
The Role of α-Enolase on the Production of Interleukin (IL)-32 in Con A-Mediated Inflammation and Rheumatoid Arthritis (RA)

 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
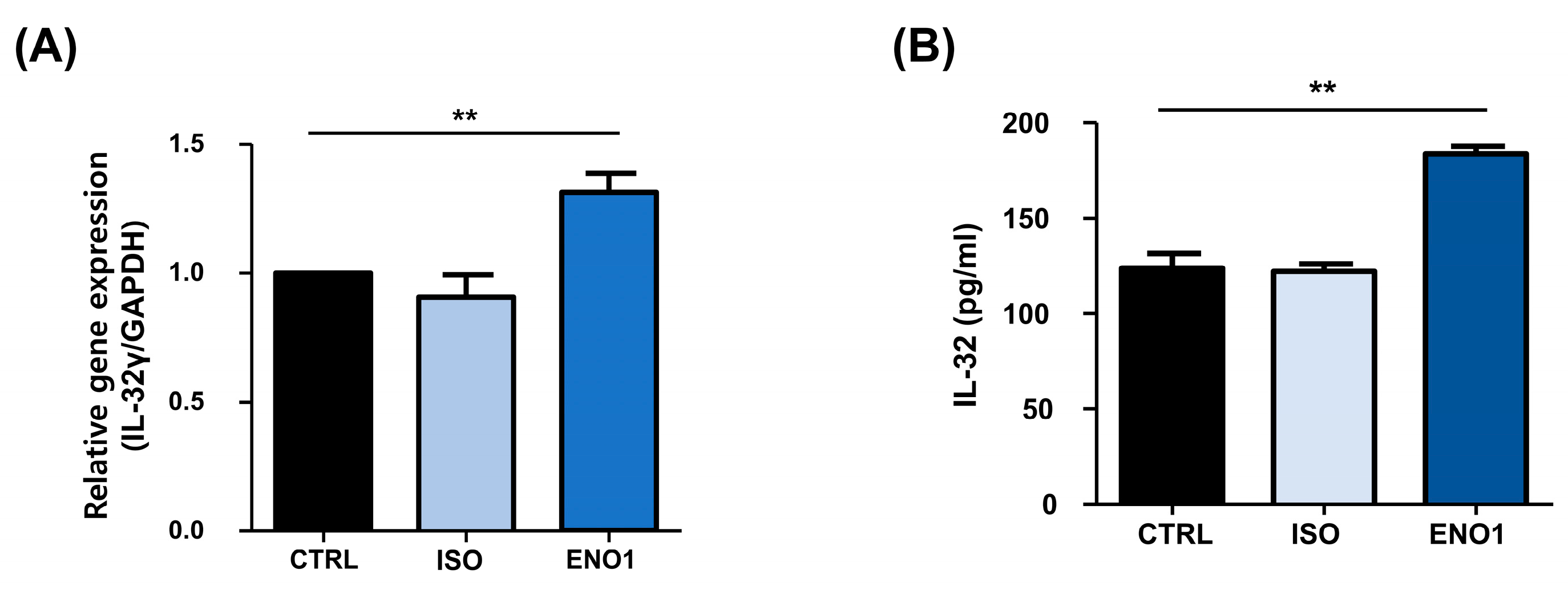
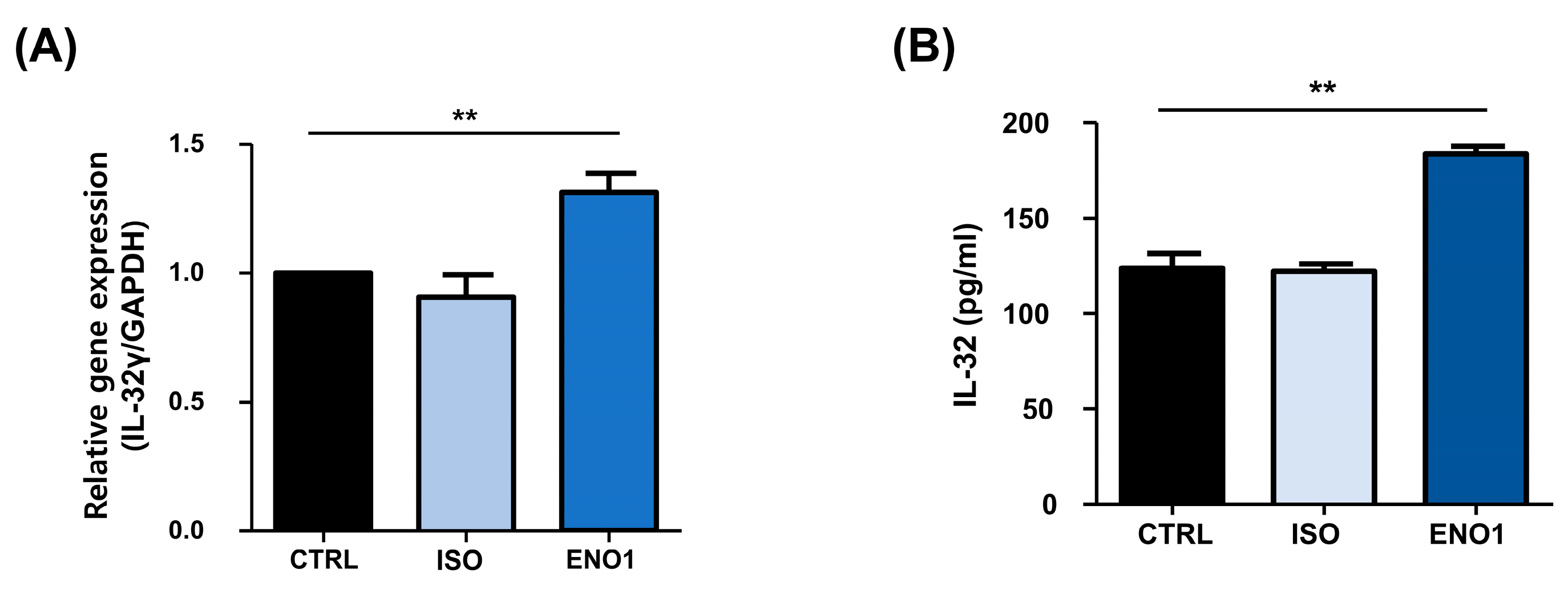
2.1. IL-32 Is Induced by ENO1 Stimulation in Con A-Activated PBMCs
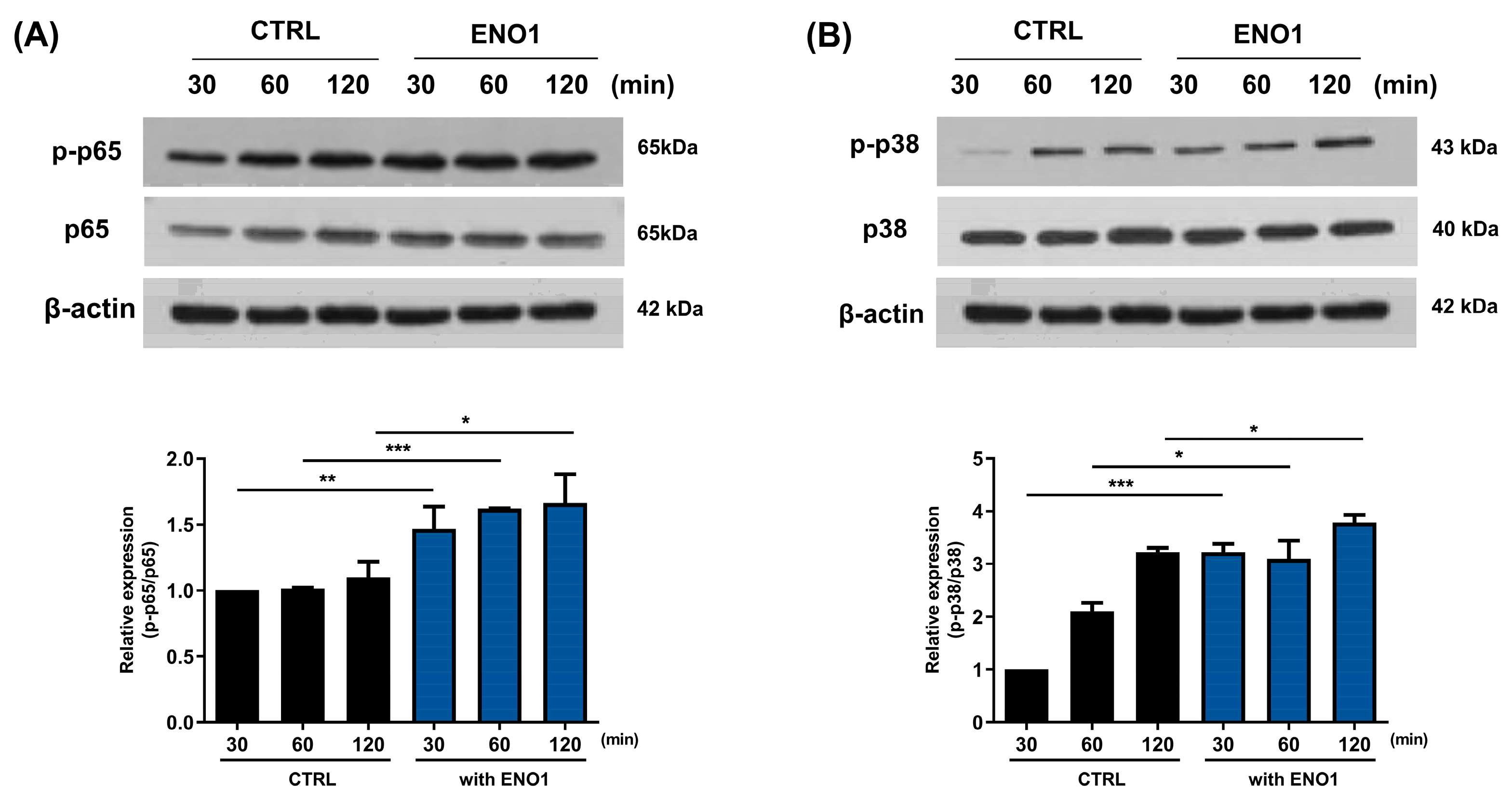
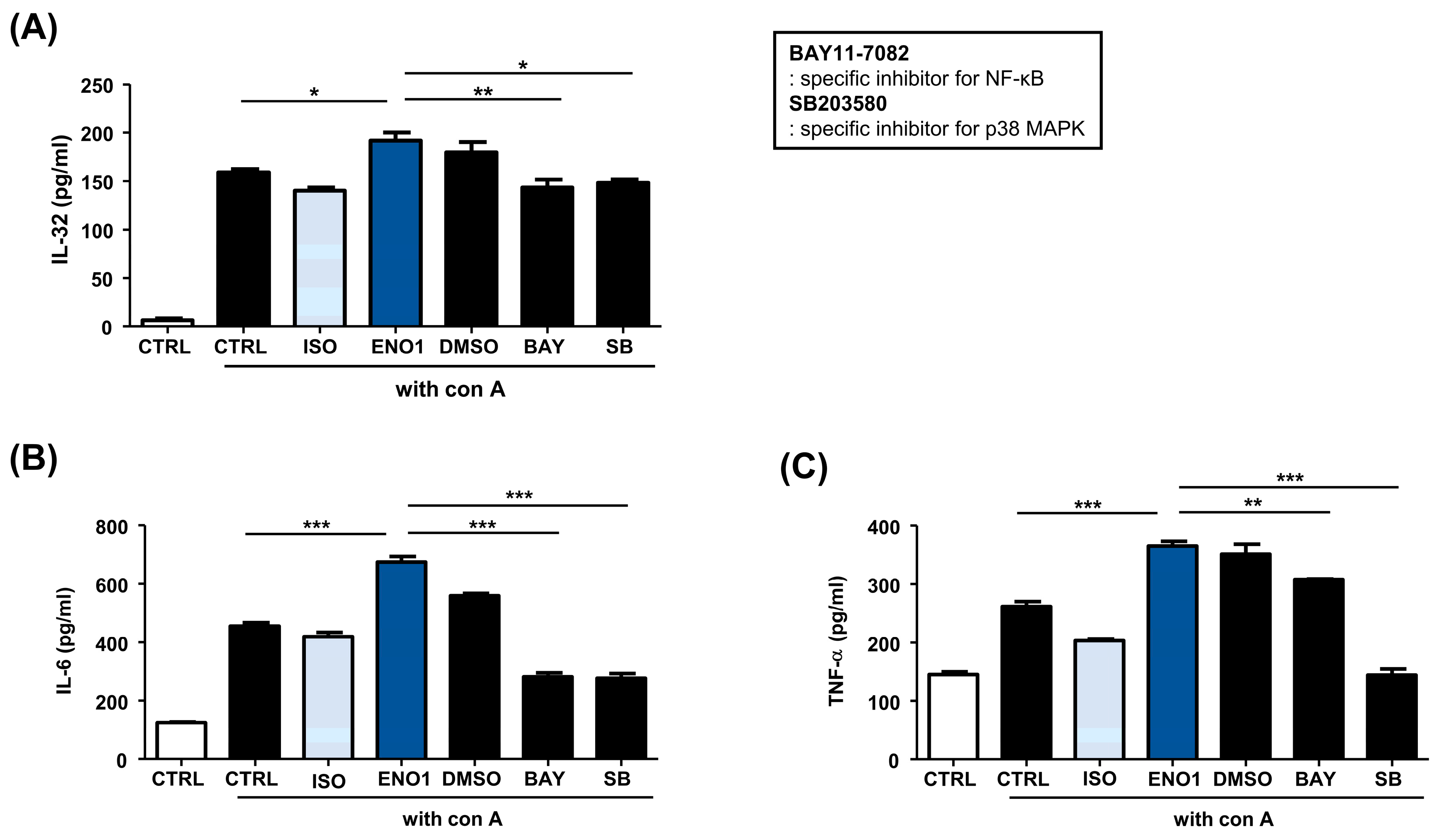
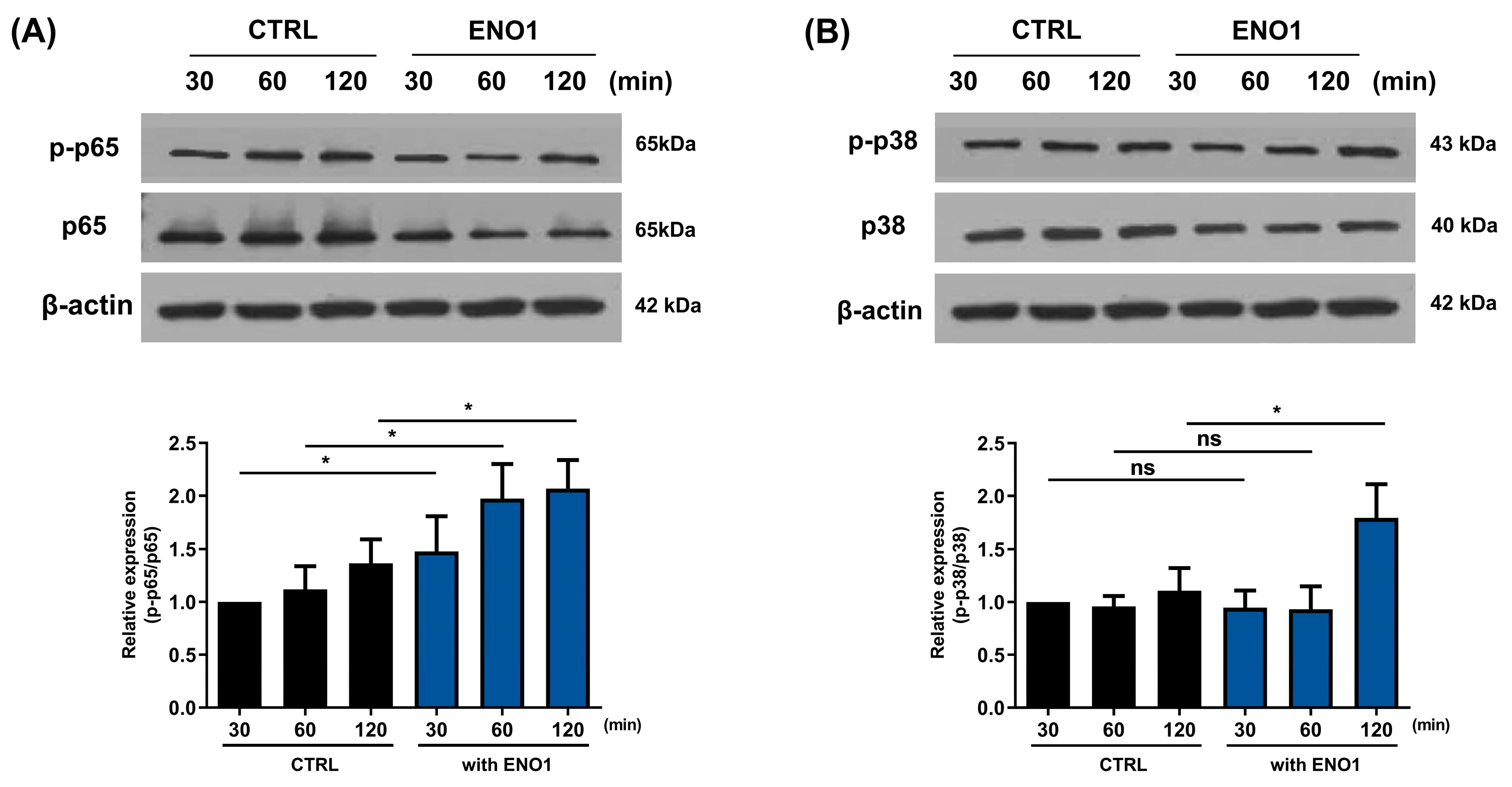
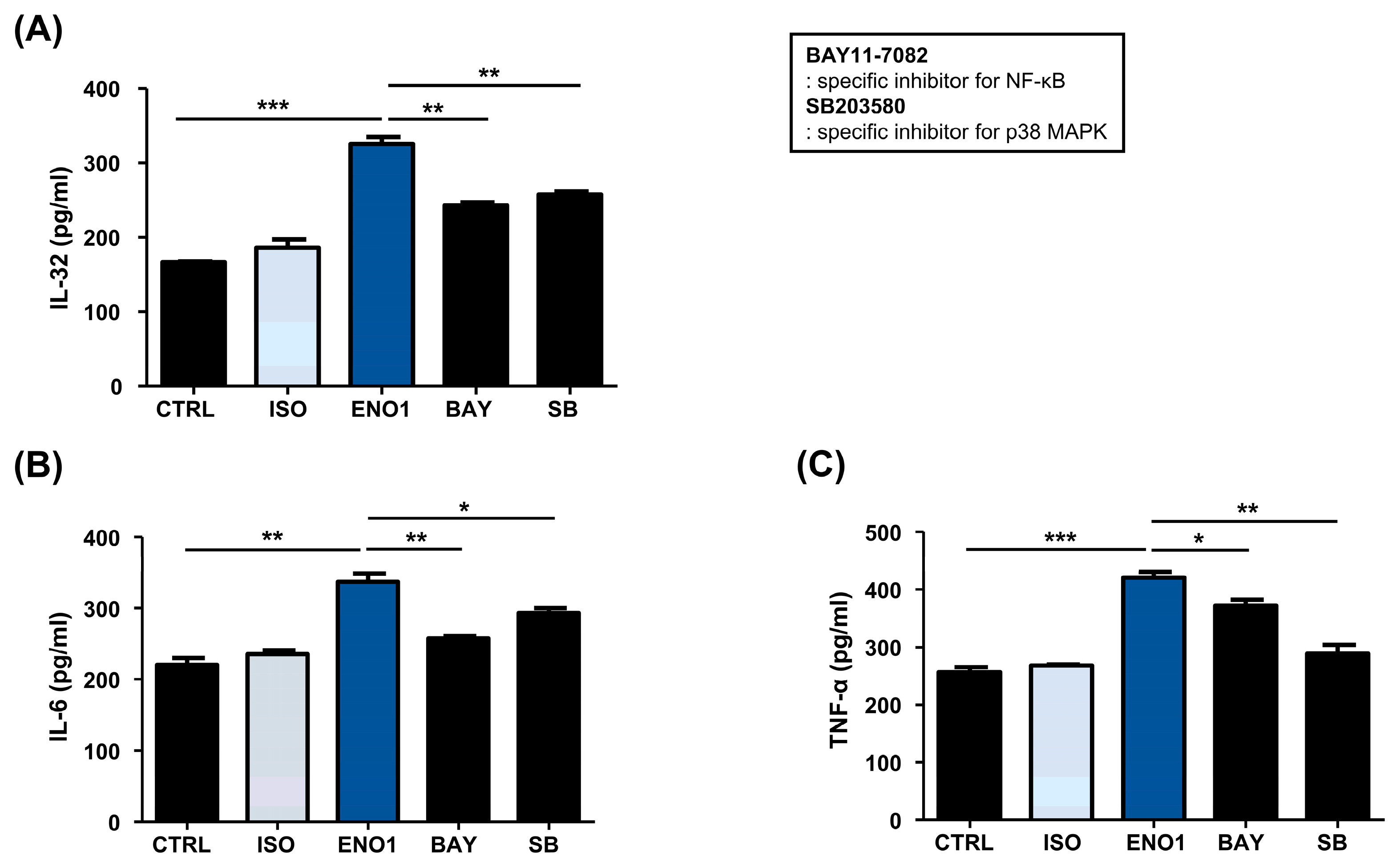
2.2. IL-32 Production Is Increased in Con A-Activated PBMCs by ENO1 Stimulation through the Activation of NF-κB and p38 MAPK
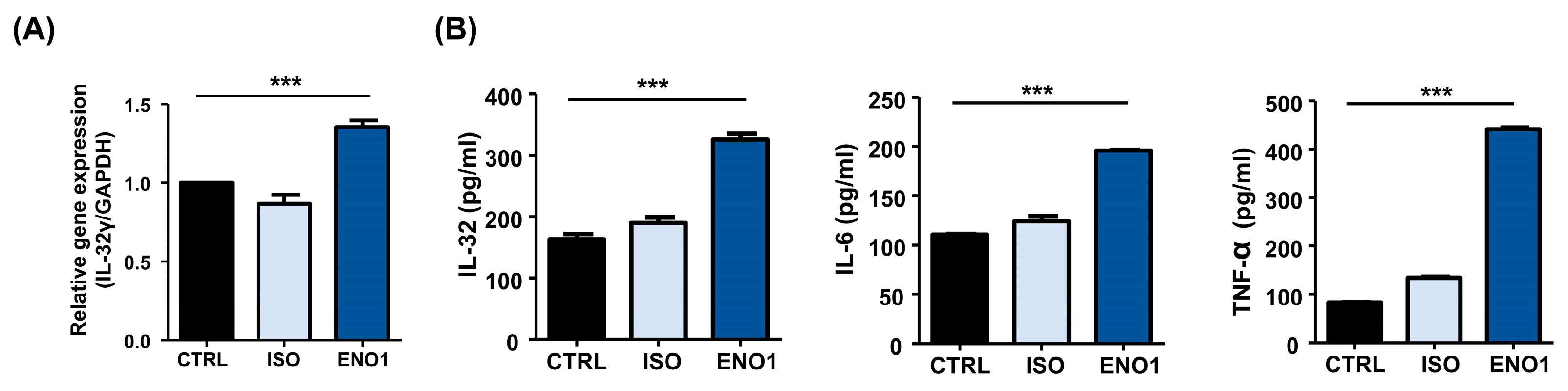
2.3. IL-32 Is Induced by ENO1 Stimulation in RA PBMCs
2.4. IL-32 Production Is Increased in RA PBMCs by ENO1 Stimulation through the Activation of NF-κB and p38 MAPK
3. Discussion
4. Materials and Methods
4.1. Isolation of PBMCs
4.2. Stimulation of PBMCs with Concanavalin A (Con A) and α-Enolase (ENO1)
4.3. Real-Time Polymerase Chain Reaction (Real-Time PCR)
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Inhibitor Study for Signal Pathway
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aass, K.R.; Kastnes, M.H.; Standal, T. Molecular interactions and functions of IL-32. J. Leucoc. Biol. 2021, 109, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Khawar, M.B.; Abbasi, M.H.; Sheikh, N. IL-32: A novel pluripotent inflammatory interleukin, towards gastric inflammation, gastric cancer, and chronic rhino sinusitis. Mediat. Inflamm. 2016, 2016, 8413768. [Google Scholar] [CrossRef] [PubMed]
