
Potential Interaction of Pinocembrin with Drug Transporters and Hepatic Drug-Metabolizing Enzymes

 and
and
Abstract

1. Introduction
2. Results
2.1. The Effect of Pinocembrin on the Function of Drug Transporters
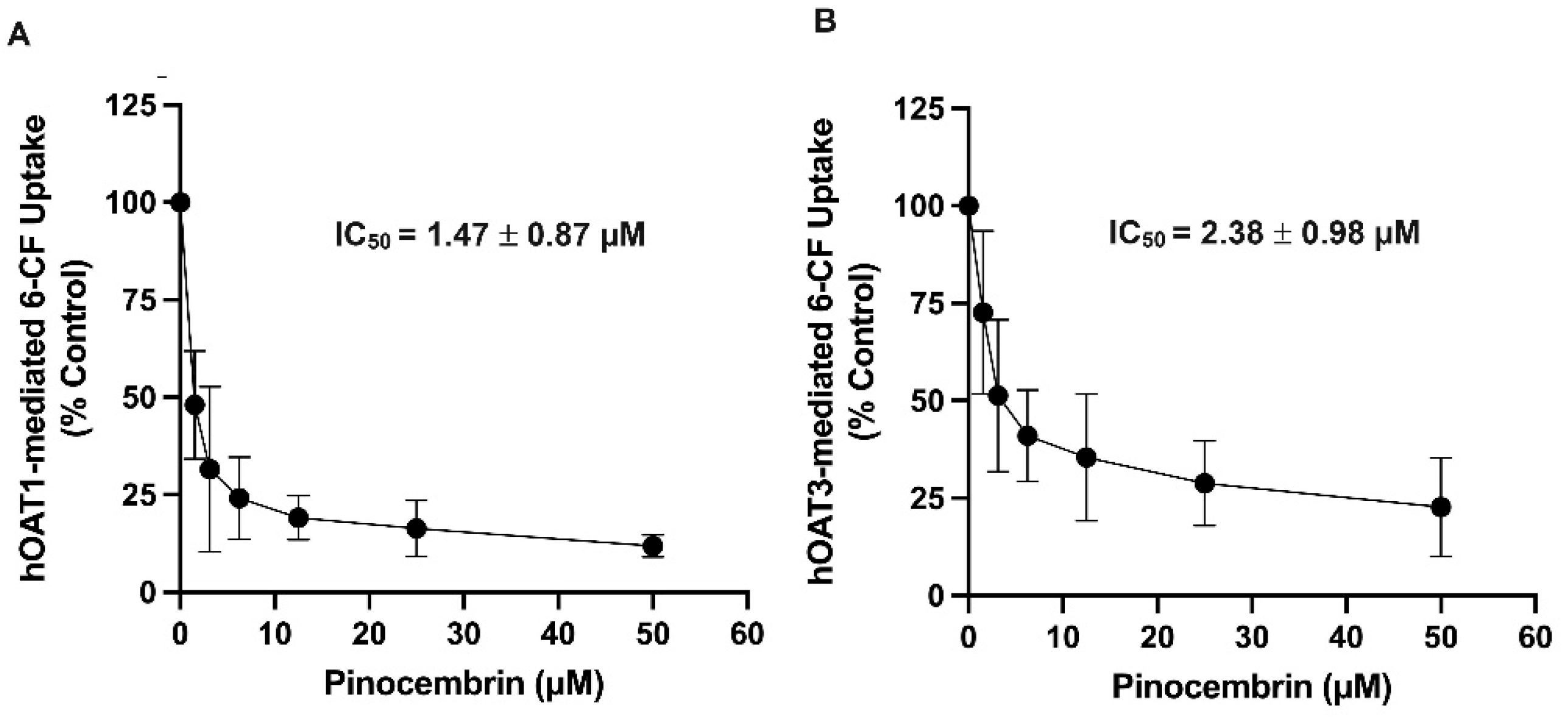
2.2. Inhibitory Potency and the Mechanism of Pinocembrin Inhibition on hOAT1 and hOAT3
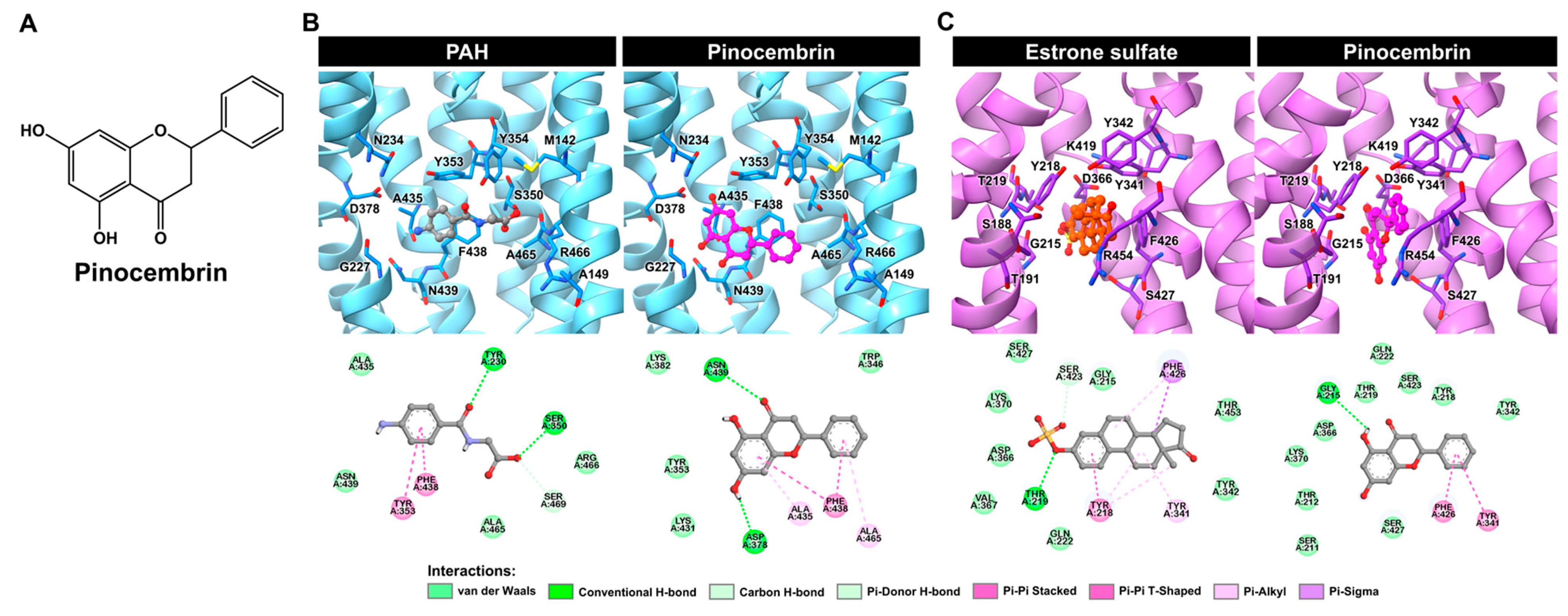
2.3. Molecular Docking of Pinocembrin with hOAT1 and hOAT3
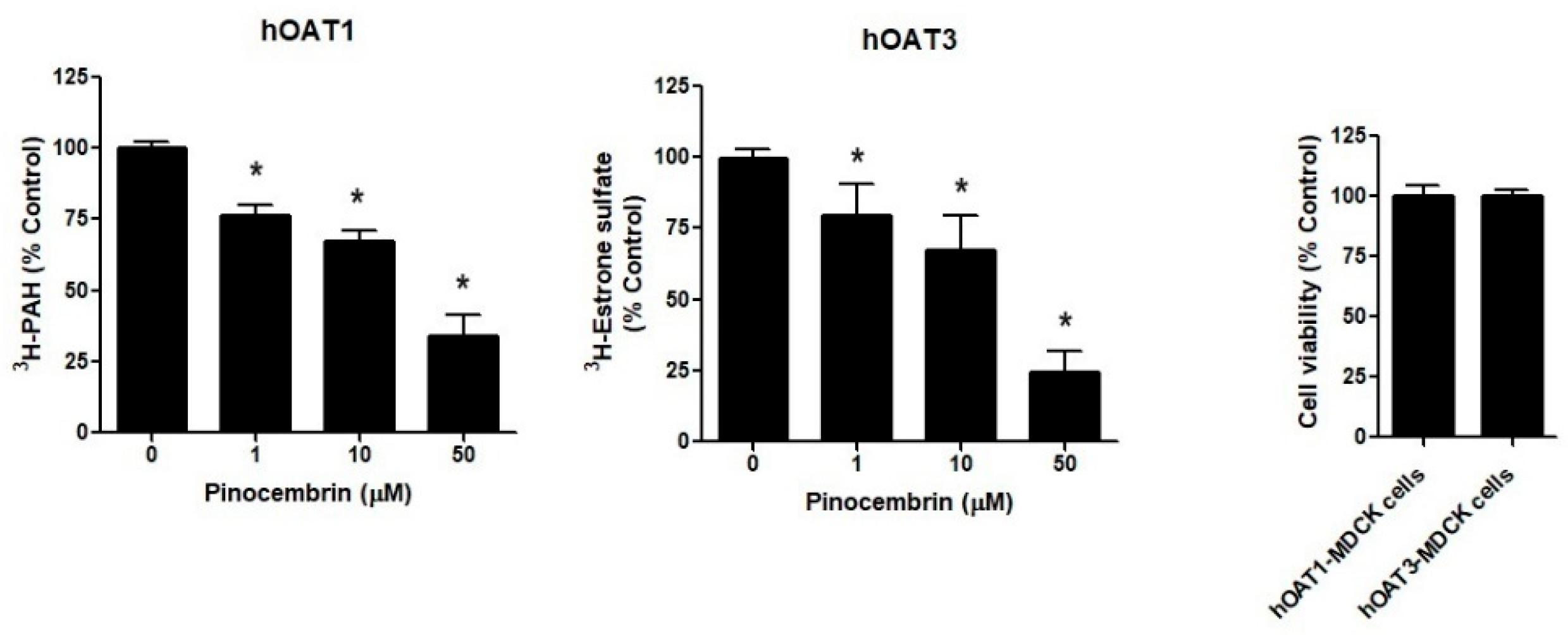
2.4. The Effect of Pinocembrin Treatment on the hOAT1 and hOAT3 Function
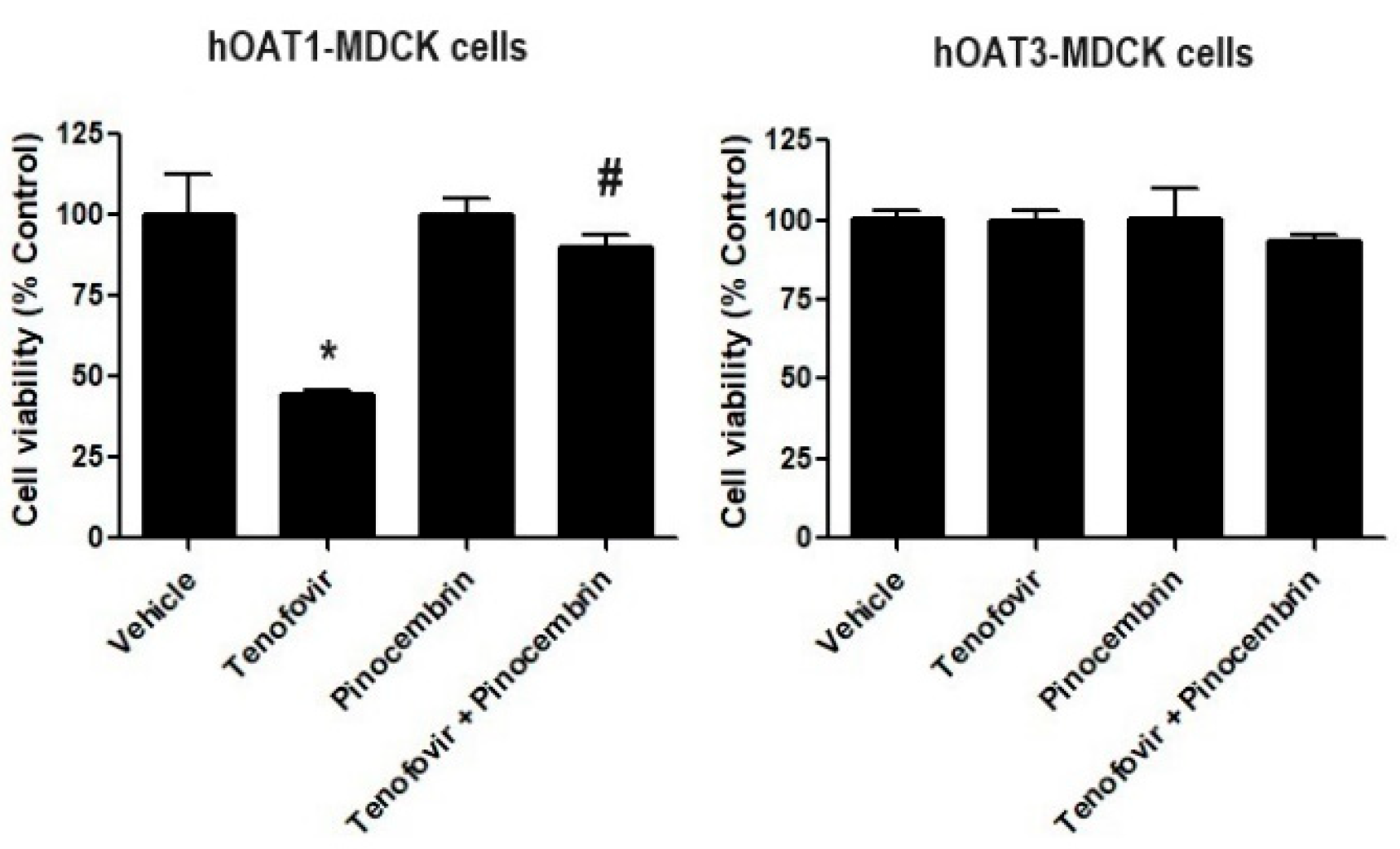
2.5. The Effect of Pinocembrin on Tenofovir Induces Toxicity in hOAT1-MDCK Cells and hOAT3-MDCK Cells
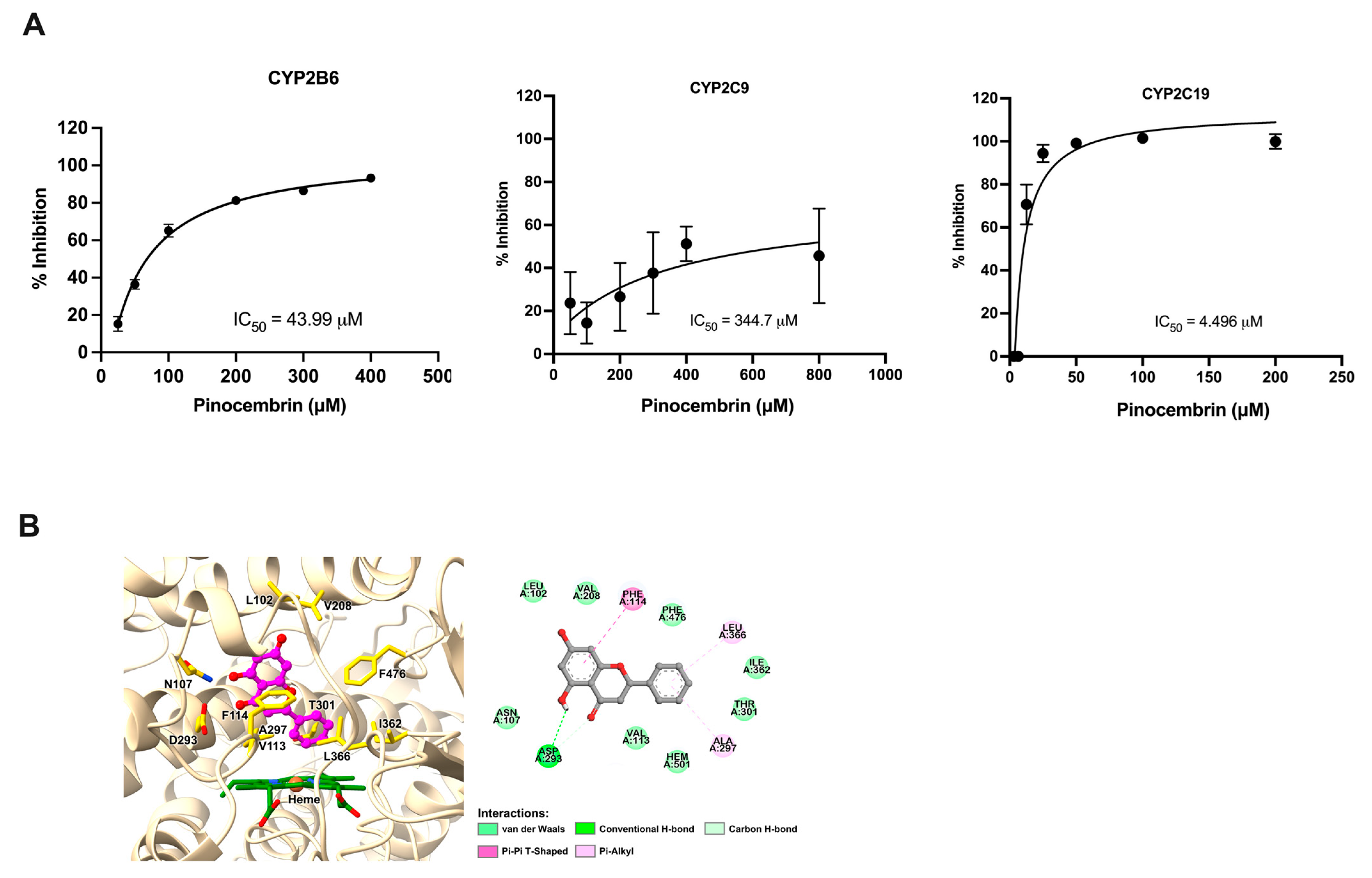
2.6. The Effect of Pinocembrin on the Activity of CYP Enzymes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Transepithelial Transport Assay
4.4. Cis-Inhibition Assay
4.5. Determining the Inhibitory Potency of Pinocembrin on OATs-Mediated Transport
4.6. Kinetics of hOAT-Mediated Transport
4.7. System Preparation and Molecular Docking
4.8. Cell-Free CYP450 Inhibition Assay
4.9. Cell Viability Assay
