


Computational Exploration of Potential Pharmacological Inhibitors Targeting the Envelope Protein of the Kyasanur Forest Disease Virus

 ,
,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
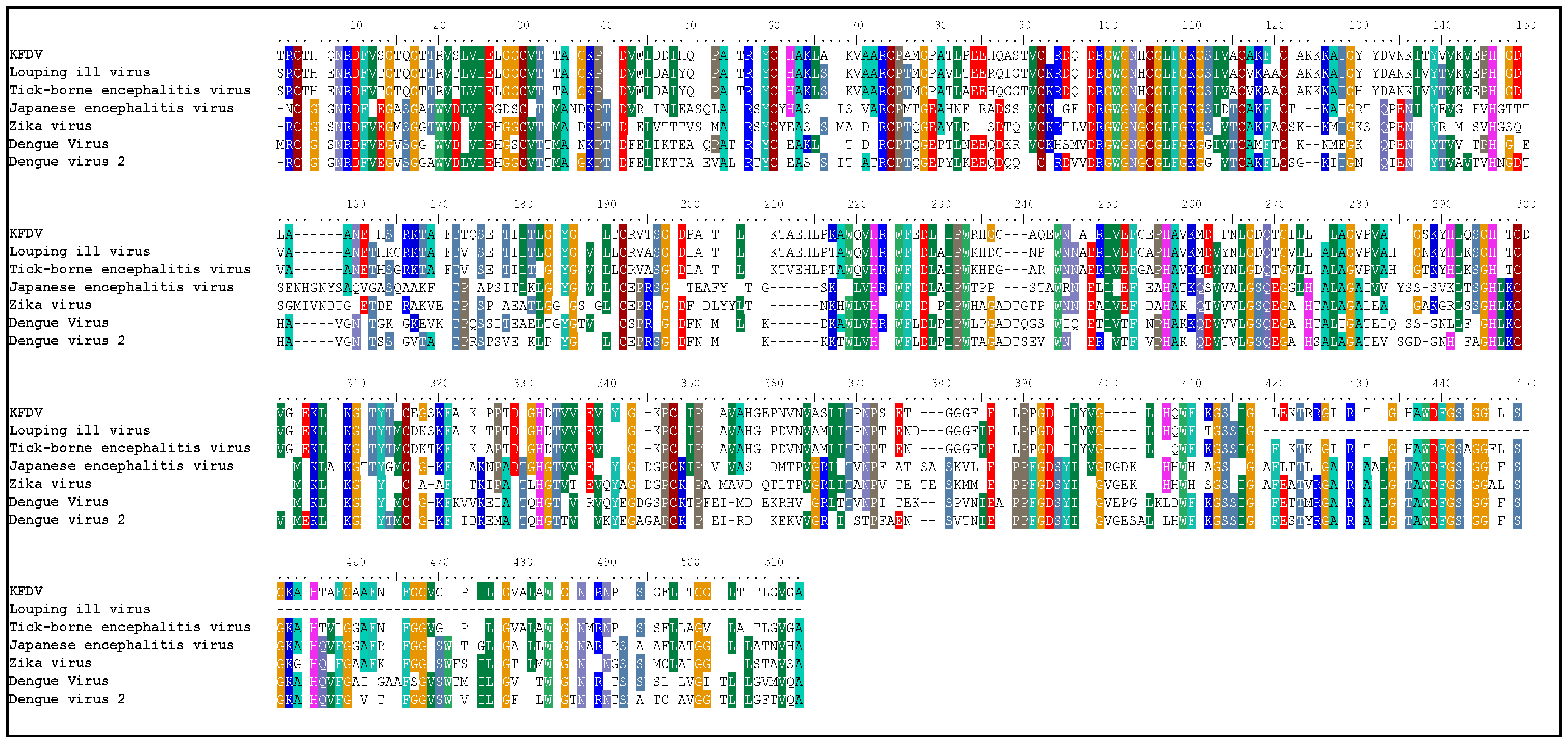
2.1. Sequence Analysis
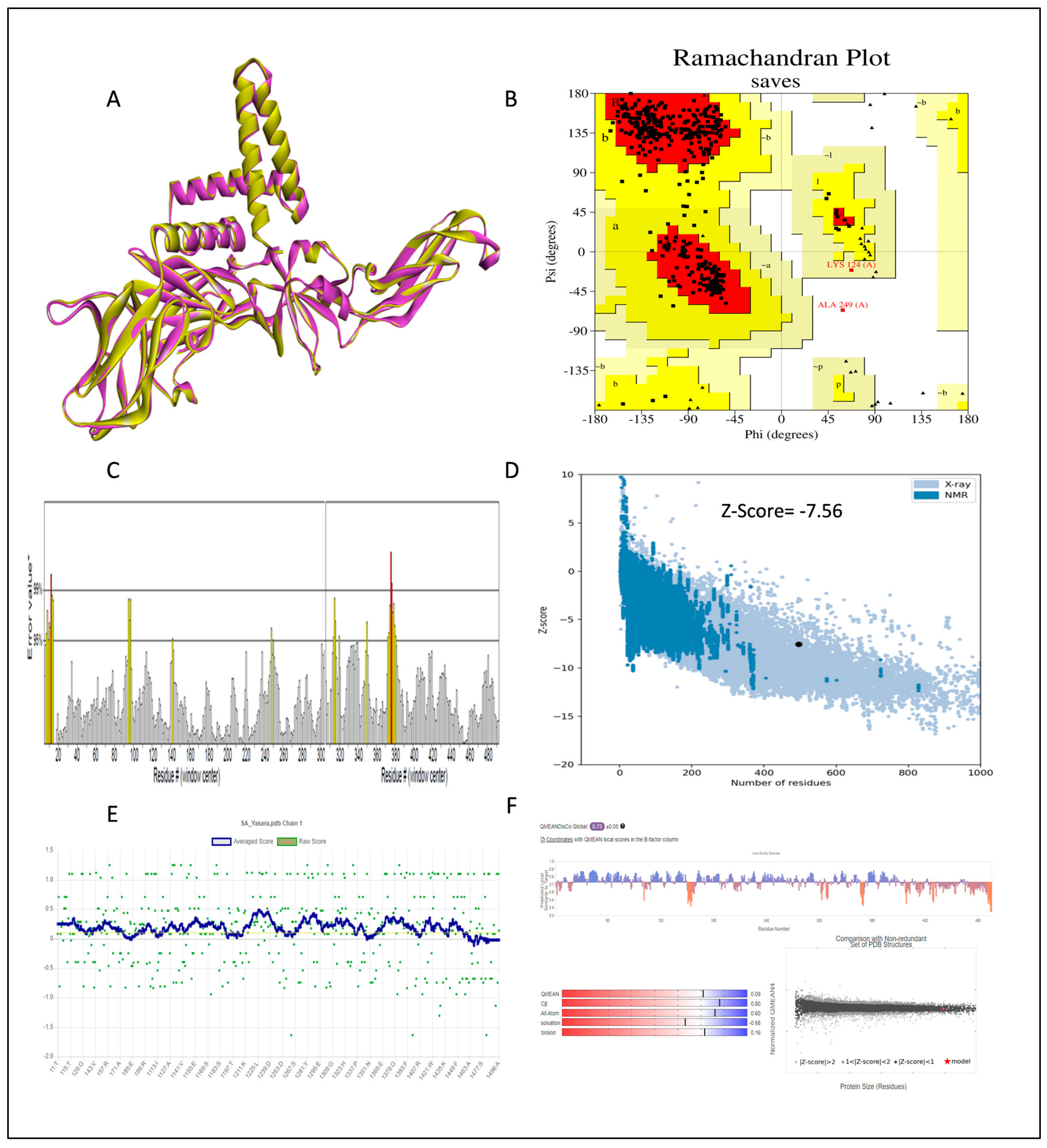
2.2. The 3D Structure Determination and Structure Validation
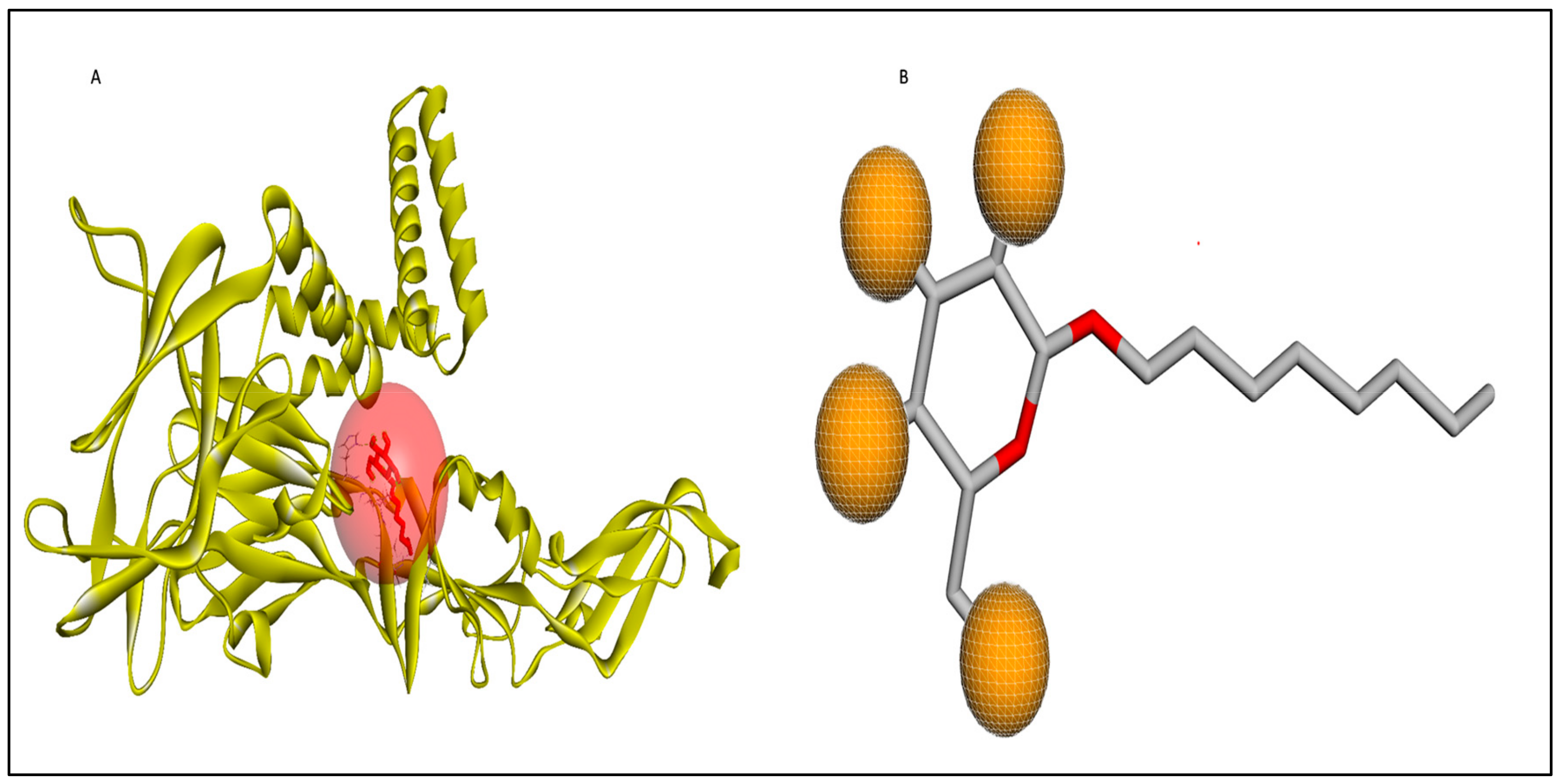
2.3. Active Site Determination
2.4. Ligand Design
2.4.1. De Novo Drug Designing
2.4.2. Pharmacophoric Screening
2.4.3. Ligand-Based Screening
2.5. Virtual Screening
2.6. Toxicity Measurement
2.7. ADME Analysis
2.8. Molecular Docking
2.9. DFT Studies
2.10. Frontier Molecular Orbital—HOMO/LUMO Calculation
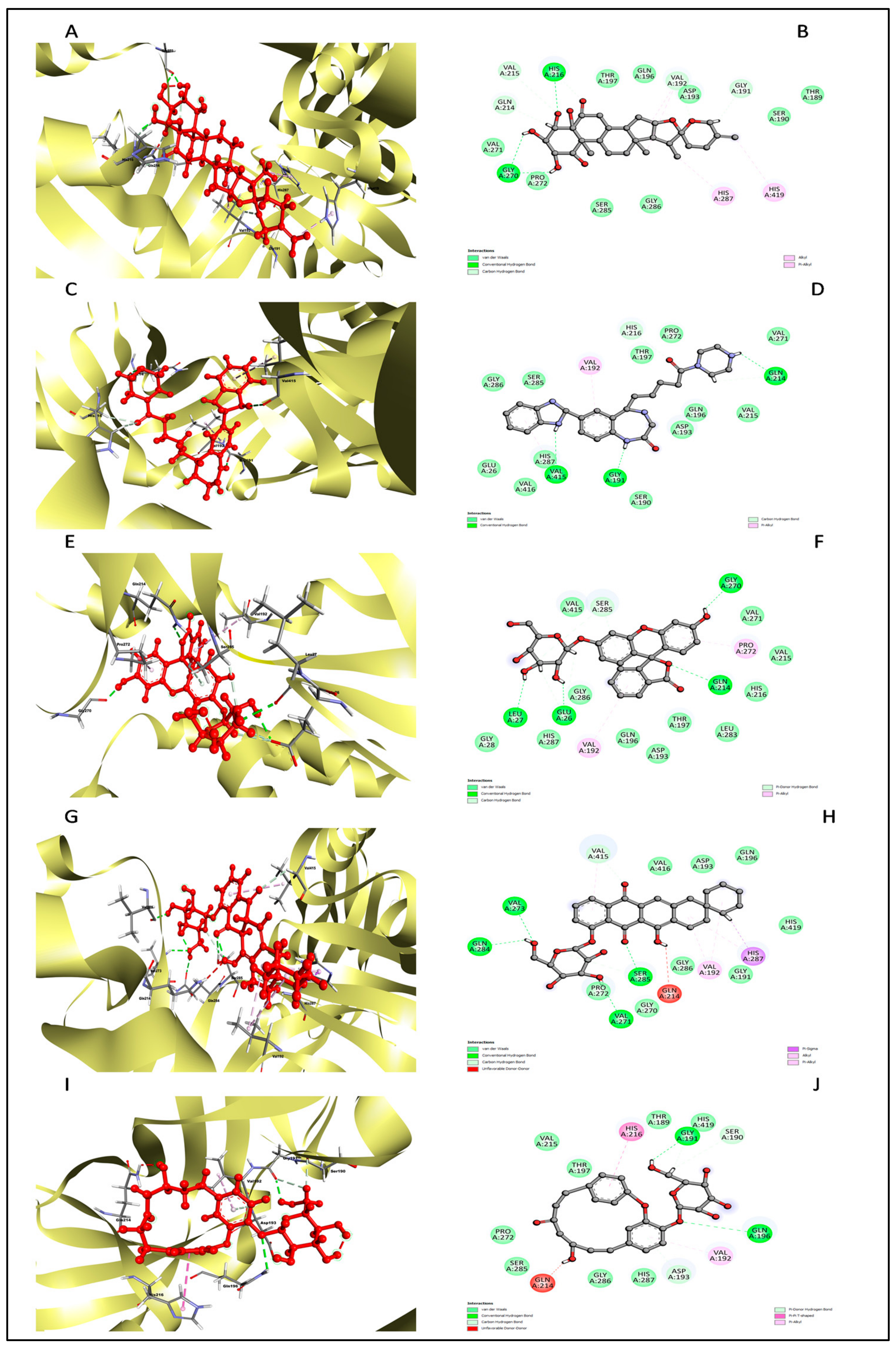
2.11. Redocking and Interaction Analysis
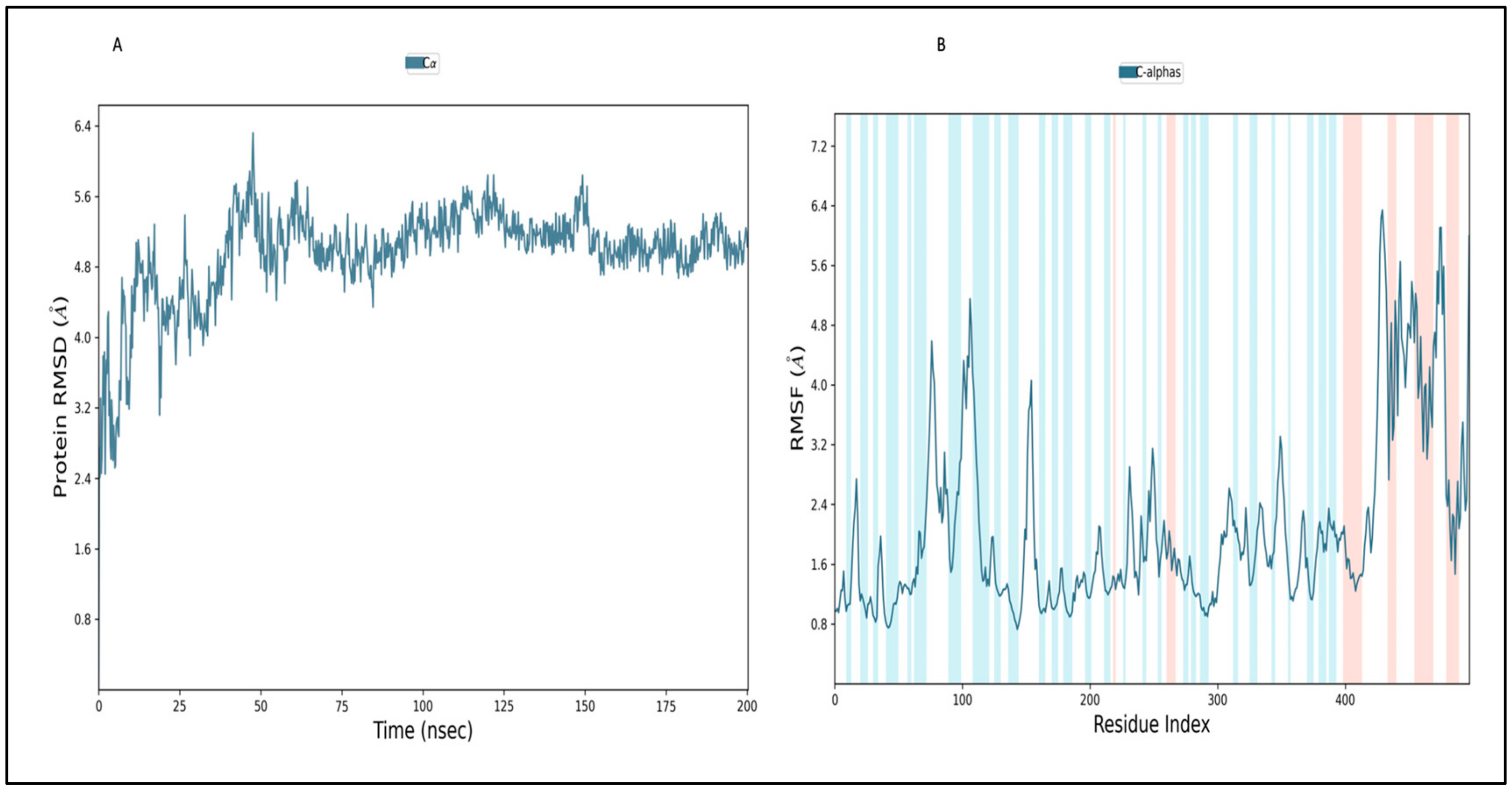
2.12. Molecular Dynamics (MD) Simulation Studies
2.12.1. Simulation of Envelope Protein
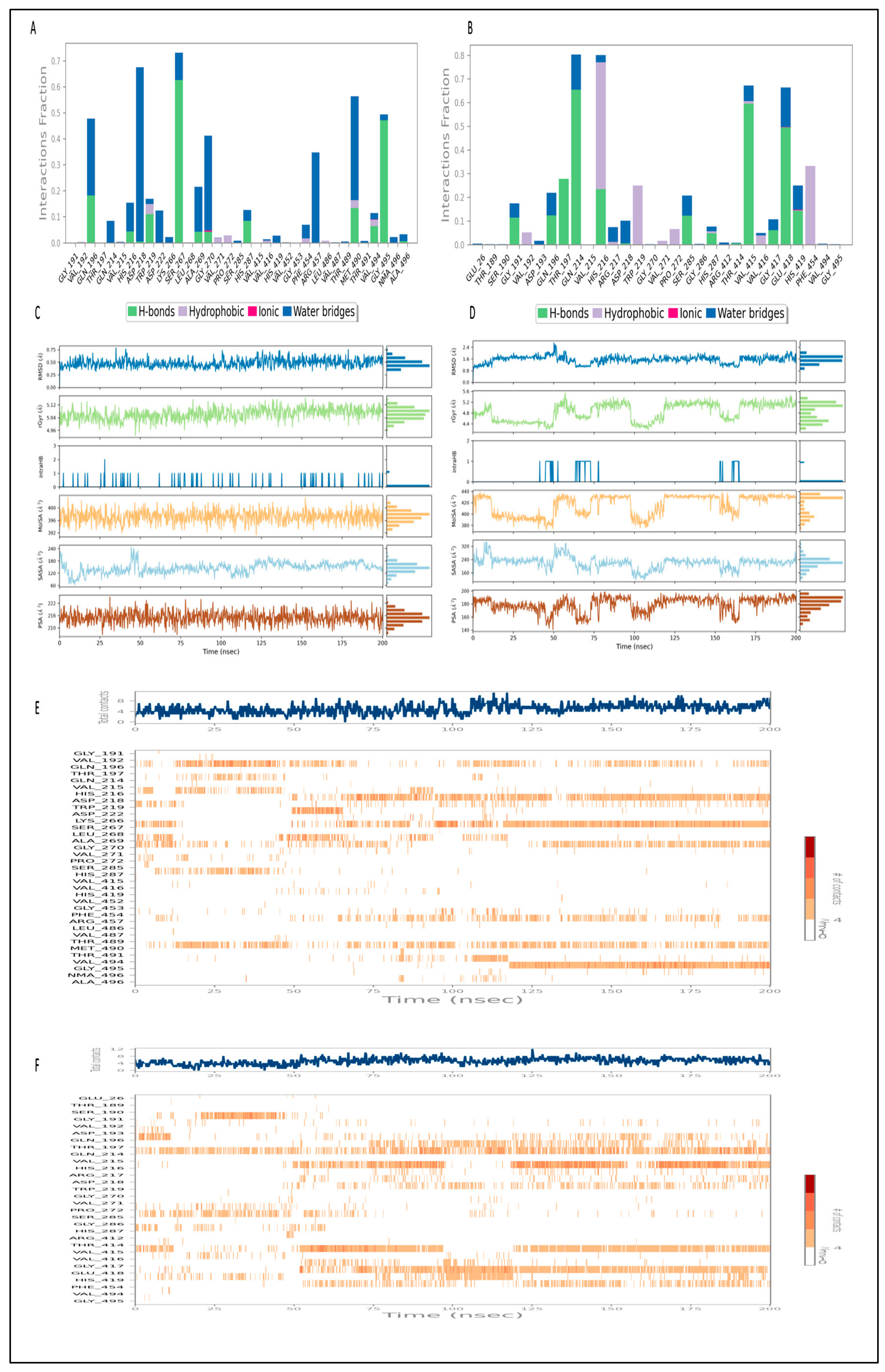
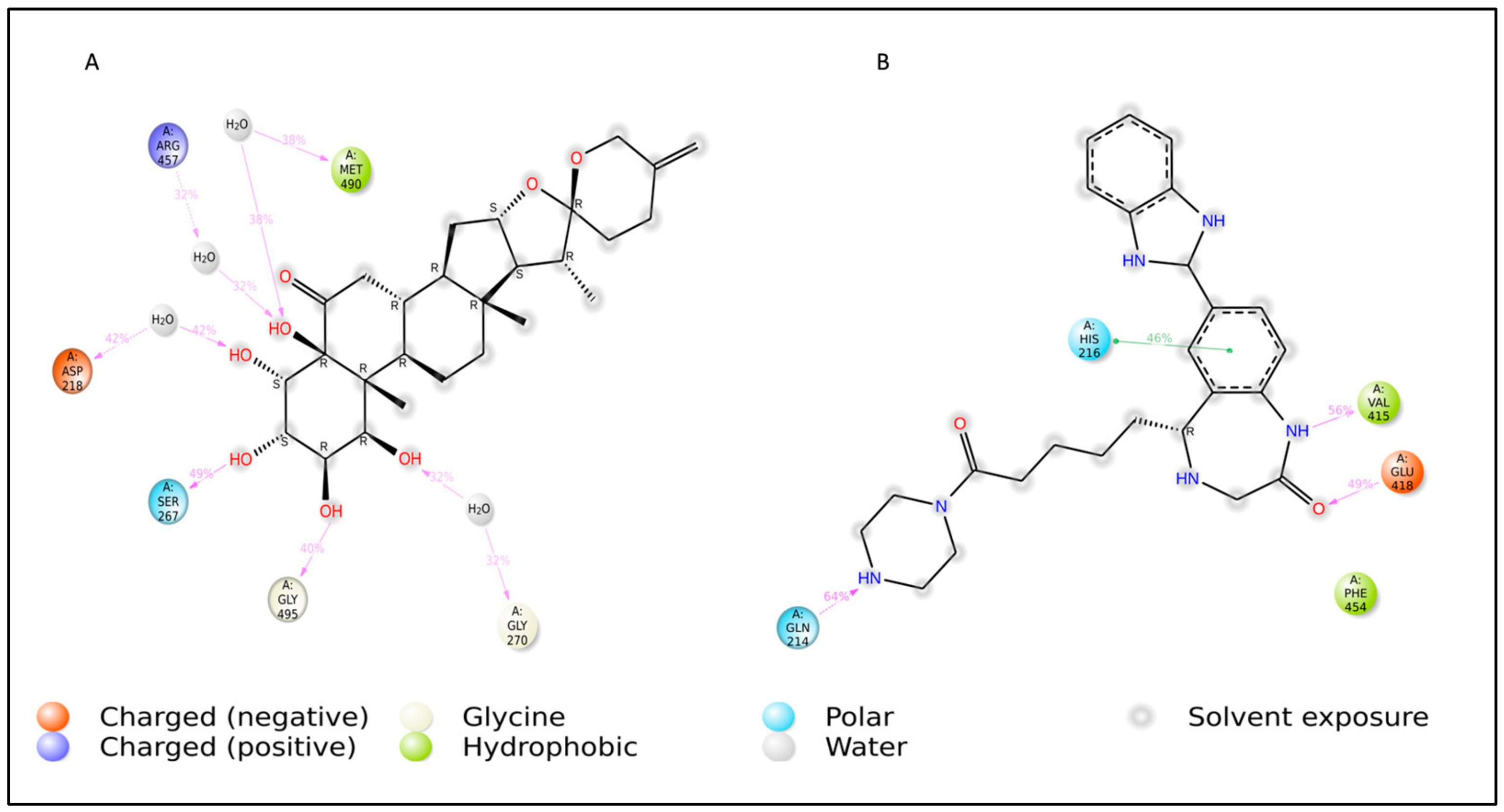
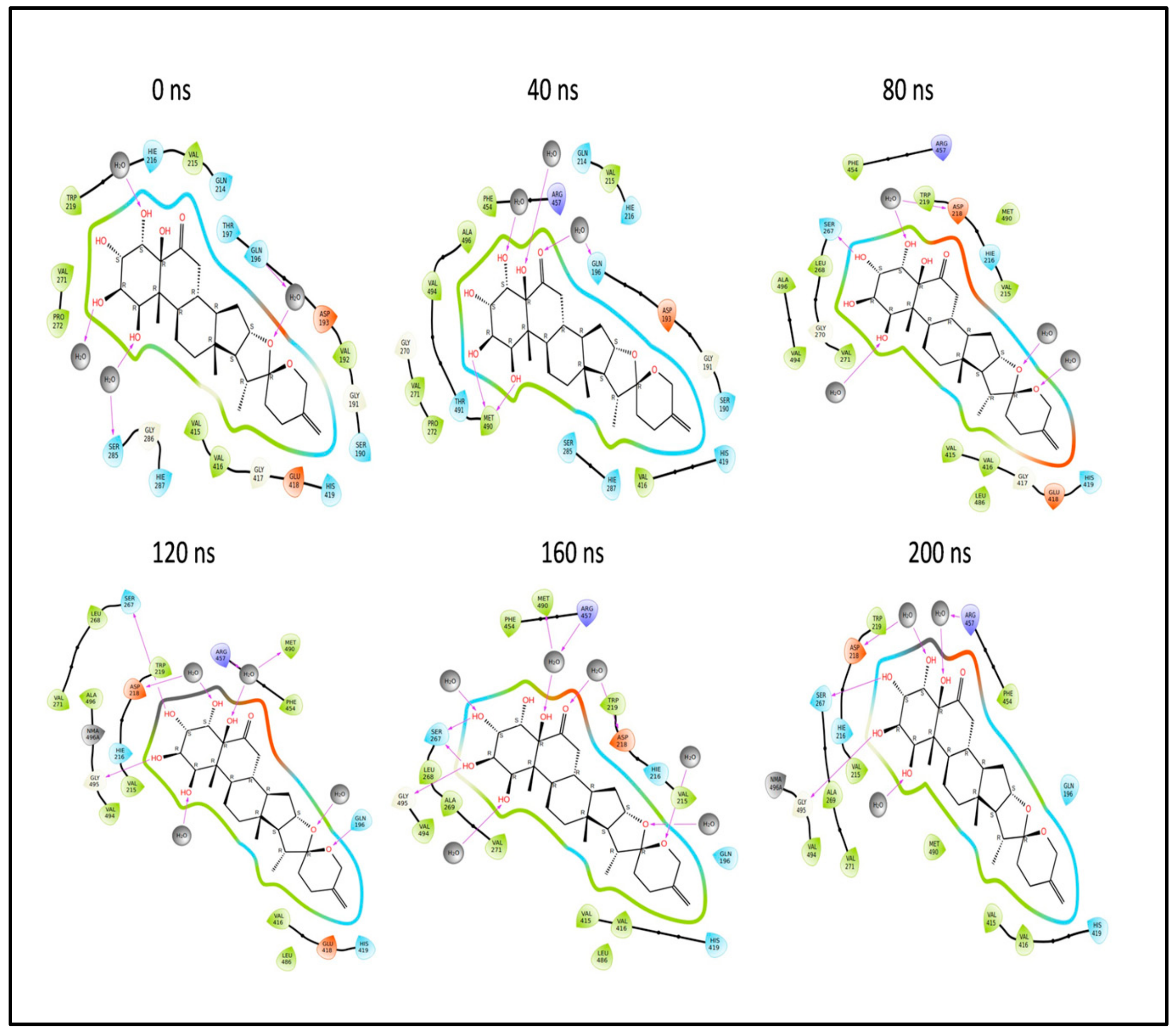
2.12.2. Simulation of L3 Complex
2.12.3. Simulation of L4 Complex
2.13. Binding Free Energy Analysis of the L3 and L4 Complex
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Sequence Retrieval and Analysis
4.2.2. Homology Modeling and Structure Refinement
4.2.3. Structure Validation
4.2.4. Binding Site/Active Site Prediction
4.2.5. Ligand Design
De Novo Design
Pharmacophore Screening
Ligand-Based Screening
4.2.6. Virtual Screening
4.2.7. ADMET Studies
4.2.8. Geometry Optimization of the Selected Ligand Compounds
4.2.9. Frontier Molecular Orbital HOMO/LUMO Calculation
4.2.10. Molecular Docking
4.2.11. Molecular Simulation Studies for Protein and Complex
4.2.12. Binding Free Energy Analysis of L3 and L4 Complex
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, B.W.; Cutts, T.A.; Court, D.A.; Theriault, S. The generation of a reverse genetics system for Kyasanur Forest Disease Virus and the ability to antagonize the induction of the antiviral state in vitro. Virus Res. 2012, 163, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Mourya, D.T.; Yadav, P.D.; Patil, D.Y.; Sahay, R.R.; Rahi, M. Experiences of Indian Council of Medical Research with tick-borne zoonotic infections: Kyasanur Forest disease & Crimean-Congo haemorrhagic fever in India with One Health focus. Indian J. Med. Res. 2021, 153, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Munivenkatappa, A.; Sahay, R.R.; Yadav, P.D.; Viswanathan, R.; Mourya, D.T. Clinical & epidemiological significance of Kyasanur forest disease. Indian J. Med. Res. 2018, 148, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.Z.; Jabbar, B.; Ahmed, N.; Rehman, A.; Nasir, H.; Nadeem, S.; Jabbar, I.; Rahman, Z.U.; Azam, S. Epidemiology, pathogenesis, and control of a tick-borne disease-Kyasanur forest disease: Current status and future directions. Front. Cell. Infect. Microbiol. 2018, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, P. Kyasanur forest disease: An epidemiological view in India. Rev. Med. Virol. 2006, 16, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Ajesh, K.; Nagaraja, B.K.; Sreejith, K. Kyasanur forest disease virus breaking the endemic barrier: An investigation into ecological effects on disease emergence and future outlook. Zoonoses Public Health 2017, 64, e73–e80. [Google Scholar] [CrossRef] [PubMed]
- ICD-10-CM Diagnosis Codes. 2017. Available online: http://www.icd10data.com/ICD10CM/Codes/A00-B99/A90-A99/A98-/A98.2 (accessed on 17 March 2017).
- Keshavamurthy, R.; Charles, L.E. Predicting Kyasanur forest disease in resource-limited settings using event-based surveillance and transfer learning. Sci. Rep. 2023, 13, 11067. [Google Scholar] [CrossRef] [PubMed]
- Asaaga, F.A.; Purse, B.V.; Rahman, M.; Srinivas, P.N.; Kalegowda, S.D.; Seshadri, T.; Young, J.C.; Oommen, M.A. The role of social vulnerability in improving interventions for neglected zoonotic diseases: The example of Kyasanur Forest Disease in India. PLoS Glob. Public Health 2023, 3, e0000758. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, R.; Verma, R.; Kumar, R.; Dhankar, M.; Bhallla, K.; Agrawal, G. Kyasanur forest disease: A rare viral hemorrhagic disease in India. Int. J. Community Med. Public Health 2018, 5, 3149–3151. [Google Scholar] [CrossRef]