- de Albuquerque, R.; Komsi, E.; Starskaia, I.; Ullah, U.; Lahesmaa, R. The role of Interleukin-32 in autoimmunity. Scand. J. Immunol. 2021, 93, e13012. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Azam, T.; Ferwerda, G.; Girardin, S.E.; Walsh, M.; Park, J.-S.; Abraham, E.; Kim, J.-M.; Yoon, D.-Y.; Dinarello, C.A. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1β and IL-6 production through a caspase 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 16309–16314. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Netea, M.G.; Kim, S.-H.; Yoon, D.-Y.; Oppers-Walgreen, B.; Radstake, T.R.; Barrera, P.; van de Loo, F.A.; Dinarello, C.A.; van den Berg, W.B. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2006, 103, 3298–3303. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Lewis, E.C.; Azam, T.; Joosten, L.A.; Jaekal, J.; Bae, S.-Y.; Dinarello, C.A.; Kim, S.-H. Interleukin-32 induces the differentiation of monocytes into macrophage-like cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3515–3520. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, V. Multifunctional α-enolase: Its role in diseases. Cell. Mol. Life Sci. CMLS 2001, 58, 902–920. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Dahlberg, C.M.; Plescia, J.; Felez, J.; Kato, K.; Plow, E.F. Role of cell-surface lysines in plasminogen binding to cells: Identification of. alpha.-enolase as a candidate plasminogen receptor. Biochemistry 1991, 30, 1682–1691. [Google Scholar] [CrossRef]
