyama, T.; Miyasaka, Y.; Ohtsuka, T.; Mizumoto, K.; Oda, Y.; Nakamura, M. Hypoxic stellate cells of pancreatic cancer stroma regulate extracellular matrix fiber organization and cancer cell motility. Cancer Lett. 2016, 372, 210–218. [Google Scholar] [CrossRef]
- Chen, J.; Ma, L.; Zhang, N.; Zhu, Y.; Zhang, K.; Xu, Z.; Wang, Q. Mesenchymal Stem Cells Promote Tumor Progression via Inducing Stroma Remodeling on Rabbit VX2 Bladder Tumor Model. Int. J. Biol. Sci. 2018, 14, 1012–1021. [Google Scholar] [CrossRef]
- Li, X.; Pan, J.; Liu, T.; Yin, W.; Miao, Q.; Zhao, Z.; Gao, Y.; Zheng, W.; Li, H.; Deng, R.; et al. Novel TCF21high pericyte subpopulation promotes colorectal cancer metastasis by remodelling perivascular matrix. Gut 2023, 72, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Papadas, A.; Deb, G.; Cicala, A.; Officer, A.; Hope, C.; Pagenkopf, A.; Flietner, E.; Morrow, Z.T.; Emmerich, P.; Wiesner, J.; et al. Stromal remodeling regulates dendritic cell abundance and activity in the tumor microenvironment. Cell Rep. 2022, 40, 111201. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liao, C.; Hu, H.; Liao, J.; Chen, Z.; Li, S.; Zeng, X.; Peng, B.; Shen, S.; Li, D.; et al. Hypoxia-driven tumor stromal remodeling and immunosuppressive microenvironment in scirrhous hepatocellular carcinoma. Hepatology 2023, 79, 780–797. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yang, T.; Zhou, L.; Li, R.; Liu, J.; Zhao, J.; Jia, R. Sig1R activates extracellular matrix-induced bladder cancer cell proliferation and angiogenesis by combing β-integrin. Aging 2023, 15, 4182–4201. [Google Scholar] [CrossRef]
- Venning, F.A.; Zornhagen, K.W.; Wullkopf, L.; Sjölund, J.; Rodriguez-Cupello, C.; Kjellman, P.; Morsing, M.; Hajkarim, M.C.; Won, K.J.; Erler, J.T.; et al. Deciphering the temporal heterogeneity of cancer-associated fibroblast subpopulations in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 175. [Google Scholar] [CrossRef]
- Heming, M.; Haessner, S.; Wolbert, J.; Lu, I.N.; Li, X.; Brokinkel, B.; Müther, M.; Holling, M.; Stummer, W.; Thomas, C.; et al. Intratumor heterogeneity and T cell exhaustion in primary CNS lymphoma. Genome Med. 2022, 14, 109. [Google Scholar] [CrossRef]
- De Palma, M.; Hanahan, D. The biology of personalized cancer medicine: Facing individual complexities underlying hallmark capabilities. Mol. Oncol. 2012, 6, 111–127. [Google Scholar] [CrossRef]
- Schütz, S.; Solé-Boldo, L.; Lucena-Porcel, C.; Hoffmann, J.; Brobeil, A.; Lonsdorf, A.S.; Rodríguez-Paredes, M.; Lyko, F. Functionally distinct cancer-associated fibroblast subpopulations establish a tumor promoting environment in squamous cell carcinoma. Nat. Commun. 2023, 14, 5413. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; DʹAvola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef]
- Fumagalli, C.; Barberis, M. Breast Cancer Heterogeneity. Diagnostics 2021, 11, 1555. [Google Scholar] [CrossRef]
- Lee, A.T.J.; Chew, W.; Wilding, C.P.; Guljar, N.; Smith, M.J.; Strauss, D.C.; Fisher, C.; Hayes, A.J.; Judson, I.; Thway, K.; et al. The adequacy of tissue microarrays in the assessment of inter- and intra-tumoural heterogeneity of infiltrating lymphocyte burden in leiomyosarcoma. Sci. Rep. 2019, 9, 14602. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, W.; Liu, X.; Zhang, Z.; Yu, L.; Li, R.; Guo, D.; Cai, W.; Quan, X.; Wu, H.; et al. Multi-omics analysis of intra-tumoural and inter-tumoural heterogeneity in pancreatic ductal adenocarcinoma. Clin. Transl. Med. 2022, 12, e670. [Google Scholar] [CrossRef] [PubMed]
- Hegde, J.V.; Margolis, D.J.; Wang, P.C.; Reiter, R.E.; Huang, J.; Steinberg, M.L.; Kamrava, M. Establishing the distribution of satellite lesions in intermediate- and high-risk prostate cancer: Implications for focused radiotherapy. Prostate Cancer Prostatic Dis. 2017, 20, 241–248. [Google Scholar] [CrossRef] [PubMed]
- van Wilpe, S.; Gorris, M.A.J.; van der Woude, L.L.; Sultan, S.; Koornstra, R.H.T.; van der Heijden, A.G.; Gerritsen, W.R.; Simons, M.; de Vries, I.J.M.; Mehra, N. Spatial and Temporal Heterogeneity of Tumor-Infiltrating Lymphocytes in Advanced Urothelial Cancer. Front. Immunol. 2021, 12, 802877. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, L.; Danilova, L.; Mo, G.; Zhu, Q.; Deshpande, A.; Bell, A.T.F.; Elisseeff, J.; Popel, A.S.; Anders, R.A.; et al. Spatial transcriptomics analysis of neoadjuvant cabozantinib and nivolumab in advanced hepatocellular carcinoma identifies independent mechanisms of resistance and recurrence. Genome Med. 2023, 15, 72. [Google Scholar] [CrossRef]
- Ooki, A.; Yamaguchi, K. The dawn of precision medicine in diffuse-type gastric cancer. Ther. Adv. Med. Oncol. 2022, 14, 17588359221083049. [Google Scholar] [CrossRef]
- Karkampouna, S.; La Manna, F.; Benjak, A.; Kiener, M.; De Menna, M.; Zoni, E.; Grosjean, J.; Klima, I.; Garofoli, A.; Bolis, M.; et al. Patient-derived xenografts and organoids model therapy response in prostate cancer. Nat. Commun. 2021, 12, 1117. [Google Scholar] [CrossRef]
- Middleton, G.; Robbins, H.; Andre, F.; Swanton, C. A state-of-the-art review of stratified medicine in cancer: Towards a future precision medicine strategy in cancer. Ann. Oncol. 2022, 33, 143–157. [Google Scholar] [CrossRef]
- Budhwani, K.I.; Patel, Z.H.; Guenter, R.E.; Charania, A.A. A hitchhiker’s guide to cancer models. Trends Biotechnol. 2022, 40, 1361–1373. [Google Scholar] [CrossRef]
- Stribbling, S.M.; Ryan, A.J. The cell-line-derived subcutaneous tumor model in preclinical cancer research. Nat. Protoc. 2022, 17, 2108–2128. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef] [PubMed]