4.10. Data Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Elbatreek, M.H.; Mahdi, I.; Ouchari, W.; Mahmoud, M.F.; Sobeh, M. Current advances on the therapeutic potential of pinocembrin: An updated review. Biomed. Pharmacother. 2023, 157, 114032. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. The Nrf2/HO-1 axis as targets for flavanones: Neuroprotection by pinocembrin, naringenin, and eriodictyol. Oxidative Med. Cell. Longev. 2019, 2019, 4724920. [Google Scholar] [CrossRef]
- Tundis, R.; Frattaruolo, L.; Carullo, G.; Armentano, B.; Badolato, M.; Loizzo, M.R.; Aiello, F.; Cappello, A.R. An ancient remedial repurposing: Synthesis of new pinocembrin fatty acid acyl derivatives as potential antimicrobial/anti-inflammatory agents. Nat. Prod. Res. 2019, 33, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Li, X.; Cui, Y.; Xiao, T.; Liu, R.; Wang, M.; Wei, Y.; Cui, M.; Ren, S.; Helian, K. Pinocembrin relieves lipopolysaccharide and bleomycin-induced lung inflammation via inhibiting TLR4-NF-κB-NLRP3 inflammasome signaling pathway. Int. Immunopharmacol. 2021, 90, 107230. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Lin, S.; Gao, Y.; Zou, X.; Zhu, J.; Chen, M.; Wan, H.; Zhu, H. Pinocembrin inhibits the proliferation and migration and promotes the apoptosis of ovarian cancer cells through down-regulating the mRNA levels of N-cadherin and GABAB receptor. Biomed. Pharmacother. 