- Huang, C.K.; Sun, Y.; Lv, L.; Ping, Y. ENO1 and Cancer. Mol. Ther.-Oncolytics 2022, 24, 288–298. [Google Scholar] [CrossRef]
- Ye, Y.; Kuhn, C.; Kösters, M.; Arnold, G.J.; Ishikawa-Ankerhold, H.; Schulz, C.; Rogenhofer, N.; Thaler, C.J.; Mahner, S.; Fröhlich, T. Anti α-enolase antibody is a novel autoimmune biomarker for unexplained recurrent miscarriages. EBioMedicine 2019, 41, 610–622. [Google Scholar] [CrossRef]
- Bae, S.; Kim, H.; Lee, N.; Won, C.; Kim, H.-R.; Hwang, Y.-i.; Song, Y.W.; Kang, J.S.; Lee, W.J. α-Enolase expressed on the surfaces of monocytes and macrophages induces robust synovial inflammation in rheumatoid arthritis. J. Immunol. 2012, 189, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kasama, T.; Soejima, M.; Kubota, T. Anti-enolase1antibodies from a patient with systemic lupus erythematosus accompanied by pulmonary arterial hypertension promote migration of pulmonary artery smooth muscle cells. Immunol. Lett. 2020, 218, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Mosca, M.; Chimenti, D.; Pratesi, F.; Baldini, C.; Anzilotti, C.; Bombardieri, S.; Migliorini, P. Prevalence and clinico-serological correlations of anti-alpha-enolase, anti-C1q, and anti-dsDNA antibodies in patients with systemic lupus erythematosus. J. Rheumatol. 2006, 33, 695–697. [Google Scholar] [PubMed]