- Fazio, M.; Ablain, J.; Chuan, Y.; Langenau, D.M.; Zon, L.I. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer 2020, 20, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Vriesendorp, H.M.; Quadri, S.M. Radiolabeled immunoglobulin therapy: Old barriers and new opportunities. Expert. Rev. Anticancer. Ther. 2001, 1, 461–478. [Google Scholar] [CrossRef]
- Wang, L.; Piao, Y.; Guo, F.; Wei, J.; Chen, Y.; Dai, X.; Zhang, X. Current progress of pig models for liver cancer research. Biomed. Pharmacother. 2023, 165, 115256. [Google Scholar] [CrossRef]
- Nascimento, C.; Ferreira, F. Tumor microenvironment of human breast cancer, and feline mammary carcinoma as a potential study model. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188587. [Google Scholar] [CrossRef]
- Zhong, L.; Huang, Y.; He, J.; Yang, N.; Xu, B.; Ma, Y.; Liu, J.; Tang, C.; Luo, C.; Wu, P.; et al. Generation of in situ CRISPR-mediated primary and metastatic cancer from monkey liver. Signal Transduct. Target. Ther. 2021, 6, 411. [Google Scholar] [CrossRef]
- Pinho, S.S.; Carvalho, S.; Cabral, J.; Reis, C.A.; Gärtner, F. Canine tumors: A spontaneous animal model of human carcinogenesis. Transl. Res. J. Lab. Clin. Med. 2012, 159, 165–172. [Google Scholar] [CrossRef]
- May, M. Cancer research with a human touch. Nature 2018, 556, 259–261. [Google Scholar] [CrossRef]
- Makino, Y.; Hikita, H.; Kato, S.; Sugiyama, M.; Shigekawa, M.; Sakamoto, T.; Sasaki, Y.; Murai, K.; Sakane, S.; Kodama, T.; et al. STAT3 is Activated by CTGF-mediated Tumor-stroma Cross Talk to Promote HCC Progression. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 99–119. [Google Scholar] [CrossRef]
- Mahmoudian, R.A.; Farshchian, M.; Abbaszadegan, M.R. Genetically engineered mouse models of esophageal cancer. Exp. Cell Res. 2021, 406, 112757. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Melani, C.; Sangaletti, S.; Barazzetta, F.M.; Werb, Z.; Colombo, M.P. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007, 67, 11438–11446. [Google Scholar] [CrossRef]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol. 2011, 6, 95–119. [Google Scholar] [CrossRef]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, X.; Chen, F.; Li, H.; Wang, T.; Liu, N.; Sun, K.; Zhou, G.; Tao, K. Modulating cancer-stroma crosstalk by a nanoparticle-based photodynamic method to pave the way for subsequent therapies. Biomaterials 2022, 289, 121813. [Google Scholar] [CrossRef]
- Watabe, T.; Liu, Y.; Kaneda-Nakashima, K.; Shirakami, Y.; Lindner, T.; Ooe, K.; Toyoshima, A.; Nagata, K.; Shimosegawa, E.; Haberkorn, U.; et al. Theranostics Targeting Fibroblast Activation Protein in the Tumor Stroma: 64Cu- and 225Ac-Labeled FAPI-04 in Pancreatic Cancer Xenograft Mouse Models. J. Nucl. Med. 2020, 61, 563–569. [Google Scholar] [CrossRef]
- Hantel, C.; Beuschlein, F. Xenograft models for adrenocortical carcinoma. Mol. Cell. Endocrinol. 2016, 421, 28–33. [Google Scholar] [CrossRef]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.A.; Ganesan, A.P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.K.; Ozcan, A.; Brenner, D.; Watanabe, N.; Legendre, M.; Thomas, D.G.; Ashwood, C.; Cummings, R.D.; Bonifant, C.; Markovitz, D.M.; et al. Novel banana lectin CAR-T cells to target pancreatic tumors and tumor-associated stroma. J. Immunother. Cancer 2023, 11, e005891. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zhang, J.; Zhang, L.; Wu, Q.; Wang, Y.; Zhang, W.; Xiao, Y.; Chen, J.; Zhan, Q. PAFR/Stat3 axis maintains the symbiotic ecosystem between tumor and stroma to facilitate tumor malignancy. Acta Pharm. Sin. B 2023, 13, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Tanaka, C.; Furihata, K.; Naganuma, S.; Ogasawara, M.; Yoshioka, R.; Taniguchi, H.; Furihata, M.; Taniuchi, K. Establishment of a mouse model of pancreatic cancer using human pancreatic cancer cell line S2-013-derived organoid. Hum. Cell 2022, 35, 735–744. [Google Scholar] [CrossRef]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef]
- Idrisova, K.F.; Simon, H.U.; Gomzikova, M.O. Role of Patient-Derived Models of Cancer in Translational Oncology. Cancers 2022, 15, 139. [Google Scholar] [CrossRef]
- Farin, H.F.; Mosa, M.H.; Ndreshkjana, B.; Grebbin, B.M.; Ritter, B.; Menche, C.; Kennel, K.B.; Ziegler, P.K.; Szabó, L.; Bollrath, J.; et al. Colorectal Cancer Organoid-Stroma Biobank Allows Subtype-Specific Assessment of Individualized Therapy Responses. Cancer Discov. 2023, 13, 2192–2211. [Google Scholar] [CrossRef]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.H.; et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells 2019, 8, 889. [Google Scholar] [CrossRef] [PubMed]
- Hajime, M.; Shuichi, Y.; Makoto, N.; Masanori, Y.; Ikuko, K.; Atsushi, K.; Mutsuo, S.; Keiichi, T. Inhibitory effect of 4-methylesculetin on hyaluronan synthesis slows the development of human pancreatic cancer in vitro and in nude mice. Int. J. Cancer 2007, 120, 2704–2709. [Google Scholar] [CrossRef]
- Hoffman, R.M. Patient-Derived Orthotopic Xenograft (PDOX) Models of Melanoma. Int. J. Mol. Sci. 2017, 18, 1875. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Duan, M.H.; Yuan, Q.E.; Li, Z.L.; Luo, C.H.; Cui, L.Y.; Li, L.C.; Xiao, Y.; Zhu, X.Y.; Zhang, H.L.; et al. Suppressive stroma-immune prognostic signature impedes immunotherapy in ovarian cancer and can be reversed by PDGFRB inhibitors. J. Transl. Med. 2023, 21, 586. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Hase, M.; Zha, R.; Feng, Y.; Li, B.Y.; Yokota, H. Suppression of breast cancer-associated bone loss with osteoblast proteomes via Hsp90ab1/moesin-mediated inhibition of TGFβ/FN1/CD44 signaling. Theranostics 2022, 12, 929–943. [Google Scholar] [CrossRef]
- Juárez, P.; Mohammad, K.S.; Yin, J.J.; Fournier, P.G.; McKenna, R.C.; Davis, H.W.; Peng, X.H.; Niewolna, M.; Javelaud, D.; Chirgwin, J.M.; et al. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res. 2012, 72, 6247–6256. [Google Scholar] [CrossRef]