- Gurav, Y.K.; Yadav, P.D.; Gokhale, M.D.; Chiplunkar, T.R.; Vishwanathan, R.; Patil, D.Y.; Jain, R.; Shete, A.M.; Patil, S.L.; Sarang, G.D.; et al. Kyasanur forest disease prevalence in western ghats proven and confirmed by recent outbreak in maharashtra, india, 2016. Vector-Borne Zoonotic Dis. 2018, 18, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.Y.; Yadav, P.D.; Shete, A.M.; Nuchina, J.; Meti, R.; Bhattad, D.; Someshwar, S.; Mourya, D.T. Occupational exposure of cashew nut workers to Kyasanur Forest disease in Goa, India. Int. J. Infect. Dis. 2017, 61, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; Fu, S.; Wang, H.; Ni, D.; Nasci, R.; Tang, Q.; Liang, G. Isolation of Kyasanur forest disease virus from febrile patient, Yunnan, China. Emerg. Infect. Dis. 2009, 15, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Kandagalla, S.; Kumbar, B.; Novak, J. Structural Modifications Introduced by NS2B Cofactor Binding to the NS3 Protease of the Kyasanur Forest Disease Virus. Int. J. Mol. Sci. 2023, 24, 10907. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, B.; Tang-Huau, T.L.; Feldmann, F.; Hanley, P.W.; Rosenke, R.; Shaia, C.; Marzi, A.; Feldmann, H. Single-dose VSV-based vaccine protects against Kyasanur Forest disease in nonhuman primates. Sci. Adv. 2023, 9, eadj1428. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, S.; Varamballi, P. In-silico design of envelope based multi-epitope vaccine candidate against Kyasanur forest disease virus. Sci. Rep. 2021, 11, 17118. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Pratibha, M.; Singh Dagur, H.; Rajakumara, E. Characterization of host receptor interaction with envelop protein of Kyasanur forest disease virus and predicting suitable epitopes for vaccine candidate. J. Biomol. Struct. Dyn. 2023, 42, 4110–4120. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Rossmann, M.G. Molecular mechanisms involved in the early steps of flavivirus cell entry. Microbes Infect. 2011, 13, 1–9. [Google Scholar] [CrossRef]
- Poonsiri, T.; Wright, G.S.; Solomon, T.; Antonyuk, S.V. Crystal structure of the Japanese encephalitis virus capsid protein. Viruses 2019, 11, 623. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Leang, S.K.; Davidson, A.; Lobigs, M. Both E protein glycans adversely affect dengue virus infectivity but are beneficial for virion release. J. Virol. 2010, 84, 5171–5180. [Google Scholar] [CrossRef]
- Klein, D.E.; Choi, J.L.; Harrison, S.C. Structure of a dengue virus envelope protein late-stage fusion intermediate. J. Virol. 2013, 87, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Rodenhuis-Zybert, I.; Wilschut, J.; Moesker, B.; Smit, J.M. Flavivirus Cell Entry and Membrane Fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Castillo, C.; Rodríguez, M.H.; Ortiz-Navarrete, V.; Alpuche-Aranda, C.M.; Martinez-Barnetche, J. Structural and immunological basis of cross-reactivity between dengue and Zika infections: Implications in serosurveillance in endemic regions. Front. Microbiol. 2023, 14, 1107496. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ragavan, R.M.; Purushothaman, I.; Swaminathan, R.; Almutairi, S.M.; Hussein, D.S.; Rasheed, R.A.; Narasingam, A. Malacitanolide, reissantin E and paclitaxel compounds as inhibitors of envelope, NS5 and NS2B/NS3 target proteins of dengue virus: Computational docking and molecular dynamics simulations studies. J. King Saud. Univ.-Sci. 2023, 35, 102868. [Google Scholar] [CrossRef]
- Srikanth, U.G.K.; Marinaik, C.B.; Gomes, A.R.; Rathnamma, D.; Byregowda, S.M.; Isloor, S.; Munivenkatarayappa, A.; Venkatesha, M.D.; Rao, S.; Rizwan, A.; et al. Evaluation of Safety and Potency of Kyasanur Forest Disease (KFD) Vaccine Inactivated with Different Concentrations of Formalin and Comparative Evaluation of In Vitro and In Vivo Methods of Virus Titration in KFD Vaccine. Biomedicines 2023, 11, 1871. [Google Scholar] [CrossRef]