- Vermeulen, N.; Arijs, I.; Joossens, S.; Vermeire, S.; Clerens, S.; Van den Bergh, K.; Michiels, G.; Arckens, L.; Schuit, F.; Van Lommel, L. Anti–α-enolase Antibodies in Patients with Inflammatory Bowel Disease. Clin. Chem. 2008, 54, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Hakoda, M.; Mogi, M.; Yano, K.; Tsuda, E.; Takahashi, K.; Furuya, T.; Ishiyama, S.; Kim, K.J. Activated human T cells directly induce osteoclastogenesis from human monocytes: Possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2001, 44, 1003–1012. [Google Scholar] [CrossRef]
- Furuzawa-Carballeda, J.; Macip-Rodriguez, P.; Cabral, A. Osteoarthritis and rheumatoid arthritis pannus have similar qualitative metabolic characteristics and pro-inflammatory cytokine response. Clin. Exp. Rheumatol. 2008, 26, 554. [Google Scholar] [PubMed]
- Sweeney, S.E.; Firestein, G.S. Rheumatoid arthritis: Regulation of synovial inflammation. Int. J. Biochem. Cell Biol. 2004, 36, 372–378. [Google Scholar] [CrossRef]
- Alsaleh, G.; Sparsa, L.; Chatelus, E.; Ehlinger, M.; Gottenberg, J.-E.; Wachsmann, D.; Sibilia, J. Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R135. [Google Scholar] [CrossRef]
- Sharma, A.R.; Jagga, S.; Chakraborty, C.; Lee, S.-S. Fibroblast-like-synoviocytes mediate secretion of pro-inflammatory cytokines via ERK and JNK MAPKs in Ti-particle-induced osteolysis. Materials 2020, 13, 3628. [Google Scholar] [CrossRef]
- Kwon, O.C.; Park, M.-C.; Kim, Y.-G. Interleukin-32 as a biomarker in rheumatic diseases: A narrative review. Front. Immunol. 2023, 14, 1140373. [Google Scholar] [CrossRef]