- Lee, N.P.; Chan, C.M.; Tung, L.N.; Wang, H.K.; Law, S. Tumor xenograft animal models for esophageal squamous cell carcinoma. J. Biomed. Sci. 2018, 25, 66. [Google Scholar] [CrossRef]
- Morton, J.J.; Keysar, S.B.; Perrenoud, L.; Chimed, T.S.; Reisinger, J.; Jackson, B.; Le, P.N.; Nieto, C.; Gomez, K.; Miller, B.; et al. Dual use of hematopoietic and mesenchymal stem cells enhances engraftment and immune cell trafficking in an allogeneic humanized mouse model of head and neck cancer. Mol. Carcinog. 2018, 57, 1651–1663. [Google Scholar] [CrossRef]
- Sun, L.; Jin, C.H.; Tan, S.; Liu, W.; Yang, Y.G. Human Immune System Mice With Autologous Tumor for Modeling Cancer Immunotherapies. Front. Immunol. 2020, 11, 591669. [Google Scholar] [CrossRef]
- Horiguchi, H.; Kadomatsu, T.; Kurahashi, R.; Hara, C.; Miyata, K.; Baba, M.; Osumi, H.; Terada, K.; Araki, K.; Takai, T.; et al. Dual functions of angiopoietin-like protein 2 signaling in tumor progression and anti-tumor immunity. Genes. Dev. 2019, 33, 1641–1656. [Google Scholar] [CrossRef] [PubMed]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; Rodenhizer, D.; Dean, T.; D’Arcangelo, E.; Xu, B.; Ailles, L.; McGuigan, A.P. A TRACER 3D Co-Culture tumour model for head and neck cancer. Biomaterials 2018, 164, 54–69. [Google Scholar] [CrossRef]
- Pape, J.; Emberton, M.; Cheema, U. 3D Cancer Models: The Need for a Complex Stroma, Compartmentalization and Stiffness. Front. Bioeng. Biotechnol. 2021, 9, 660502. [Google Scholar] [CrossRef]
- Karakas, H.E.; Kim, J.; Park, J.; Oh, J.M.; Choi, Y.; Gozuacik, D.; Cho, Y.K. A microfluidic chip for screening individual cancer cells via eavesdropping on autophagy-inducing crosstalk in the stroma niche. Sci. Rep. 2017, 7, 2050. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.T.; Hattersley, M.M.; Scarpitti, M.; Yang, B.; Devereaux, E.; Ramachandran, V.; Arumugam, T.; Ji, B.; Logsdon, C.D.; et al. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol. Cancer Res. 2012, 10, 1147–1157. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef]
- Hogstrom, J.M.; Cruz, K.A.; Selfors, L.M.; Ward, M.N.; Mehta, T.S.; Kanarek, N.; Philips, J.; Dialani, V.; Wulf, G.; Collins, L.C.; et al. Simultaneous isolation of hormone receptor-positive breast cancer organoids and fibroblasts reveals stroma-mediated resistance mechanisms. J. Biol. Chem. 2023, 299, 105021. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e1624. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef]
- Yu, L.S.; Jhunjhunwala, M.; Hong, S.Y.; Yu, L.Y.; Lin, W.R.; Chen, C.S. Tissue Architecture Influences the Biological Effectiveness of Boron Neutron Capture Therapy in In Vitro/In Silico Three-Dimensional Self-Assembly Cell Models of Pancreatic Cancers. Cancers 2021, 13, 4058. [Google Scholar] [CrossRef]
- Yang, M.; Qin, C.; Tao, L.; Cheng, G.; Li, J.; Lv, F.; Yang, N.; Xing, Z.; Chu, X.; Han, X.; et al. Synchronous targeted delivery of TGF-β siRNA to stromal and tumor cells elicits robust antitumor immunity against triple-negative breast cancer by comprehensively remodeling the tumor microenvironment. Biomaterials 2023, 301, 122253. [Google Scholar] [CrossRef]
- Keller, F.; Bruch, R.; Schneider, R.; Meier-Hubberten, J.; Hafner, M.; Rudolf, R. A Scaffold-Free 3-D Co-Culture Mimics the Major Features of the Reverse Warburg Effect In Vitro. Cells 2020, 9, 1900. [Google Scholar] [CrossRef]
- Neufeld, L.; Yeini, E.; Reisman, N.; Shtilerman, Y.; Ben-Shushan, D.; Pozzi, S.; Madi, A.; Tiram, G.; Eldar-Boock, A.; Ferber, S.; et al. Microengineered perfusable 3D-bioprinted glioblastoma model for in vivo mimicry of tumor microenvironment. Sci. Adv. 2021, 7, eabi9119. [Google Scholar] [CrossRef]
- Chi, C.W.; Lao, Y.H.; Ahmed, A.H.R.; Benoy, E.C.; Li, C.; Dereli-Korkut, Z.; Fu, B.M.; Leong, K.W.; Wang, S. High-Throughput Tumor-on-a-Chip Platform to Study Tumor-Stroma Interactions and Drug Pharmacokinetics. Adv. Healthc. Mater. 2020, 9, e2000880. [Google Scholar] [CrossRef]
- Berger Fridman, I.; Kostas, J.; Gregus, M.; Ray, S.; Sullivan, M.R.; Ivanov, A.R.; Cohen, S.; Konry, T. High-throughput microfluidic 3D biomimetic model enabling quantitative description of the human breast tumor microenvironment. Acta Biomater. 2021, 132, 473–488. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Chan, A.S.; Lai, F.P.; Leung, S.Y. Organoid cultures for cancer modeling. Cell Stem Cell 2023, 30, 917–937. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Mostafa, A.; Morton, J.P.; Hawinkels, L.; Prakash, J. Translating complexity and heterogeneity of pancreatic tumor: 3D in vitro to in vivo models. Adv. Drug Deliv. Rev. 2021, 174, 265–293. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lõhmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Schwörer, S.; Cimino, F.V.; Ros, M.; Tsanov, K.M.; Ng, C.; Lowe, S.W.; Carmona-Fontaine, C.; Thompson, C.B. Hypoxia Potentiates the Inflammatory Fibroblast Phenotype Promoted by Pancreatic Cancer Cell-Derived Cytokines. Cancer Res. 2023, 83, 1596–1610. [Google Scholar] [CrossRef]
- Sailer, V.; von Amsberg, G.; Duensing, S.; Kirfel, J.; Lieb, V.; Metzger, E.; Offermann, A.; Pantel, K.; Schuele, R.; Taubert, H.; et al. Experimental in vitro, ex vivo and in vivo models in prostate cancer research. Nat. Rev. Urol. 2023, 20, 158–178. [Google Scholar] [CrossRef]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Zou, Z.; Lin, Z.; Wu, C.; Tan, J.; Zhang, J.; Peng, Y.; Zhang, K.; Li, J.; Wu, M.; Zhang, Y. Micro-Engineered Organoid-on-a-Chip Based on Mesenchymal Stromal Cells to Predict Immunotherapy Responses of HCC Patients. Adv. Sci. 2023, 10, e2302640. [Google Scholar] [CrossRef]