- Hafeez, S.; Achur, R.; Kiran, S.K.; Thippeswamy, N.B. Computational prediction of B and T-cell epitopes of Kyasanur Forest Disease virus marker proteins towards the development of precise diagnosis and potent subunit vaccine. J. Biomol. Struct. Dyn. 2023, 41, 9157–9176. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2003, 100, 6986–6991. [Google Scholar] [CrossRef]
- Kantardjieff, K.A.; Rupp, B. Protein isoelectric point as a predictor for increased crystallization screening efficiency. Bioinformatics 2004, 20, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [CrossRef]
- Guruprasad, K.; Reddy, B.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved protein–ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W438–W442. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schneider, G. De novo design at the edge of chaos: Miniperspective. J. Med. Chem. 2016, 59, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de novo drug design: From conventional to machine learning methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef] [PubMed]
- Nagamalla, L.; Kumar, J.S.; Shaik, M.R.; Sanjay, C.; Alsamhan, A.M.; Kasim, M.A.; Alwarthan, A. Identification of Novel AXL Kinase Inhibitors Using Ligand-Based Pharmacophore Screening and Molecular Dynamics Simulations. Crystals 2022, 12, 1158. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, W.; Fang, H.; Perkins, R.; Tong, W.; Hong, H. Homology modeling, molecular docking, and molecular dynamics simulations elucidated alpha-fetoprotein binding modes. BMC Bioinform. 2013, 14 (Suppl. S14), S6. [Google Scholar] [CrossRef] [PubMed]
- Oduselu, G.O.; Afolabi, R.; Ademuwagun, I.; Vaughan, A.; Adebiyi, E. Structure-based pharmacophore modeling, virtual screening, and molecular dynamics simulation studies for identification of Plasmodium falciparum 5-aminolevulinate synthase inhibitors. Front. Med. 2023, 9, 1022429. [Google Scholar] [CrossRef]
- Kochnev, Y.; Hellemann, E.; Cassidy, K.C.; Durrant, J.D. Webina: An open-source library and web app that runs AutoDockVina entirely in the web browser. Bioinformatics 2020, 36, 4513–4515. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- van den Anker, J.; Reed, M.D.; Allegaert, K.; Kearns, G.L. Developmental changes in pharmacokinetics and pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Baidya, A.T.; Mathew, A.T.; Yadav, A.K.; Kumar, R. Structural modification aimed for improving solubility of lead compounds in early phase drug discovery. Bioorganic Med. Chem. 2022, 56, 116614. [Google Scholar] [CrossRef] [PubMed]
- Sawale, R.T.; Kalyankar, T.M.; George, R.; Deosarkar, S.D. Molar Refraction Polarizability of Antiemetic drug 4-amino-5-chloro-N-(2-(diethylamino) ethyl)-2 methoxybenzamide hydrochloride monohydrate in {Aqueous-Sodium or Lithium Chloride} Solutions at 30 °C. J. Appl. Pharm. Sci. 2016, 6, 120–124. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P., Jr. In silico strategies to support fragment-to-lead optimization in drug discovery. Front. Chem. 2020, 8, 93. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Luer, M.S.; Penzak, S.R. Pharmacokinetic properties. In Applied Clinical Pharmacokinetics and Pharmacodynamics of Psychopharmacological Agents; Adis: Cham, Switzerland, 2016; pp. 3–27. [Google Scholar]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef] [PubMed]
- Löbenberg, R.; Amidon, G.L.; Ferraz, H.G.; Bou-Chacra, N. Mechanism of gastrointestinal drug absorption and application in therapeutic drug delivery. In Therapeutic Delivery Methods: A Concise Overview of Emerging Areas; Future Science Ltd.: London, UK, 2013. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, V. P-glycoprotein transporter in drug development. EXCLI J. 2016, 15, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef] [PubMed]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Arya, R.; Kumari, S.; Pandey, B.; Mistry, H.; Bihani, S.C.; Das, A.; Prashar, V.; Gupta, G.D.; Panicker, L.; Kumar, M. Structural insights into SARS-CoV-2 proteins. J. Mol. Biol. 2021, 433, 166725. [Google Scholar] [CrossRef] [PubMed]