- Damen, M.S.; Popa, C.D.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A. Interleukin-32 in chronic inflammatory conditions is associated with a higher risk of cardiovascular diseases. Atherosclerosis 2017, 264, 83–91. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, J.S.; Kwon, J.S.; Jeong, M.H.; Cho, J.G.; Park, J.C.; Kang, J.C.; Ahn, Y. BAY 11-7082, a nuclear factor-κB inhibitor, reduces inflammation and apoptosis in a rat cardiac ischemia-reperfusion injury model. Int. Heart J. 2010, 51, 348–353. [Google Scholar] [CrossRef]
- Mori, N.; Yamada, Y.; Ikeda, S.; Yamasaki, Y.; Tsukasaki, K.; Tanaka, Y.; Tomonaga, M.; Yamamoto, N.; Fujii, M. Bay 11-7082 inhibits transcription factor NF-κB and induces apoptosis of HTLV-I–infected T-cell lines and primary adult T-cell leukemia cells. Blood J. Am. Soc. Hematol. 2002, 100, 1828–1834. [Google Scholar]
- Avci, N.G.; Ebrahimzadeh-Pustchi, S.; Akay, Y.M.; Esquenazi, Y.; Tandon, N.; Zhu, J.-J.; Akay, M. NF-κB inhibitor with Temozolomide results in significant apoptosis in glioblastoma via the NF-κB (p65) and actin cytoskeleton regulatory pathways. Sci. Rep. 2020, 10, 13352. [Google Scholar] [CrossRef]
- Hu, S.; Luo, Q.; Cun, B.; Hu, D.; Ge, S.; Fan, X.; Chen, F. The pharmacological NF-κB inhibitor BAY11-7082 induces cell apoptosis and inhibits the migration of human uveal melanoma cells. Int. J. Mol. Sci. 2012, 13, 15653–15667. [Google Scholar] [CrossRef]