2019, 120, 109505. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wan, G.; Yang, B.; Gu, X.; Lin, J. Cardioprotective natural compound pinocembrin attenuates acute ischemic myocardial injury via enhancing glycolysis. Oxidative Med. Cell. Longev. 2020, 2020, 4850328. [Google Scholar] [CrossRef] [PubMed]
- Izuta, H.; Shimazawa, M.; Tazawa, S.; Araki, Y.; Mishima, S.; Hara, H. Protective effects of Chinese propolis and its component, chrysin, against neuronal cell death via inhibition of mitochondrial apoptosis pathway in SH-SY5Y cells. J. Agric. Food Chem. 2008, 56, 8944–8953. [Google Scholar] [CrossRef]
- Campbell, B.C.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Luo, X.; Yang, Z. Advances in biosynthesis, pharmacology, and pharmacokinetics of pinocembrin, a promising natural small-molecule drug. Molecules 2019, 24, 2323. [Google Scholar] [CrossRef]
- Cao, G.; Ying, P.; Yan, B.; Xue, W.; Li, K.; Shi, A.; Sun, T.; Yan, J.; Hu, X. Pharmacokinetics, safety, and tolerability of single and multiple-doses of pinocembrin injection administered intravenously in healthy subjects. J. Ethnopharmacol. 2015, 168, 31–36. [Google Scholar] [CrossRef]