- Kasabi, G.S.; Murhekar, M.V.; Sandhya, V.K.; Raghunandan, R.; Kiran, S.K.; Channabasappa, G.H.; Mehendale, S.M. Coverage and effectiveness of Kyasanur forest disease (KFD) vaccine in Karnataka, South India, 2005–2010. PLoS Neglected Trop. Dis. 2013, 7, e2025. [Google Scholar] [CrossRef]
- Kiran, S.K.; Pasi, A.; Kumar, S.; Kasabi, G.S.; Gujjarappa, P.; Shrivastava, A.; Mehendale, S.; Chauhan, L.S.; Laserson, K.F.; Murhekar, M. Kyasanur Forest disease outbreak and vaccination strategy, Shimoga District, India, 2013–2014. Emerg. Infect. Dis. 2015, 21, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Cook, B.W.; Ranadheera, C.; Nikiforuk, A.M.; Cutts, T.A.; Kobasa, D.; Court, D.A.; Theriault, S.S. Limited effects of Type I interferons on Kyasanur Forest Disease Virus in cell culture. PLoS Neglected Trop. Dis. 2016, 10, e0004871. [Google Scholar] [CrossRef] [PubMed]
- Rajak, A.; Kumar, J.S.; Dhankher, S.; Sandhya, V.K.; Kiran, S.K.; Golime, R.; Dash, P.K. Development and application of a recombinant Envelope Domain III protein based indirect human IgM ELISA for Kyasanur forest disease virus. Acta Tropica 2022, 235, 106623. [Google Scholar] [CrossRef] [PubMed]
- Bhutkar, M.; Singh, V.; Dhaka, P.; Tomar, S. Virus-host protein-protein interactions as molecular drug targets for arboviral infections. Front. Virol. 2022, 2, 959586. [Google Scholar] [CrossRef]
- Shil, P.; Yadav, P.D.; Patil, A.A.; Balasubramanian, R.; Mourya, D.T. Bioinformatics characterization of envelope glycoprotein from Kyasanur Forest disease virus. Indian J. Med. Res. 2018, 147, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Oladejo, D.O.; Oduselu, G.O.; Dokunmu, T.M.; Isewon, I.; Oyelade, J.; Okafor, E.; Adebiyi, E. In silico structure prediction, molecular docking, and dynamic simulation of Plasmodium falciparum AP2-I transcription factor. Bioinform. Biol. Insights 2023, 17. [Google Scholar] [CrossRef] [PubMed]
- Bachtiar, Z.; Mustopa, A.Z.; Astuti, R.I.; Fauziyah, F.; Fatimah, F.; Rozirwan, R.; Wulandari, T.N.M.; Wijaya, D.P.; Agustriani, F.; Arwansyah, A.; et al. Production of codon-optimized Factor C fragment from Tachypleus gigas in the Pichia pastoris GS115 expression system for endotoxin detection. J. Genet. Eng. Biotechnol. 2023, 21, 103. [Google Scholar] [CrossRef] [PubMed]
- Kabiraj, A.; Laha, A.; Panja, A.S.; Bandopadhyay, R. In silico comparative structural and functional analysis of arsenite methyltransferase from bacteria, fungi, fishes, birds, and mammals. J. Genet. Eng. Biotechnol. 2023, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Khaliq, M.; Suk, J.E.; Patkar, C.; Li, L.; Kuhn, R.J.; Post, C.B. Antiviral compounds discovered by virtual screening of small–molecule libraries against dengue virus E protein. ACS Chem. Biol. 2008, 3, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.A.; Heinz, F.X.; Mandl, C.; Kunz, C.; Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 1995, 375, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Umamaheswari, A.; Kumar, M.M.; Pradhan, D.; Marisetty, H. Docking studies towards exploring antiviral compounds against envelope protein of yellow fever virus. Interdiscip. Sci. Comput. Life Sci. 2011, 3, 64–77. [Google Scholar] [CrossRef]
- Rey, F.A. Dengue virus envelope glycoprotein structure: New insight into its interactions during viral entry. Proc. Natl. Acad. Sci. USA 2003, 100, 6899–6901. [Google Scholar] [CrossRef] [PubMed]
- Hengphasatporn, K.; Garon, A.; Wolschann, P.; Langer, T.; Yasuteru, S.; Huynh, T.N.T.; Chavasiri, W.; Saelee, T.; Boonyasuppayakorn, S.; Rungrotmongkol, T. Multiple virtual screening strategies for the discovery of novel compounds active against dengue virus: A hit identification study. Sci. Pharm. 2020, 88, 2. [Google Scholar] [CrossRef]