- Lee, J.; Rhee, M.H.; Kim, E.; Cho, J.Y. BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediat. Inflamm. 2012, 2012, 416036. [Google Scholar] [CrossRef]
- Lali, F.V.; Hunt, A.E.; Turner, S.J.; Foxwell, B.M. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J. Biol. Chem. 2000, 275, 7395–7402. [Google Scholar]
- Ji, M.; Wang, Z.; Chen, J.; Gu, L.; Chen, M.; Ding, Y.; Liu, T. Up-regulated ENO1 promotes the bladder cancer cell growth and proliferation via regulating β-catenin. Biosci. Rep. 2019, 39, BSR20190503. [Google Scholar] [CrossRef]
- Almaguel, F.A.; Casiano, C.A. Alpha-enolase: Emerging tumor-associated antigen, cancer biomarker, and oncotherapeutic target. Front. Genet. 2021, 11, 614726. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, L.; Hao, Y.; Suo, C.; Shen, S.; Wei, H.; Ma, W.; Zhang, P.; Wang, T.; Gu, X. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat. Cancer 2022, 3, 75–89. [Google Scholar] [CrossRef]
- Yan, H.; He, D.; Huang, X.; Zhang, E.; Chen, Q.; Xu, R.; Liu, X.; Zi, F.; Cai, Z. Role of interleukin-32 in cancer biology. Oncol. Lett. 2018, 16, 41–47. [Google Scholar] [CrossRef]
- Sloot, Y.J.; Smit, J.W.; Joosten, L.A.; Netea-Maier, R.T. Insights into the role of IL-32 in cancer. Semi Immunol. 2018, 38, 24–32. [Google Scholar] [CrossRef]
- Seo, E.-H.; Kang, J.-W.; Kim, K.-H.; Cho, M.-C.; Lee, S.-J.; Kim, H.-J.; Kim, J.-H.; Kim, E.-J.; Park, D.-K.; Kim, S.-H. Detection of expressed IL-32 in human stomach cancer using ELISA and immunostaining. J. Microbiol. Biotechnol. 2008, 18, 1606–1612. [Google Scholar]