- Food and Drug Administration. Guidance for Industry: In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme-and Transporter-Mediated Drug Interactions; Center for Drug Evaluation and Research, FDA: Silver Spring, MD, USA, 2020. [Google Scholar]
- Burckhardt, G. Drug transport by organic anion transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef]
- Slaughter, R.L.; Edwards, D.J. Recent advances: The cytochrome P450 enzymes. Ann. Pharmacother. 1995, 29, 619–624. [Google Scholar] [CrossRef]
- Navrátilová, L.; Ramos Mandíková, J.; Pávek, P.; Mladěnka, P.; Trejtnar, F. Honey flavonoids inhibit hOATP2B1 and hOATP1A2 transporters and hOATP-mediated rosuvastatin cell uptake in vitro. Xenobiotica 2018, 48, 745–755. [Google Scholar] [CrossRef]
- Yang, Z.H.; Sun, X.; Qi, Y.; Mei, C.; Sun, X.B.; Du, G.H. Uptake characteristics of pinocembrin and its effect on p-glycoprotein at the blood-brain barrier in in vitro cell experiments. J. Asian Nat. Prod. Res. 2012, 14, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.W.; Li, Y.; Paxton, J.W.; Birch, N.P.; Scheepens, A. Identification of novel dietary phytochemicals inhibiting the efflux transporter breast cancer resistance protein (BCRP/ABCG2). Food Chem. 2013, 138, 2267–2274. [Google Scholar] [CrossRef]