- McGrath, J.; O’Doherty, L.; Conlon, N.; Dunne, J.; Brady, G.; Ibrahim, A.; McCormack, W.; Walsh, C.; Domegan, L.; Walsh, S.; et al. Point of care detection of SARS-CoV-2 antibodies and neutralisation capacity—Lateral flow immunoassay evaluation compared to commercial assay to inform potential role in therapeutic and surveillance practices. Front. Public Health 2023, 11, 1245464. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.I.; Saloa, S.; Mahfuj, S.; Islam, M.J.; Mou, M.J. Computer-aided drug design of Azadirachtaindica compounds against nervous necrosis virus by targeting grouper heat shock cognate protein 70 (GHSC70): Quantum mechanics calculations and molecular dynamic simulation approaches. Genom. Inform. 2022, 20, e33. [Google Scholar] [CrossRef] [PubMed]
- Uzzaman, M.; Shawon, J.; Siddique, Z.A. Molecular docking, dynamics simulation and ADMET prediction of Acetaminophen and its modified derivatives based on quantum calculations. SN Appl. Sci. 2019, 1, 1437. [Google Scholar] [CrossRef]
- Shawon, J.; Khan, A.M.; Shahriar, I.; Halim, M.A. Improving the binding affinity and interaction of 5-Pentyl-2-Phenoxyphenol against Mycobacterium Enoyl ACP reductase by computational approach. Inform. Med. Unlocked 2021, 23, 100528. [Google Scholar] [CrossRef]
- Umar, A.K.; Zothantluanga, J.H.; Luckanagul, J.A.; Limpikirati, P.; Sriwidodo, S. Structure-based computational screening of 470 natural quercetin derivatives for identification of SARS-CoV-2 Mpro inhibitor. PeerJ 2023, 11, e14915. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, J.; Patel, R.; Goswami, D.; Saraf, M.; Rawal, R.M. Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Comput. Biol. Med. 2021, 135, 104568. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Gogoi, N.; Chetia, D.; Khan, J.; Banwas, S.; Alshehri, B.; Alaidarous, M.A.; Laddha, U.D.; Khairnar, S.J.; Walode, S.G. Repurposing of phytomedicine-derived bioactive compounds with promising anti-SARS-CoV-2 potential: Molecular docking, MD simulation and drug-likeness/ADMET studies. Saudi J. Biol. Sci. 2022, 29, 2432–2446. [Google Scholar] [CrossRef]
- Umar, A.K.; Zothantluanga, J.H.; Aswin, K.; Maulana, S.; SulaimanZubair, M.; Lalhlenmawia, H.; Rudrapal, M.; Chetia, D. Antiviral phytocompounds “ellagic acid” and “(+)-sesamin” of Brideliaretusa identified as potential inhibitors of SARS-CoV-2 3CL pro using extensive molecular docking, molecular dynamics simulation studies, binding free energy calculations, and bioactivity prediction. Struct. Chem. 2022, 33, 1445–1465. [Google Scholar] [CrossRef] [PubMed]
- Zothantluanga, J.H.; Abdalla, M.; Rudrapal, M.; Tian, Q.; Chetia, D.; Li, J. Computational investigations for identification of bioactive molecules from Baccaurearamiflora and Bergeniaciliata as inhibitors of SARS-CoV-2 Mpro. Polycycl. Aromat. Compd. 2023, 43, 2459–2487. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASyserver. In The Proteomics Protocols Handbook; Springer Protocols Handbooks; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36 (Suppl. S2), W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Geourjon, C.; Deleage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32 (Suppl. S2), W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Tambunan, U.S.; Zahroh, H.; Parikesit, A.A.; Idrus, S.; Kerami, D. Screening analogs of β-OG pocket binder as fusion inhibitor of dengue virus 2. Drug Target Insights 2015, 9, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, T.; Yennamalli, R.; Campbell, P.; Stoermer, M.J.; Fairlie, D.P.; Kobe, B.; Young, P.R. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antivir. Res. 2009, 84, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Douguet, D.; Munier-Lehmann, H.; Labesse, G.; Pochet, S. LEA3D: A computer-aided ligand design for structure-based drug design. J. Med. Chem. 2005, 48, 2457–2468. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology: Methods and Protocols; Humana Press: New York, NY, USA, 2015; pp. 243–250. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Andrienko, G.A. Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations, version 1.8, Build 682. Available online: https://www.chemcraftprog.com (accessed on 20 December 2023).