- Wang, Y.; Jeon, H. 3D cell cultures toward quantitative high-throughput drug screening. Trends Pharmacol. Sci. 2022, 43, 569–581. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef]
- Choi, S.Y.; Shim, J.; Gu, D.E.; Kim, S.Y.; Kim, H.J.; Shin, D.Y.; Chung, M.K. Clonal evolution of long-term expanding head and neck cancer organoid: Impact on treatment response for personalized therapeutic screening. Oral. Oncol. 2023, 146, 106571. [Google Scholar] [CrossRef] [PubMed]
- Wörthmüller, J.; Rüegg, C. MAGI1, a Scaffold Protein with Tumor Suppressive and Vascular Functions. Cells 2021, 10, 1494. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, J.S.; Nobre, A.R.; Mondal, C.; Taha, I.; Farias, E.F.; Fertig, E.J.; Naba, A.; Aguirre-Ghiso, J.A.; Bravo-Cordero, J.J. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat. Cancer 2022, 3, 90–107. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.; Dong, Y.; Su, Y.; Leslie, F.; Sun, M.; Wang, F. Local scaffold-assisted delivery of immunotherapeutic agents for improved cancer immunotherapy. Adv. Drug Deliv. Rev. 2022, 185, 114308. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Parihar, V.K.; Singh, N.; Hatware, K.; Page, A.; Sharma, M.; Prajapati, M.K.; Kanugo, A.; Pawde, D.; Maru, S.; et al. Targeted delivery of panitumumab-scaffold bosutinib-encapsulated polycaprolactone nanoparticles for EGFR-overexpressed colorectal cancer. Nanomedicine 2023, 18, 713–741. [Google Scholar] [CrossRef]
- Hope, A.; Wade, S.J.; Aghmesheh, M.; Vine, K.L. Localized delivery of immunotherapy via implantable scaffolds for breast cancer treatment. J. Control. Release 2022, 341, 399–413. [Google Scholar] [CrossRef]
- Shahriar, S.M.S.; Andrabi, S.M.; Islam, F.; An, J.M.; Schindler, S.J.; Matis, M.P.; Lee, D.Y.; Lee, Y.K. Next-Generation 3D Scaffolds for Nano-Based Chemotherapeutics Delivery and Cancer Treatment. Pharmaceutics 2022, 14, 2712. [Google Scholar] [CrossRef]
- Weiden, J.; Tel, J.; Figdor, C.G. Synthetic immune niches for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 212–219. [Google Scholar] [CrossRef]
- Sun, W.; Luo, Z.; Lee, J.; Kim, H.J.; Lee, K.; Tebon, P.; Feng, Y.; Dokmeci, M.R.; Sengupta, S.; Khademhosseini, A. Organ-on-a-Chip for Cancer and Immune Organs Modeling. Adv. Healthc. Mater. 2019, 8, e1801363. [Google Scholar] [CrossRef]
- Farran, B.; Nagaraju, G.P. The dynamic interactions between the stroma, pancreatic stellate cells and pancreatic tumor development: Novel therapeutic targets. Cytokine Growth Factor Rev. 2019, 48, 11–23. [Google Scholar] [CrossRef]
- Lee, Y.C.; Lam, H.M.; Rosser, C.; Theodorescu, D.; Parks, W.C.; Chan, K.S. The dynamic roles of the bladder tumour microenvironment. Nat. Rev. Urol. 2022, 19, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.V.; Zhang, Y.S.; Gaspar, V.M.; Mano, J.F. 3D-bioprinted cancer-on-a-chip: Level-up organotypic in vitro models. Trends Biotechnol. 2022, 40, 432–447. [Google Scholar] [CrossRef] [PubMed]
- Del Piccolo, N.; Shirure, V.S.; Bi, Y.; Goedegebuure, S.P.; Gholami, S.; Hughes, C.C.W.; Fields, R.C.; George, S.C. Tumor-on-chip modeling of organ-specific cancer and metastasis. Adv. Drug Deliv. Rev. 2021, 175, 113798. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, Z.; Yu, Y.; Zhang, P. HIF1α lactylation enhances KIAA1199 transcription to promote angiogenesis and vasculogenic mimicry in prostate cancer. Int. J. Biol. Macromol. 2022, 222, 2225–2243. [Google Scholar] [CrossRef]
- Firatligil-Yildirir, B.; Bati-Ayaz, G.; Tahmaz, I.; Bilgen, M.; Pesen-Okvur, D.; Yalcin-Ozuysal, O. On-chip determination of tissue-specific metastatic potential of breast cancer cells. Biotechnol. Bioeng. 2021, 118, 3799–3810. [Google Scholar] [CrossRef]
- Hou, Q.; Zhu, L.; Wang, L.; Liu, X.; Xiao, F.; Xie, Y.; Zheng, W.; Jiang, X. Screening on-chip fabricated nanoparticles for penetrating the blood-brain barrier. Nanoscale 2022, 14, 3234–3241. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q.; Zhang, H.; Han, S.; Liu, N.; Ren, H.; Guo, H.; Xu, F. Construction of cancer-on-a-chip for drug screening. Drug Discov. Today 2021, 26, 1875–1890. [Google Scholar] [CrossRef]
- Haase, K.; Offeddu, G.S.; Gillrie, M.R.; Kamm, R.D. Endothelial Regulation of Drug Transport in a 3D Vascularized Tumor Model. Adv. Funct. Mater. 2020, 30, 2002444. [Google Scholar] [CrossRef]
- Williams, S.T.; Wells, G.; Conroy, S.; Gagg, H.; Allen, R.; Rominiyi, O.; Helleday, T.; Hullock, K.; Pennington, C.E.W.; Rantala, J.; et al. Precision oncology using ex vivo technology: A step towards individualised cancer care? Expert Rev. Mol. Med. 2022, 24, e39. [Google Scholar] [CrossRef]
- Pence, K.A.; Mishra, D.K.; Thrall, M.; Dave, B.; Kim, M.P. Breast cancer cells form primary tumors on ex vivo four-dimensional lung model. J. Surg. Res. 2017, 210, 181–187. [Google Scholar] [CrossRef]
- He, L.; Deng, C. Recent advances in organotypic tissue slice cultures for anticancer drug development. Int. J. Biol. Sci. 2022, 18, 5885–5896. [Google Scholar] [CrossRef] [PubMed]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Suckert, T.; Rassamegevanon, T.; Müller, J.; Dietrich, A.; Graja, A.; Reiche, M.; Löck, S.; Krause, M.; Beyreuther, E.; von Neubeck, C. Applying Tissue Slice Culture in Cancer Research-Insights from Preclinical Proton Radiotherapy. Cancers 2020, 12, 1589. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, R.; Chan, M.; Shin, J.S.; Nishida-Aoki, N.; Kenerson, H.L.; Elemento, O.; Beltran, H.; Yeung, R.; Gujral, T.S. Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery. Oncoimmunology 2019, 8, e1670019. [Google Scholar] [CrossRef]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Jiang, X.; Guha, P.; Lausted, C.; Carter, J.A.; Hsu, C.; Labadie, K.P.; Kohli, K.; Kenerson, H.L.; Daniel, S.K.; et al. Blockade of interleukin 10 potentiates antitumour immune function in human colorectal cancer liver metastases. Gut 2023, 72, 325–337. [Google Scholar] [CrossRef]