- Kondža, M.; Bojić, M.; Tomić, I.; Maleš, Ž.; Rezić, V.; Ćavar, I. Characterization of the CYP3A4 enzyme inhibition potential of selected flavonoids. Molecules 2021, 26, 3018. [Google Scholar] [CrossRef]
- Kondža, M.; Rimac, H.; Maleš, Ž.; Turčić, P.; Ćavar, I.; Bojić, M. Inhibitory effect of acacetin, apigenin, chrysin and pinocembrin on human cytochrome P450 3A4. Croat. Chem. Acta 2020, 93, 33–39. [Google Scholar] [CrossRef]
- Bhatt, S.; Dhiman, S.; Kumar, V.; Gour, A.; Manhas, D.; Sharma, K.; Ojha, P.K.; Nandi, U. Assessment of the CYP1A2 inhibition-mediated drug interaction potential for pinocembrin using in silico, in vitro, and in vivo approaches. ACS Omega 2022, 7, 20321–20331. [Google Scholar]
- Boonnop, R.; Meetam, P.; Siangjong, L.; Tuchinda, P.; Thongphasuk, P.; Soodvilai, S.; Soodvilai, S. Black ginger extract and its active compound, 5,7-dimethoxyflavone, increase intestinal drug absorption via efflux drug transporter inhibitions. Drug Metab. Pharmacokinet. 2023, 50, 100500. [Google Scholar] [CrossRef]
- Dou, T.; Lian, T.; Shu, S.; He, Y.; Jiang, J. The substrate and inhibitor binding mechanism of polyspecific transporter OAT1 revealed by high-resolution cryo-EM. Nat. Struct. Mol. Biol. 2023, 30, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.T.; Wu, W.; Lun, C.; Nigam, S. The drug transporter OAT3 (SLC22A8) and endogenous metabolite communication via the gut-liver-kidney axis. J. Biol. Chem. 2017, 292, 15789–15803. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.M.; Neary, M.; Owen, A. The role of drug transporters in the kidney: Lessons from tenofovir. Front. Pharmacol. 2014, 5, 248. [Google Scholar] [CrossRef]
- Zou, L.; Matsson, P.; Stecula, A.; Ngo, H.X.; Zur, A.A.; Giacomini, K.M. Drug Metabolites Potently Inhibit Renal Organic Anion Transporters, OAT1 and OAT3. J. Pharm. Sci. 2021, 110, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Sayre, C.L.; Alrushaid, S.; Martinez, S.E.; Anderson, H.D.; Davies, N.M. Pre-clinical pharmacokinetic and pharmacodynamic characterization of selected chiral flavonoids: Pinocembrin and pinostrobin. J. Pharm. Pharm. Sci. 2015, 18, 368–395. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 inhibition: Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Padmanabhan, S. Handbook of Pharmacogenomics and Stratified Medicine; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter | IC50 (μM) | Ki (μM) | Km (μM) | Vmax (nmol/min/cm2) | ||
|---|---|---|---|---|---|---|
| No Pinocembrin | With Pinocembrin | No Pinocembrin | With Pinocembrin | |||
| hOAT1 | 1.47 ± 0.87 | 0.81 ± 0.12 | 12.33 ± 1.32 | 42.55 ± 2.98 * | 5.43 ± 0.25 | 6.03 ± 1.02 |