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Bouback, T.A.; Pokhrel, S.; Albeshri, A.; Aljohani, A.M.; Samad, A.; Alam, R.; Hossen, M.S.; Al-Ghamdi, K.; Talukder, M.E.K.; Ahammad, F.; et al. Pharmacophore-based virtual screening quantum mechanics calculations molecular dynamics simulation approaches identified potential natural antiviral drug candidates against MERS-CoV S1-NTD. Molecules 2021, 26, 4961. [Google Scholar] [CrossRef]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; pp. 84–es. [Google Scholar] [CrossRef]
- Chow, E.; Rendleman, C.A.; Bowers, K.J.; Dror, R.O.; Hughes, D.H.; Gullingsrud, J.; Sacerdoti, F.D.; Shaw, D.E. Desmond Performance on a Cluster of Multicore Processors; DE Shaw Research Technical Report DESRES/TR--2008-01; D. E. Shaw Research: New York, NY, USA, 2008. [Google Scholar]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Toukmaji, A.Y.; Board, J.A., Jr. Ewald summation techniques in perspective: A survey. Comput. Phys. Commun. 1996, 95, 73–92. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Organism | PDB ID | Max Score | Query Cover | Identity % | e-Value |
|---|---|---|---|---|---|---|
| 01 | Tick-borne encephalitis virus (strain HYPR) | 5O6A | 851 | 100% | 80.24 | 0 |
| 02 | Louping ill virus | 6J5C | 689 | 80% | 80.8 | 0 |
| 03 | Dengue virus 2 | 4CBF | 394 | 99% | 40.99 | 9.00 × 10−133 |
| 04 | Zika virus | 5GZR | 377 | 99% | 39.06 | 1.00 × 10−125 |
| 05 | Japanese encephalitis virus | 5WSN | 375 | 99% | 39.45 | 5.00 × 10−125 |
| Features | Remark |
|---|---|
| Protein | Envelope protein |
| Accession number | D7RF80.1|POLG_KFDV:282–777 |
| Length of sequence | 496 aa |
| pI (theoretical) | 7.26 |
| Molecular mass | 53,633.08 Da |
| Index (aliphatic) | 83.10 |
| GRAVY | −0.16 |
| Index (instability)(II) | 29.41Stable |
| Type of Secondary Structure | No. of Amino Acids |
|---|---|
| Alpha helix (Hh) | 95 (19.15%) |
| Extended strand (Ee) | 167 (33.67%) |
| Pi helix (Ii) | 0 (0.00%) |
| Bend region (Ss) | 0 (0.00%) |
| Random coil (Cc) | 203 (40.93%) |
| Beta turn (Tt) | 31 (6.25%) |
| Beta bridge (Bb) | 0 (0.00%) |
| 310 helix (Gg) | 0 (0.00%) |
| Location of Domain | Sequence Position | |
|---|---|---|
| Envelope protein | Extracellular region | 1–446 |
| Transmembrane | 447–469 | |
| Cytoplasmic region | 470–475 | |
| TM helix | 476–495 |
| Validation Method | Robetta Model | I-Tasser Model | Refined Model |
|---|---|---|---|
| ERRAT score | 89.85 | 91.51 | 94.19 |
| Procheck | |||
| Most favored region | 93.1% | 75.4% | 92.3 |
| Additionally allowed regions | 6.5% | 20.3% | 7.2 |
| Generously allowed region | 0.2% | 3.1% | 0.2 |
| Disallowed region | 0.2% | 1.2% | 0.2 |
| ProSA Web score | −7.91 | −7.47 | −7.56 |
| Verify 3D | 89.11% | 78.63% | 76.81% |
| QMean Disco Global | 0.72 ± 0.05 | 0.65 ± 0.05 | 0.73 ± 0.05 |
| QmeanZscore | 1.03 | −7.11 | 0.09 |
| Qmean all-atom | 0.56 | −0.43 | 0.60 |
| Qmean torsion | 1.30 | −6.10 | 0.16 |
| Qmean Solvation | −0.72 | −2.78 | 0.68 |
| Qmean Cβ | −0.09 | −1.29 | 0.80 |
| Predicted Binding Site | c-Value | Docking Energy (kcal/mol) | Active Site Grid Box (x, y, z) | Amino Acid Residue |
|---|---|---|---|---|
| 1 | 0.09 | −4.6 | 15.707, 0.627, 11.566 | Ile48, His49, Gln50, Pro194, Val215, Val273, Ala274, Gly286 |
| 2 | 0.07 | −1.3 | 11.537, −25.032, −2.164 | Asp149, Tyr150, Asn154, Ser158, Asn159 |
| 3 | 0.07 | −5.6 | 11.120, −9.669, −8.399 | Arg9, Thr32 |
| 4 | 0.05 | −4.1 | 10.638, −25.498, 2.086 | Ala152, Ser156 |
| 5 | 0.04 | −3.9 | 23.933, 10.562, −16.247 | Leu430, Val433, Leu437 |
| Pharmacophore Features | Coordinates of Center | Radius (Å) | ||
|---|---|---|---|---|
| x | y | z | ||
| HBD | 14.99 | 3.66 | 2.38 | 0.5 |
| HBD | 17.58 | 3.28 | 3.18 | 0.5 |
| HBD | 18.62 | 6.15 | 4.05 | 0.5 |
| HBD | 13.11 | 4.53 | 4.83 | 0.5 |
| HBA | 14.99 | 3.66 | 2.38 | 0.5 |
| HBA | 17.58 | 3.28 | 3.18 | 0.5 |
| HBA | 18.62 | 6.15 | 4.05 | 0.5 |
| HBA | 13.11 | 4.53 | 4.83 | 0.5 |
| Ligand ID | Formula | Structure | Binding Energy (kcal/mol) |
|---|---|---|---|