- Naipal, K.A.; Verkaik, N.S.; Sánchez, H.; van Deurzen, C.H.; den Bakker, M.A.; Hoeijmakers, J.H.; Kanaar, R.; Vreeswijk, M.P.; Jager, A.; van Gent, D.C. Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer 2016, 16, 78. [Google Scholar] [CrossRef]
- Elsesy, M.E.; Oh-Hohenhorst, S.J.; Oing, C.; Eckhardt, A.; Burdak-Rothkamm, S.; Alawi, M.; Müller, C.; Schüller, U.; Maurer, T.; von Amsberg, G.; et al. Preclinical patient-derived modeling of castration-resistant prostate cancer facilitates individualized assessment of homologous recombination repair deficient disease. Mol. Oncol. 2023, 17, 1129–1147. [Google Scholar] [CrossRef]
- Zottel, A.; Jovčevska, I.; Šamec, N. Non-animal glioblastoma models for personalized treatment. Heliyon 2023, 9, e21070. [Google Scholar] [CrossRef]
- Weitz, J.R.; Tiriac, H.; de Mendoza, T.H.; Wascher, A.; Lowy, A.M. Using Organotypic Tissue Slices to Investigate the Microenvironment of Pancreatic Cancer: Pharmacotyping and Beyond. Cancers 2021, 13, 4991. [Google Scholar] [CrossRef]
- Misra, S.; Moro, C.F.; Del Chiaro, M.; Pouso, S.; Sebestyén, A.; Löhr, M.; Björnstedt, M.; Verbeke, C.S. Ex vivo organotypic culture system of precision-cut slices of human pancreatic ductal adenocarcinoma. Sci. Rep. 2019, 9, 2133. [Google Scholar] [CrossRef] [PubMed]
- Koerfer, J.; Kallendrusch, S.; Merz, F.; Wittekind, C.; Kubick, C.; Kassahun, W.T.; Schumacher, G.; Moebius, C.; Gaßler, N.; Schopow, N.; et al. Organotypic slice cultures of human gastric and esophagogastric junction cancer. Cancer Med. 2016, 5, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- van de Merbel, A.F.; van der Horst, G.; van der Mark, M.H.; van Uhm, J.I.M.; van Gennep, E.J.; Kloen, P.; Beimers, L.; Pelger, R.C.M.; van der Pluijm, G. An ex vivo Tissue Culture Model for the Assessment of Individualized Drug Responses in Prostate and Bladder Cancer. Front. Oncol. 2018, 8, 400. [Google Scholar] [CrossRef]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef]
- Voabil, P.; de Bruijn, M.; Roelofsen, L.M.; Hendriks, S.H.; Brokamp, S.; van den Braber, M.; Broeks, A.; Sanders, J.; Herzig, P.; Zippelius, A.; et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 2021, 27, 1250–1261. [Google Scholar] [CrossRef]
- Ciraku, L.; Moeller, R.A.; Esquea, E.M.; Gocal, W.A.; Hartsough, E.J.; Simone, N.L.; Jackson, J.G.; Reginato, M.J. An Ex Vivo Brain Slice Model to Study and Target Breast Cancer Brain Metastatic Tumor Growth. J. Vis. Exp. 2021, 175, 10.3791/62617. [Google Scholar] [CrossRef]
- Zhou, J.B.; Tang, D.; He, L.; Lin, S.; Lei, J.H.; Sun, H.; Xu, X.; Deng, C.X. Machine learning model for anti-cancer drug combinations: Analysis, prediction, and validation. Pharmacol. Res. 2023, 194, 106830. [Google Scholar] [CrossRef]
- Martin, S.Z.; Wagner, D.C.; Hörner, N.; Horst, D.; Lang, H.; Tagscherer, K.E.; Roth, W. Ex vivo tissue slice culture system to measure drug-response rates of hepatic metastatic colorectal cancer. BMC Cancer 2019, 19, 1030. [Google Scholar] [CrossRef]
- Jordão, G.; Tavares, J.N. Mathematical models in cancer therapy. Biosystems 2017, 162, 12–23. [Google Scholar] [CrossRef]
- Hong, M.; Tao, S.; Zhang, L.; Diao, L.T.; Huang, X.; Huang, S.; Xie, S.J.; Xiao, Z.D.; Zhang, H. RNA sequencing: New technologies and applications in cancer research. J. Hematol. Oncol. 2020, 13, 166. [Google Scholar] [CrossRef]
- Popilski, H.; Stepensky, D. Mathematical modeling analysis of intratumoral disposition of anticancer agents and drug delivery systems. Expert. Opin. Drug Metab. Toxicol. 2015, 11, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, B.; Lee, J.S.; Mutasa-Gottgens, E.; Naughton, B.; Bacon, W.; Manning, J.; Wang, Y.; Pollard, J.; Mendez, M.; Hill, J.; et al. Applications of single-cell RNA sequencing in drug discovery and development. Nature reviews. Drug Discov. 2023, 22, 496–520. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, J.K.; Negi, S.; Kashyap, M.; Nizamuddin, S.; Singh, A.; Khattri, A. Pan-Cancer Analysis Shows Enrichment of Macrophages, Overexpression of Checkpoint Molecules, Inhibitory Cytokines, and Immune Exhaustion Signatures in EMT-High Tumors. Front. Oncol. 2021, 11, 793881. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, N.; Ghoshdastider, U.; Baruah, P.; Kulshrestha, T.; Skanderup, A.J. A pan-cancer metabolic atlas of the tumor microenvironment. Cell Rep. 2022, 39, 110800. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.L.D.; Moraes, M.C.; Pérez-de-Oliveira, M.E.; Silva, V.M.D.; Saldivia-Siracusa, C.; Pedroso, C.M.; Lopes, M.A.; Vargas, P.A.; Kochanny, S.; Pearson, A.; et al. Machine learning for the prediction of toxicities from head and neck cancer treatment: A systematic review with meta-analysis. Oral. Oncol. 2023, 140, 106386. [Google Scholar] [CrossRef]
- Mandair, D.; Reis-Filho, J.S.; Ashworth, A. Biological insights and novel biomarker discovery through deep learning approaches in breast cancer histopathology. npj Breast Cancer 2023, 9, 21. [Google Scholar] [CrossRef]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. Semin. Cancer Biol. 2021, 68, 132–142. [Google Scholar] [CrossRef]
- Tanoli, Z.; Vähä-Koskela, M.; Aittokallio, T. Artificial intelligence, machine learning, and drug repurposing in cancer. Expert. Opin. Drug Discov. 2021, 16, 977–989. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Ma, A.; Wang, Q.E.; Liu, B.; Li, L.; Xu, D.; Ma, Q. Deep transfer learning of cancer drug responses by integrating bulk and single-cell RNA-seq data. Nat. Commun. 2022, 13, 6494. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Greenman, C.D.; Lagergren, J. Computational Cancer Biology: An Evolutionary Perspective. PLoS Comput. Biol. 2016, 12, e1004717. [Google Scholar] [CrossRef]