- Tsai, C.-Y.; Wang, C.-S.; Tsai, M.-M.; Chi, H.-C.; Cheng, W.-L.; Tseng, Y.-H.; Chen, C.-Y.; Lin, C.D.; Wu, J.-I.; Wang, L.-H. Interleukin-32 increases human gastric cancer cell invasion associated with tumor progression and metastasis. Clin. Cancer Res. 2014, 20, 2276–2288. [Google Scholar] [CrossRef]
- Takagi, K.; Shimomura, A.; Imura, J.; Mori, H.; Noguchi, A.; Tanaka, S.; Minamisaka, T.; Nishida, T.; Hatta, H.; Nakajima, T. Interleukin-32 regulates downstream molecules and promotes the invasion of pancreatic cancer cells. Oncol. Lett. 2022, 23, 14. [Google Scholar] [CrossRef]
- Takagi, K.; Imura, J.; Shimomura, A.; Noguchi, A.; Minamisaka, T.; Tanaka, S.; Nishida, T.; Hatta, H.; Nakajima, T. Establishment of highly invasive pancreatic cancer cell lines and the expression of IL-32. Oncol. Lett. 2020, 20, 2888–2896. [Google Scholar] [CrossRef]
- Takikita, M.; Altekruse, S.; Lynch, C.F.; Goodman, M.T.; Hernandez, B.Y.; Green, M.; Cozen, W.; Cockburn, M.; Sibug Saber, M.; Topor, M. Associations between selected biomarkers and prognosis in a population-based pancreatic cancer tissue microarray. Cancer Res. 2009, 69, 2950–2955. [Google Scholar] [CrossRef]
- Yun, J.; Park, M.H.; Son, D.J.; Nam, K.T.; Moon, D.B.; Ju, J.H.; Hwang, O.K.; Choi, J.S.; Kim, T.H.; Jung, Y.S. IL-32 gamma reduces lung tumor development through upregulation of TIMP-3 overexpression and hypomethylation. Cell Death Dis. 2018, 9, 306. [Google Scholar] [CrossRef]