| 161783612 | C17 H30 O9 | 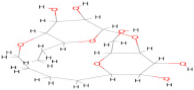 | −10.20 |
| CNP0187513.6 | C26 H30 N2 O8 |  | −8.80 |
| CNP0247967 | C27 H40 O8 |  | −8.80 |
| SA8 | C25 H28 N6 O2 |  | −8.80 |
| 101929509 | C14 H26 O9 |  | −8.60 |
| ZINC000028541549 | C26 H30 N2 O8 | 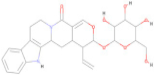 | −8.60 |
| ZINC000100052673 | C26 H22 O10 | 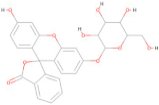 | −8.60 |
| CNP0097629.2 | C29 H34 O9 |  | −8.50 |
| CNP0178494.1 | C24 H20 O12 |  | −8.50 |
| CNP0247704.2 | C25 H30 O9 |  | −8.50 |
| SA28 | C33 H34 N4 O3 |  | −8.50 |
| SA29 | C33 H34 N4 O3 | 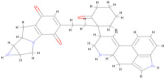 | −8.50 |
| Name of Drug | Oral Toxicity | Organ Toxicity-Hepatotoxicity | Carcinogenicity | Mutagenicity | Cytotoxicity | |
|---|---|---|---|---|---|---|
| LD50 | Class | |||||
| 161783612 (L1) | 2000 | 4 | IA | IA | IA | IA |
| CNP0187513.6 (L2) | 300 | 3 | IA | IA | IA | IA |
| CNP0247967(L3) | 10000 | 6 | IA | IA | IA | A |
| SA8(L4) | 2000 | 4 | IA | IA | IA | IA |
| 101929509(L5) | 51 | 3 | IA | IA | IA | IA |
| ZINC000028541549 (L6) | 300 | 3 | IA | IA | IA | IA |
| ZINC0001000052673(L7) | 2190 | 5 | IA | IA | IA | IA |
| CNP0097629.2(L8) | 3000 | 5 | IA | IA | IA | IA |
| CNP0178494.1(L9) | 5000 | 5 | IA | IA | A | IA |
| CNP0247704.2(L10) | 4000 | 5 | IA | IA | IA | IA |
| SA28(L11) | 400 | 4 | IA | IA | A | IA |
| SA29(L12) | 400 | 4 | IA | IA | A | IA |
| Molecule | 161783612 (L1) | SA8 (L4) | CNP0247967 (L3) | ZINC0001000052673 (L7) | CNP0097629.2 (L8) | CNP0247704.2 (L10) |
|---|---|---|---|---|---|---|
| Formula | C17H30O9 | C25H32N6O2 | C27H40O8 | C26H22O10 | C29H34O9 | C25H30O9 |
| Physicochemical properties | ||||||
| Weight (molecular) (Da) | 378.41 | 448.56 | 492.6 | 494.45 | 526.57 | 474.5 |
| #Heavy atoms | 26 | 33 | 35 | 36 | 38 | 34 |
| #Rotatable bonds | 2 | 7 | 0 | 3 | 3 | 3 |
| #Aromatic heavy atoms | 0 | 12 | 0 | 18 | 6 | 12 |
| #H-bond donors | 5 | 5 | 5 | 5 | 5 | 5 |
| #H-bond acceptors | 9 | 4 | 8 | 10 | 9 | 9 |
| MR | 87.64 | 150.18 | 126.48 | 120.87 | 135.38 | 120.32 |
| TPSA | 138.07 | 97.53 | 136.68 | 155.14 | 153.75 | 145.91 |
| Lipophilicity Consensus log P | −0.96 | 1.54 | 1.11 | 1.29 | 1.83 | 0.87 |
| Water solubility ESOL log S | −0.96 | −3.39 | −3.24 | −4.1 | −4.46 | −3.48 |
| Water solubility ESOL class | Very soluble | Soluble | Soluble | Moderately soluble | Moderately soluble | Soluble |
| Pharmacokinetics | ||||||
| GI absorption | Low | High | High | Low | Low | Low |
| log Kp (cm/s) skin permeation | −9.84 | −7.96 | −8.91 | −8.17 | −7.89 | −8.48 |
| BBB permeant | No | No | No | No | No | No |
| Drug likeness | ||||||
| Lipinski #violations | 0 | 0 | 0 | 0 | 1 | 0 |
| Veber #violations | 0 | 0 | 0 | 1 | 1 | 1 |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| PAINS #alerts | 0 | 0 | 0 | 0 | 1 | 0 |
| Lead likeness #violations | 1 | 1 | 1 | 1 | 1 | 1 |
| Synthetic accessibility | 7.32 | 4.06 | 6.93 | 5.78 | 6.72 | 6.76 |
| Metabolism | ||||||
| Pgp substrate | Yes | Yes | Yes | No | Yes | Yes |
| CYP2D6 inhibitor | No | Yes | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No | No |
| CYP2C19 inhibitor | No | Yes | No | No | Yes | No |
| CYP1A2 inhibitor | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | Yes | No | No |
| Compound ID | Name | Binding Affinity (kcal/mol) | Inhibition Constant Ki |
|---|---|---|---|
| 161783612 (L1) | (1S,3R,5R,6S,12R,14R,15R,16R,17R,18R)-5-ethyl-14-(hydroxymethyl)-2,4,7,13-etraoxatricyclo [10.2.2.23,6]octadecane-15,16,17,18-tetrol | −4.58 | 436.03 µM |
| CNP0247967 (L3) | Tupichigenin C | −7.26 | 4.79 µM |
| SA28 (L4) | De novo design | −7.82 | 1.86 µM |
| ZINC0001000052673 (L7) | Fluorescein beta-D-galactopyranoside | −7.65 | 2.48 µM |
| CNP0097629.2 (L8) | 5′-hydroxy-7′-{[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-3′,4′,5′a,6′,11′,11′a-hexahydro-1′H-spiro[cyclohexane-1,2′-tetracene]-6′,11′-dione | −7.04 | 6.91 µM |
| CNP0247704.2 (L10) | 10-hydroxy-4-{[3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-2-oxatricyclo [13.2.2.13,7]icosa-1(17),3(20),4,6,15,18-hexane-12-one | −5.72 | 64.51 µM |