- Chen, J.; Weihs, D.; Vermolen, F.J. Computational modeling of therapy on pancreatic cancer in its early stages. Biomech. Model. Mechanobiol. 2020, 19, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Haeno, H.; Gonen, M.; Davis, M.B.; Herman, J.M.; Iacobuzio-Donahue, C.A.; Michor, F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 2012, 148, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Weihs, D.; Vermolen, F.J. A model for cell migration in non-isotropic fibrin networks with an application to pancreatic tumor islets. Biomech. Model. Mechanobiol. 2018, 17, 367–386. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.D. The 3R principle: Advancing clinical application of human pluripotent stem cells. Stem Cell Res. Ther. 2013, 4, 21. [Google Scholar] [CrossRef]
- Beck, J.A.; Lloyd, S.; Hafezparast, M.; Lennon-Pierce, M.; Eppig, J.T.; Festing, M.F.; Fisher, E.M. Genealogies of mouse inbred strains. Nat. Genet. 2000, 24, 23–25. [Google Scholar] [CrossRef]
- Workman, P. The NCI-60 Human Tumor Cell Line Screen: A Catalyst for Progressive Evolution of Models for Discovery and Development of Cancer Drugs. Cancer Res. 2023, 83, 3170–3173. [Google Scholar] [CrossRef]
- Zushin, P.H.; Mukherjee, S.; Wu, J.C. FDA Modernization Act 2.0: Transitioning beyond animal models with human cells, organoids, and AI/ML-based approaches. J. Clin. Investig. 2023, 133, e175824. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Li, Z.; Li, W.; Huang, W. Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J. Transl. Med. 2021, 19, 40. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, W.; Maskey, N.; Yang, F.; Zheng, Z.; Li, C.; Wang, R.; Wu, P.; Mao, S.; Zhang, J.; et al. Urological cancer organoids, patients’ avatars for precision medicine: Past, present and future. Cell Biosci. 2022, 12, 132. [Google Scholar] [CrossRef]
- Chan, W.S.; Mo, X.; Ip, P.P.C.; Tse, K.Y. Patient-derived organoid culture in epithelial ovarian cancers-Techniques, applications, and future perspectives. Cancer Med. 2023, 12, 19714–19731. [Google Scholar] [CrossRef]
- Arrigoni, C.; Bersini, S.; Gilardi, M.; Moretti, M. In Vitro Co-Culture Models of Breast Cancer Metastatic Progression towards Bone. Int. J. Mol. Sci. 2016, 17, 1405. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, Y.; Yu, T.; Yang, H.; Li, Y.; Xiao, J.; Li, B. A Promising transwell co-culture cell model for silicosis. Toxicol. Vitr. Int. J. Publ. Assoc. 2022, 81, 105318. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Shang, M.; Zhang, Y.G.; Jiao, Y.; Xia, Y.; Garrett, S.; Bakke, D.; Bäuerl, C.; Martinez, G.P.; Kim, C.H.; et al. Lactic Acid Bacteria Isolated From Korean Kimchi Activate the Vitamin D Receptor-autophagy Signaling Pathways. Inflamm. Bowel Dis. 2020, 26, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, X.; Yu, P.; Xie, F.; Kwan, J.S.H.; Chan, W.N.; Fang, C.; Zhang, J.; Cheung, A.H.K.; Chow, C.; et al. H. pylori-induced NF-κB-PIEZO1-YAP1-CTGF axis drives gastric cancer progression and cancer-associated fibroblast-mediated tumour microenvironment remodelling. Clin. Transl. Med. 2023, 13, e1481. [Google Scholar] [CrossRef]
- Zhao, H.; Jiang, E.; Shang, Z. 3D Co-culture of Cancer-Associated Fibroblast with Oral Cancer Organoids. J. Dent. Res. 2021, 100, 201–208. [Google Scholar] [CrossRef]
- Zhang, M.; Fang, Y.; Fu, X.; Liu, J.; Liu, Y.; Zhu, Z.; Ni, Y.; Yao, M.; Pan, Q.; Cao, W.; et al. Cancer-associated fibroblasts nurture LGR5 marked liver tumor-initiating cells and promote their tumor formation, growth, and metastasis. Cancer Med. 2023, 12, 18032–18049. [Google Scholar] [CrossRef]
- Schuth, S.; Le Blanc, S.; Krieger, T.G.; Jabs, J.; Schenk, M.; Giese, N.A.; Büchler, M.W.; Eils, R.; Conrad, C.; Strobel, O. Patient-specific modeling of stroma-mediated chemoresistance of pancreatic cancer using a three-dimensional organoid-fibroblast co-culture system. J. Exp. Clin. Cancer Res. 2022, 41, 312. [Google Scholar] [CrossRef]
- Mosa, M.H.; Michels, B.E.; Menche, C.; Nicolas, A.M.; Darvishi, T.; Greten, F.R.; Farin, H.F. A Wnt-Induced Phenotypic Switch in Cancer-Associated Fibroblasts Inhibits EMT in Colorectal Cancer. Cancer Res. 2020, 80, 5569–5582. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e122. [Google Scholar] [CrossRef]
- Woolston, A.; Khan, K.; Spain, G.; Barber, L.J.; Griffiths, B.; Gonzalez-Exposito, R.; Hornsteiner, L.; Punta, M.; Patil, Y.; Newey, A.; et al. Genomic and Transcriptomic Determinants of Therapy Resistance and Immune Landscape Evolution during Anti-EGFR Treatment in Colorectal Cancer. Cancer Cell 2019, 36, 35–50.e39. [Google Scholar] [CrossRef] [PubMed]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Melzer, C.; Ohe, J.V.; Hass, R. Altered Tumor Plasticity after Different Cancer Cell Fusions with MSC. Int. J. Mol. Sci. 2020, 21, 8347. [Google Scholar] [CrossRef]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Weng, X.; Sharma, S.; Awate, P.; Liu, Z.; Gao, D.; Mitsiades, N.; Schwab, J.H.; et al. Pleiotropic Mechanisms Drive Endocrine Resistance in the Three-Dimensional Bone Microenvironment. Cancer Res. 2021, 81, 371–383. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Francois, M.; Boivin, M.N.; Martineau, D.; Galipeau, J. Neo-organoid of marrow mesenchymal stromal cells secreting interleukin-12 for breast cancer therapy. Cancer Res. 2008, 68, 4810–4818. [Google Scholar] [CrossRef]
- Mertz, D.R.; Parigoris, E.; Sentosa, J.; Lee, J.H.; Lee, S.; Kleer, C.G.; Luker, G.; Takayama, S. Triple-negative breast cancer cells invade adipocyte/preadipocyte-encapsulating geometrically inverted mammary organoids. Integr. Biol. 2023, 15, zyad004. [Google Scholar] [CrossRef]