- Sorrentino, C.; Di Carlo, E. Expression of IL-32 in human lung cancer is related to the histotype and metastatic phenotype. Am. J. Respir. Crit. Care Med. 2009, 180, 769–779. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, H.; Shin, S.; Agura, T.; Jeong, S.; Ahn, H.; Lee, J.; Kim, Y.; Kang, J.S. The Role of α-Enolase on the Production of Interleukin (IL)-32 in Con A-Mediated Inflammation and Rheumatoid Arthritis (RA). Pharmaceuticals 2024, 17, 531. https://doi.org/10.3390/ph17040531
Jo H, Shin S, Agura T, Jeong S, Ahn H, Lee J, Kim Y, Kang JS. The Role of α-Enolase on the Production of Interleukin (IL)-32 in Con A-Mediated Inflammation and Rheumatoid Arthritis (RA). Pharmaceuticals. 2024; 17(4):531. https://doi.org/10.3390/ph17040531
Chicago/Turabian StyleJo, Hyejung, Seulgi Shin, Tomoyo Agura, Seoyoun Jeong, Hyovin Ahn, Junmyung Lee, Yejin Kim, and Jae Seung Kang. 2024. "The Role of α-Enolase on the Production of Interleukin (IL)-32 in Con A-Mediated Inflammation and Rheumatoid Arthritis (RA)" Pharmaceuticals 17, no. 4: 531. https://doi.org/10.3390/ph17040531
APA StyleJo, H., Shin, S., Agura, T., Jeong, S., Ahn, H., Lee, J., Kim, Y., & Kang, J. S. (2024). The Role of α-Enolase on the Production of Interleukin (IL)-32 in Con A-Mediated Inflammation and Rheumatoid Arthritis (RA). Pharmaceuticals, 17(4), 531. https://doi.org/10.3390/ph17040531