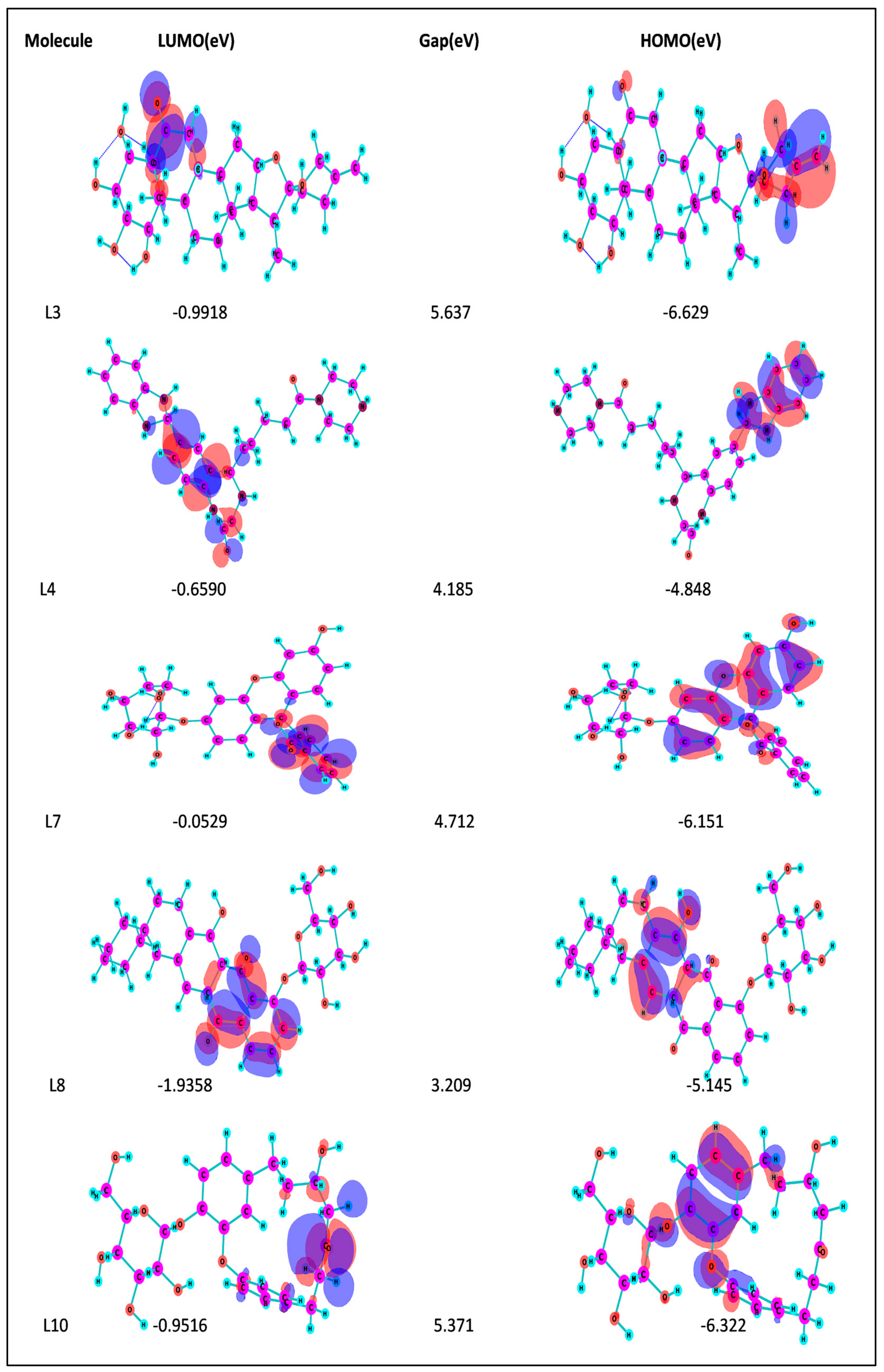
| Ligand | Optimized Energy (eV) | HOMO (eV) | LUMO (eV) | Energy Gap (eV) |
|---|---|---|---|---|
| L3 | −44,966 | −6.63 | −0.99 | 5.63 |
| L4 | −39,428 | −4.85 | −0.66 | 4.18 |
| L7 | −47,727 | −6.15 | −0.05 | 4.71 |
| L8 | −48,985 | −5.15 | −1.93 | 3.20 |
| L10 | −44,777 | −6.32 | −0.95 | 5.37 |
| Compound ID | Binding Affinity (kcal/mol) | |
|---|---|---|
| Before Optimization | After Optimization | |
| CNP0247967 (L3) | −7.26 | −7.58 |
| SA28 (L4) | −7.82 | −8.91 |
| ZINC0001000052673 (L7) | −7.65 | −6.43 |
| CNP0097629.2 (L8) | −7.04 | −6.12 |
| CNP0247704.2 (L10) | −5.72 | −6.02 |
| Complex | Amino Acid Residue | Bond Distance (Å) | Bond Category | Type of Bond |
|---|---|---|---|---|
| L3 | His216 | 1.95 | HB | CHB |
| Gly270 | 1.65 | HB | CHB | |
| Gly270 | 2.11 | HB | CHB | |
| Val192 | 2.67 | HB | Carbon H–B | |
| Val215 | 2.99 | HB | Carbon H–B | |
| His216 | 2.42 | HB | Carbon H–B | |
| Gln214 | 2.86 | HB | Carbon H–B | |
| Val192 | 4.89 | HP | Alkyl | |
| Val192 | 3.73 | HP | Alkyl | |
| His287 | 4.19 | HP | Pi–Alkyl | |
| His419 | 4.34 | HP | Pi–Alkyl | |
| L4 | Gly191 | 1.77 | HB | CHB |
| Gln214 | 1.75 | HB | CHB | |
| Val415 | 2.55 | HB | CHB | |
| His216 | 2.75 | HB | Carbon H–B | |
| His216 | 2.75 | HB | Carbon H–B | |
| Val192 | 4.93 | HP | Pi–Alkyl | |
| Val415 | 5.24 | HP | Pi–Alkyl | |
| L7 | Gln214 | 2.10 | HB | CHB |
| Glu26 | 2.10 | HB | CHB | |
| Leu27 | 2.38 | HB | CHB | |
| Gly270 | 1.67 | HB | CHB | |
| Ser285 | 2.83 | HB | Pi–Donor H–B | |
| Val192 | 5.23 | HP | Pi–Alkyl | |
| Pro272 | 3.90 | HP | Pi–Alkyl | |
| L8 | Val273 | 2.39 | HB | CHB |
| Ser285 | 2.15 | HB | CHB | |
| Gln284 | 2.27 | HB | CHB | |
| Val271 | 2.28 | HB | CHB | |
| Ser285 | 2.87 | HB | Carbon H–B | |
| Val415 | 2.77 | HB | Carbon H–B | |
| His287 | 3.71 | HP | Pi–Sigma | |
| Val192 | 5.29 | HP | Alkyl | |
| Val192 | 4.37 | HP | Alkyl | |
| His287 | 4.93 | HP | Pi–Alkyl | |
| Val415 | 5.18 | HP | Pi–Alkyl | |
| L10 | Gln196 | 2.79 | HB | CHB |
| Gly191 | 2.13 | HB | CHB | |
| Asp193 | 2.68 | HB | Pi–Donor H–B | |
| His216 | 4.57 | HP | Pi–Pi T-shaped | |
| Val192 | 4.60 | HP | Pi–Alkyl |
| Energies (kcal/mol) | L3 Complex | L4 Complex |
|---|---|---|
| ΔGbind | −85.26 ± 4.63 | −66.60 ± 2.92 |
| ΔGbindLipo | −32.51 ± 1.64 | −19.96 ± 0.96 |
| ΔGbindvdW | −70.63 ± 3.57 | −53.79 ± 1.23 |
| ΔGbindCoulomb | −43.66 ± 4.48 | −23.60 ± 1.52 |
| ΔGbindHbond | −1.87 ± 0.25 | −0.58 ± 0.01 |
| ΔGbindSolvGB | 60.54 ± 3.45 | 33.55 ± 2.75 |
| ΔGbindCovalent | 4.22 ± 1.66 | 0.31 ± 0.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achappa, S.; Aldabaan, N.A.; Desai, S.V.; Muddapur, U.M.; Shaikh, I.A.; Mahnashi, M.H.; Alshehri, A.A.; Mannasaheb, B.A.; Khan, A.A. Computational Exploration of Potential Pharmacological Inhibitors Targeting the Envelope Protein of the Kyasanur Forest Disease Virus. Pharmaceuticals 2024, 17, 884. https://doi.org/10.3390/ph17070884
Achappa S, Aldabaan NA, Desai SV, Muddapur UM, Shaikh IA, Mahnashi MH, Alshehri AA, Mannasaheb BA, Khan AA. Computational Exploration of Potential Pharmacological Inhibitors Targeting the Envelope Protein of the Kyasanur Forest Disease Virus. Pharmaceuticals. 2024; 17(7):884. https://doi.org/10.3390/ph17070884
Chicago/Turabian StyleAchappa, Sharanappa, Nayef Abdulaziz Aldabaan, Shivalingsarj V. Desai, Uday M. Muddapur, Ibrahim Ahmed Shaikh, Mater H. Mahnashi, Abdullateef A. Alshehri, Basheerahmed Abdulaziz Mannasaheb, and Aejaz Abdullatif Khan. 2024. "Computational Exploration of Potential Pharmacological Inhibitors Targeting the Envelope Protein of the Kyasanur Forest Disease Virus" Pharmaceuticals 17, no. 7: 884. https://doi.org/10.3390/ph17070884
APA StyleAchappa, S., Aldabaan, N. A., Desai, S. V., Muddapur, U. M., Shaikh, I. A., Mahnashi, M. H., Alshehri, A. A., Mannasaheb, B. A., & Khan, A. A. (2024). Computational Exploration of Potential Pharmacological Inhibitors Targeting the Envelope Protein of the Kyasanur Forest Disease Virus. Pharmaceuticals, 17(7), 884. https://doi.org/10.3390/ph17070884