- Choi, J.I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53. [Google Scholar] [CrossRef]
- Lim, J.T.C.; Kwang, L.G.; Ho, N.C.W.; Toh, C.C.M.; Too, N.S.H.; Hooi, L.; Benoukraf, T.; Chow, P.K.; Dan, Y.Y.; Chow, E.K.; et al. Hepatocellular carcinoma organoid co-cultures mimic angiocrine crosstalk to generate inflammatory tumor microenvironment. Biomaterials 2022, 284, 121527. [Google Scholar] [CrossRef]
- Zhou, Z.; Van der Jeught, K.; Li, Y.; Sharma, S.; Yu, T.; Moulana, I.; Liu, S.; Wan, J.; Territo, P.R.; Opyrchal, M.; et al. A T Cell-Engaging Tumor Organoid Platform for Pancreatic Cancer Immunotherapy. Adv. Sci. 2023, 10, e2300548. [Google Scholar] [CrossRef]
- Zhou, G.; Lieshout, R.; van Tienderen, G.S.; de Ruiter, V.; van Royen, M.E.; Boor, P.P.C.; Magré, L.; Desai, J.; Köten, K.; Kan, Y.Y.; et al. Modelling immune cytotoxicity for cholangiocarcinoma with tumour-derived organoids and effector T cells. Br. J. Cancer 2022, 127, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Bi, D.; Xie, R.; Li, M.; Guo, J.; Liu, H.; Guo, X.; Fang, J.; Ding, T.; Zhu, H.; et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 398. [Google Scholar] [CrossRef] [PubMed]
- Holokai, L.; Chakrabarti, J.; Broda, T.; Chang, J.; Hawkins, J.A.; Sundaram, N.; Wroblewski, L.E.; Peek, R.M., Jr.; Wang, J.; Helmrath, M.; et al. Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection. PLoS Pathog. 2019, 15, e1007468. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.V.; Ferreira, L.P.; Rocha, M.; Gaspar, V.M.; Mano, J.F. Advances in bioengineering pancreatic tumor-stroma physiomimetic Biomodels. Biomaterials 2022, 287, 121653. [Google Scholar] [CrossRef]
- Zhang, J.; Tavakoli, H.; Ma, L.; Li, X.; Han, L.; Li, X. Immunotherapy discovery on tumor organoid-on-a-chip platforms that recapitulate the tumor microenvironment. Adv. Drug Deliv. Rev. 2022, 187, 114365. [Google Scholar] [CrossRef]
- Haque, M.R.; Wessel, C.R.; Leary, D.D.; Wang, C.; Bhushan, A.; Bishehsari, F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst. Nanoeng. 2022, 8, 36. [Google Scholar] [CrossRef]
- Levy, R.J.; Paşca, S.P. What Have Organoids and Assembloids Taught Us About the Pathophysiology of Neuropsychiatric Disorders? Biol. Psychiatry 2023, 93, 632–641. [Google Scholar] [CrossRef]
- Kim, E.; Choi, S.; Kang, B.; Kong, J.; Kim, Y.; Yoon, W.H.; Lee, H.R.; Kim, S.; Kim, H.M.; Lee, H.; et al. Creation of bladder assembloids mimicking tissue regeneration and cancer. Nature 2020, 588, 664–669. [Google Scholar] [CrossRef]
- Langer, E.M.; Allen-Petersen, B.L.; King, S.M.; Kendsersky, N.D.; Turnidge, M.A.; Kuziel, G.M.; Riggers, R.; Samatham, R.; Amery, T.S.; Jacques, S.L.; et al. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell Rep. 2019, 26, 608–623.e606. [Google Scholar] [CrossRef]
- Kaur, S.; Kaur, I.; Rawal, P.; Tripathi, D.M.; Vasudevan, A. Non-matrigel scaffolds for organoid cultures. Cancer Lett. 2021, 504, 58–66. [Google Scholar] [CrossRef]
- Varinelli, L.; Guaglio, M.; Brich, S.; Zanutto, S.; Belfiore, A.; Zanardi, F.; Iannelli, F.; Oldani, A.; Costa, E.; Chighizola, M.; et al. Decellularized extracellular matrix as scaffold for cancer organoid cultures of colorectal peritoneal metastases. J. Mol. Cell Biol. 2023, 14, mjac064. [Google Scholar] [CrossRef] [PubMed]
- van Tienderen, G.S.; Rosmark, O.; Lieshout, R.; Willemse, J.; de Weijer, F.; Rendin, L.E.; Westergren-Thorsson, G.; Doukas, M.; Groot Koerkamp, B.; van Royen, M.E.; et al. Extracellular matrix drives tumor organoids toward desmoplastic matrix deposition and mesenchymal transition. Acta Biomater. 2023, 158, 115–131. [Google Scholar] [CrossRef] [PubMed]
- van Tienderen, G.S.; van Beek, M.E.A.; Schurink, I.J.; Rosmark, O.; Roest, H.P.; Tieleman, J.; Demmers, J.; Muntz, I.; Conboy, J.; Westergren-Thorsson, G.; et al. Modelling metastatic colonization of cholangiocarcinoma organoids in decellularized lung and lymph nodes. Front. Oncol. 2022, 12, 1101901. [Google Scholar] [CrossRef]
- Shan, H.; Chen, M.; Zhao, S.; Wei, X.; Zheng, M.; Li, Y.; Lin, Q.; Jiang, Z.; Chen, Z.; Fei, C.; et al. Acoustic virtual 3D scaffold for direct-interacting tumor organoid-immune cell coculture systems. Sci. Adv. 2024, 10, eadr4831. [Google Scholar] [CrossRef] [PubMed]
- Öhlinger, K.; Kolesnik, T.; Meindl, C.; Gallé, B.; Absenger-Novak, M.; Kolb-Lenz, D.; Fröhlich, E. Air-liquid interface culture changes surface properties of A549 cells. Toxicol. Vitr. 2019, 60, 369–382. [Google Scholar] [CrossRef]
- Lamers, M.M.; van der Vaart, J.; Knoops, K.; Riesebosch, S.; Breugem, T.I.; Mykytyn, A.Z.; Beumer, J.; Schipper, D.; Bezstarosti, K.; Koopman, C.D.; et al. An organoid-derived bronchioalveolar model for SARS-CoV-2 infection of human alveolar type II-like cells. EMBO J. 2021, 40, e105912. [Google Scholar] [CrossRef]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef]
- Yin, S.; Xi, R.; Wu, A.; Wang, S.; Li, Y.; Wang, C.; Tang, L.; Xia, Y.; Yang, D.; Li, J.; et al. Patient-derived tumor-like cell clusters for drug testing in cancer therapy. Sci. Transl. Med. 2020, 12, eaaz1723. [Google Scholar] [CrossRef]
- Bae, J.; Choi, Y.S.; Cho, G.; Jang, S.J. The Patient-Derived Cancer Organoids: Promises and Challenges as Platforms for Cancer Discovery. Cancers 2022, 14, 2144. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Chen, X.; Li, R.; Zhao, H.; Wang, X.; Shao, Z.; Shang, Z. Phenotype transition of fibroblasts incorporated into patient-derived oral carcinoma organoids. Oral. Dis. 2023, 29, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, R.; Chen, Y.; Yang, X.; Shang, Z. Stromal nicotinamide N-methyltransferase orchestrates the crosstalk between fibroblasts and tumour cells in oral squamous cell carcinoma: Evidence from patient-derived assembled organoids. Oncogene 2023, 42, 1166–1180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-organoid culture supports differentiation of human CAR+ iPSCs into highly functional CAR T cells. Cell Stem Cell 2022, 29, 515–527.e518. [Google Scholar] [CrossRef]