| hOAT3 | 2.38 ± 0.98 | 1.30 ± 0.24 | 11.95 ± 1.58 | 30.20 ± 3.21 * | 9.88 ± 1.05 | 11.32 ± 2.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sangkapat, S.; Boonnop, R.; Pimta, J.; Chabang, N.; Nutho, B.; Jutabha, P.; Soodvilai, S. Potential Interaction of Pinocembrin with Drug Transporters and Hepatic Drug-Metabolizing Enzymes. Pharmaceuticals 2025, 18, 42. https://doi.org/10.3390/ph18010042
Sangkapat S, Boonnop R, Pimta J, Chabang N, Nutho B, Jutabha P, Soodvilai S. Potential Interaction of Pinocembrin with Drug Transporters and Hepatic Drug-Metabolizing Enzymes. Pharmaceuticals. 2025; 18(1):42. https://doi.org/10.3390/ph18010042
Chicago/Turabian StyleSangkapat, Sirima, Rattiporn Boonnop, Jeerawat Pimta, Napason Chabang, Bodee Nutho, Promsuk Jutabha, and Sunhapas Soodvilai. 2025. "Potential Interaction of Pinocembrin with Drug Transporters and Hepatic Drug-Metabolizing Enzymes" Pharmaceuticals 18, no. 1: 42. https://doi.org/10.3390/ph18010042
APA StyleSangkapat, S., Boonnop, R., Pimta, J., Chabang, N., Nutho, B., Jutabha, P., & Soodvilai, S. (2025). Potential Interaction of Pinocembrin with Drug Transporters and Hepatic Drug-Metabolizing Enzymes. Pharmaceuticals, 18(1), 42. https://doi.org/10.3390/ph18010042