- Mason, J.; Öhlund, D. Key aspects for conception and construction of co-culture models of tumor-stroma interactions. Front. Bioeng. Biotechnol. 2023, 11, 1150764. [Google Scholar] [CrossRef]
- Benton, G.; Arnaoutova, I.; George, J.; Kleinman, H.K.; Koblinski, J. Matrigel: From discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev. 2014, 79–80, 3–18. [Google Scholar] [CrossRef]
- Kozlowski, M.T.; Crook, C.J.; Ku, H.T. Towards organoid culture without Matrigel. Commun. Biol. 2021, 4, 1387. [Google Scholar] [CrossRef]
- Dominijanni, A.; Devarasetty, M.; Soker, S. Manipulating the Tumor Microenvironment in Tumor Organoids Induces Phenotypic Changes and Chemoresistance. iScience 2020, 23, 101851. [Google Scholar] [CrossRef]
- Sung, S.Y.; Hsieh, C.L.; Law, A.; Zhau, H.E.; Pathak, S.; Multani, A.S.; Lim, S.; Coleman, I.M.; Wu, L.C.; Figg, W.D.; et al. Coevolution of prostate cancer and bone stroma in three-dimensional coculture: Implications for cancer growth and metastasis. Cancer Res. 2008, 68, 9996–10003. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, W.; Zuo, L.; Xu, M.; Wu, Y.; Huang, J.; Zhang, X.; Li, Y.; Wang, J.; Chen, J.; et al. The Fibrillin-1/VEGFR2/STAT2 signaling axis promotes chemoresistance via modulating glycolysis and angiogenesis in ovarian cancer organoids and cells. Cancer Commun. 2022, 42, 245–265. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor organoid-T-cell coculture systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin αvβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e110. [Google Scholar] [CrossRef] [PubMed]
- Kassis, T.; Hernandez-Gordillo, V.; Langer, R.; Griffith, L.G. OrgaQuant: Human Intestinal Organoid Localization and Quantification Using Deep Convolutional Neural Networks. Sci. Rep. 2019, 9, 12479. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Schuster, B.; Kashaf, S.S.; Liu, P.; Ben-Yishay, R.; Ishay-Ronen, D.; Izumchenko, E.; Shen, L.; Weber, C.R.; Bielski, M.; et al. OrganoID: A versatile deep learning platform for tracking and analysis of single-organoid dynamics. PLoS Comput. Biol. 2022, 18, e1010584. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Lee, H.; Kim, D.; Han, S.K.; Ha, D.; Shin, K.; Kim, S. Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nat. Commun. 2020, 11, 5485. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gieniec, K.A.; Lannagan, T.R.M.; Wang, T.; Asai, N.; Mizutani, Y.; Iida, T.; Ando, R.; Thomas, E.M.; Sakai, A.; et al. The Origin and Contribution of Cancer-Associated Fibroblasts in Colorectal Carcinogenesis. Gastroenterology 2022, 162, 890–906. [Google Scholar] [CrossRef]
- Marsee, A.; Roos, F.J.M.; Verstegen, M.M.A.; Gehart, H.; de Koning, E.; Lemaigre, F.; Forbes, S.J.; Peng, W.C.; Huch, M.; Takebe, T.; et al. Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell 2021, 28, 816–832. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.; Wang, Z.; Liu, Y.; Yu, J.; Wang, W.; Chen, S.; Wu, W.; Wang, J.; Qian, G.; et al. Standardization of organoid culture in cancer research. Cancer Med. 2023, 12, 14375–14386. [Google Scholar] [CrossRef]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef]
- Primac, I.; Maquoi, E.; Blacher, S.; Heljasvaara, R.; Van Deun, J.; Smeland, H.Y.; Canale, A.; Louis, T.; Stuhr, L.; Sounni, N.E.; et al. Stromal integrin α11 regulates PDGFR-β signaling and promotes breast cancer progression. J. Clin. Investig. 2019, 129, 4609–4628. [Google Scholar] [CrossRef]
- Shukla, P.; Yeleswarapu, S.; Heinrich, M.A.; Prakash, J.; Pati, F. Mimicking tumor microenvironment by 3D bioprinting: 3D cancer modeling. Biofabrication 2022, 14, 032002. [Google Scholar] [CrossRef]
- Juraski, A.C.; Sharma, S.; Sparanese, S.; da Silva, V.A.; Wong, J.; Laksman, Z.; Flannigan, R.; Rohani, L.; Willerth, S.M. 3D bioprinting for organ and organoid models and disease modeling. Expert. Opin. Drug Discov. 2023, 18, 1043–1059. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef]
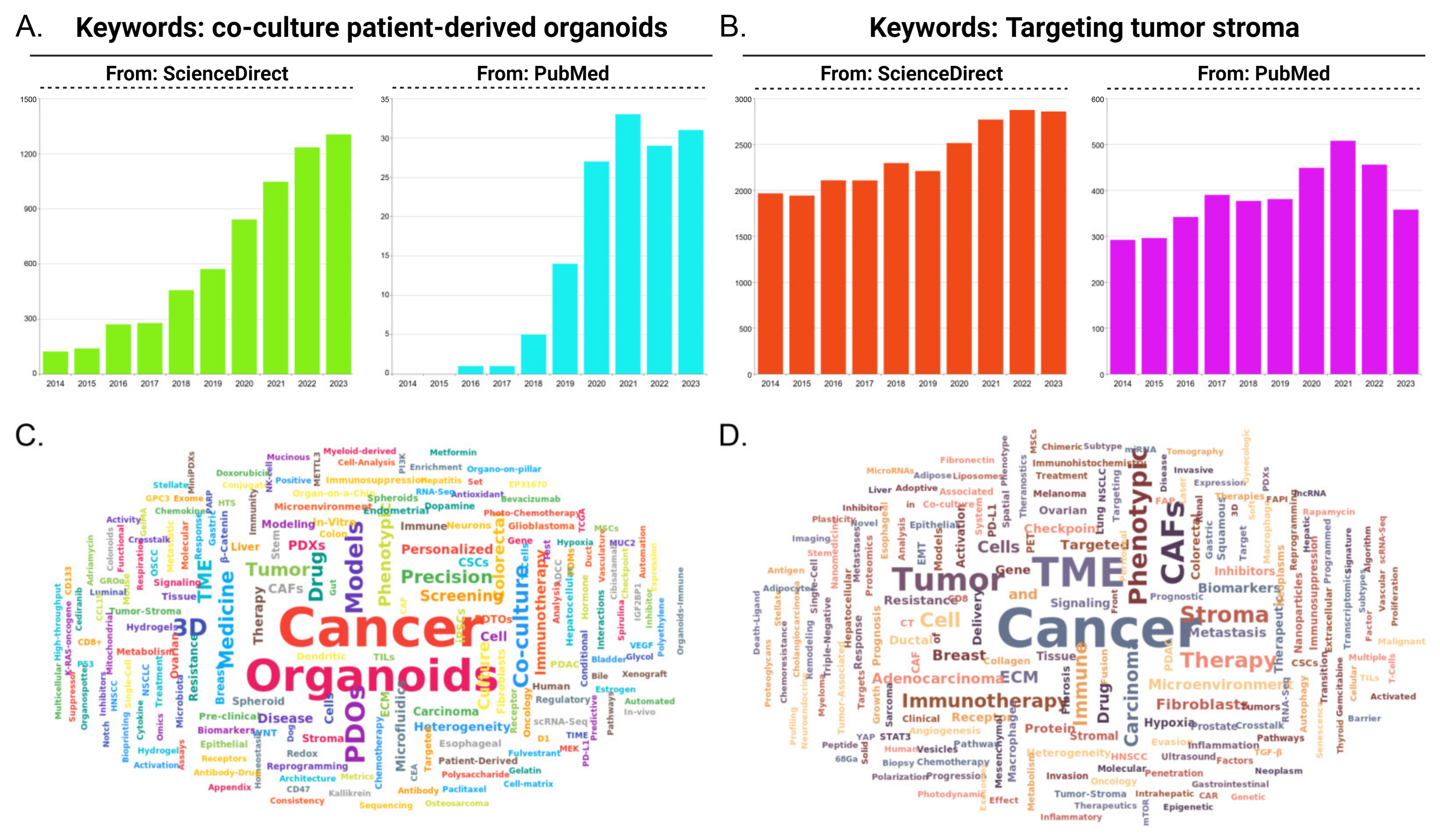



{kind=link}
{kind=link}
{kind=link}
| Model Type | Advantage | Disadvantage |
|---|---|---|
| In vivo models |
|
|
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
| ||
| ||
| In vitro models |
|
|
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
| ||
| ||
| Ex vivo models |
|
|
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
| ||
| ||
| In silico models |
|
|
|
| |
|
| |
|
| |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Q.-S.; Shan, X.-F.; Yau, V.; Cai, Z.-G.; Xie, S. Facilitation of Tumor Stroma-Targeted Therapy: Model Difficulty and Co-Culture Organoid Method. Pharmaceuticals 2025, 18, 62. https://doi.org/10.3390/ph18010062
Feng Q-S, Shan X-F, Yau V, Cai Z-G, Xie S. Facilitation of Tumor Stroma-Targeted Therapy: Model Difficulty and Co-Culture Organoid Method. Pharmaceuticals. 2025; 18(1):62. https://doi.org/10.3390/ph18010062
Chicago/Turabian StyleFeng, Qiu-Shi, Xiao-Feng Shan, Vicky Yau, Zhi-Gang Cai, and Shang Xie. 2025. "Facilitation of Tumor Stroma-Targeted Therapy: Model Difficulty and Co-Culture Organoid Method" Pharmaceuticals 18, no. 1: 62. https://doi.org/10.3390/ph18010062
APA StyleFeng, Q.-S., Shan, X.-F., Yau, V., Cai, Z.-G., & Xie, S. (2025). Facilitation of Tumor Stroma-Targeted Therapy: Model Difficulty and Co-Culture Organoid Method. Pharmaceuticals, 18(1), 62. https://doi.org/10.3390/ph18010062