
PolySialic Acid Nanoparticles Actuate Complement-Factor-H-Mediated Inhibition of the Alternative Complement Pathway: A Safer Potential Therapy for Age-Related Macular Degeneration

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
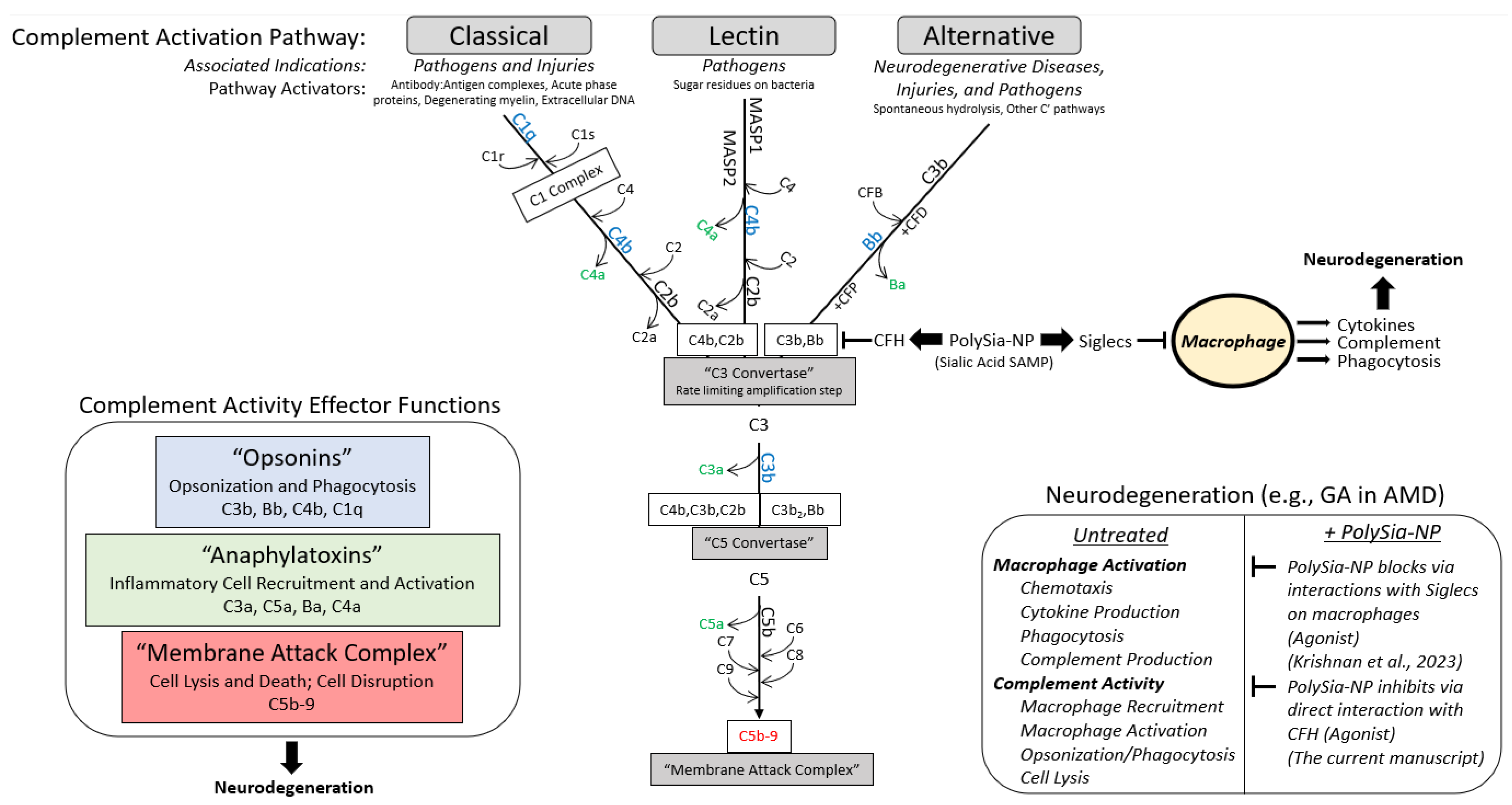
1. Introduction
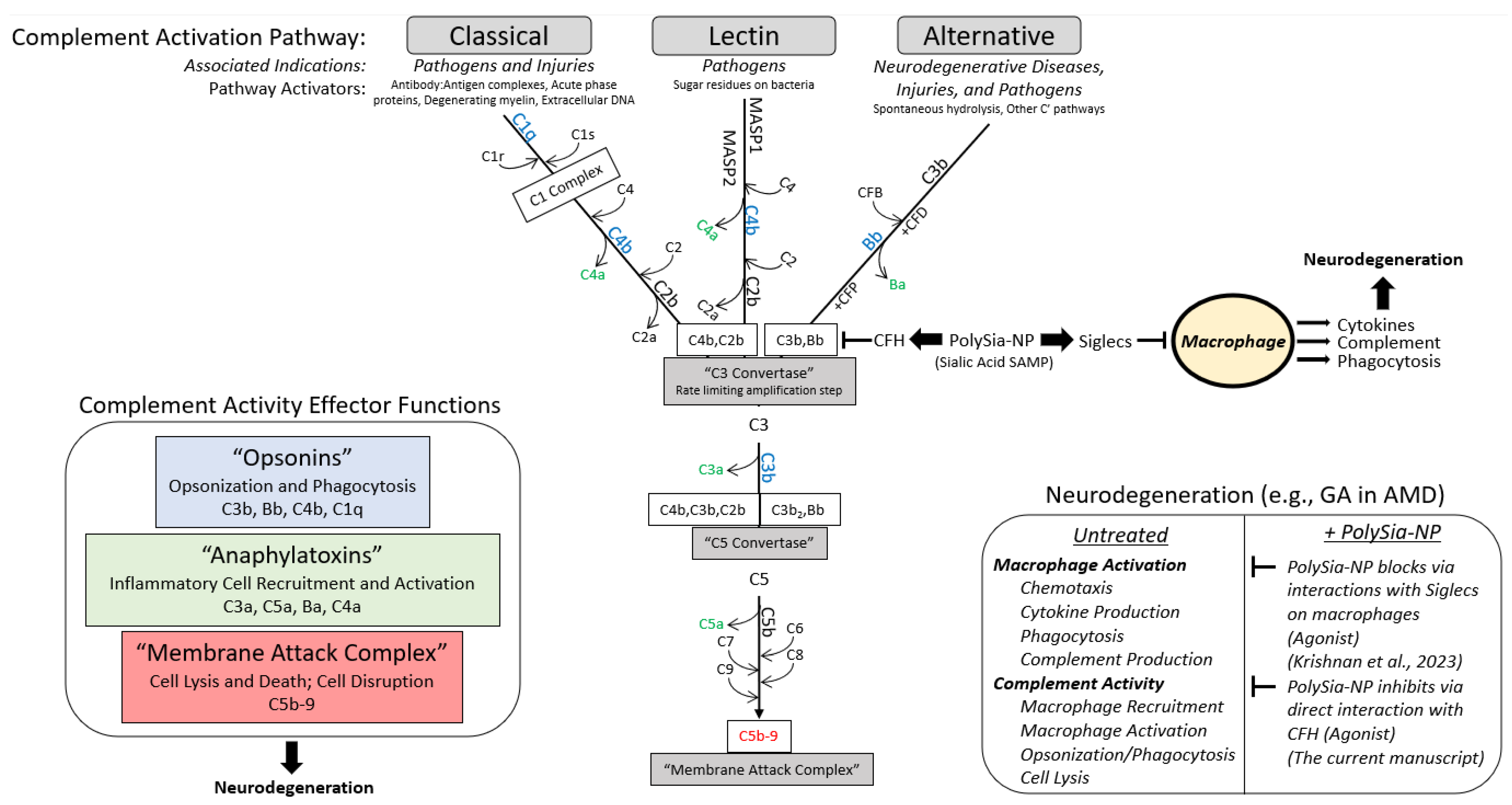
2. Results
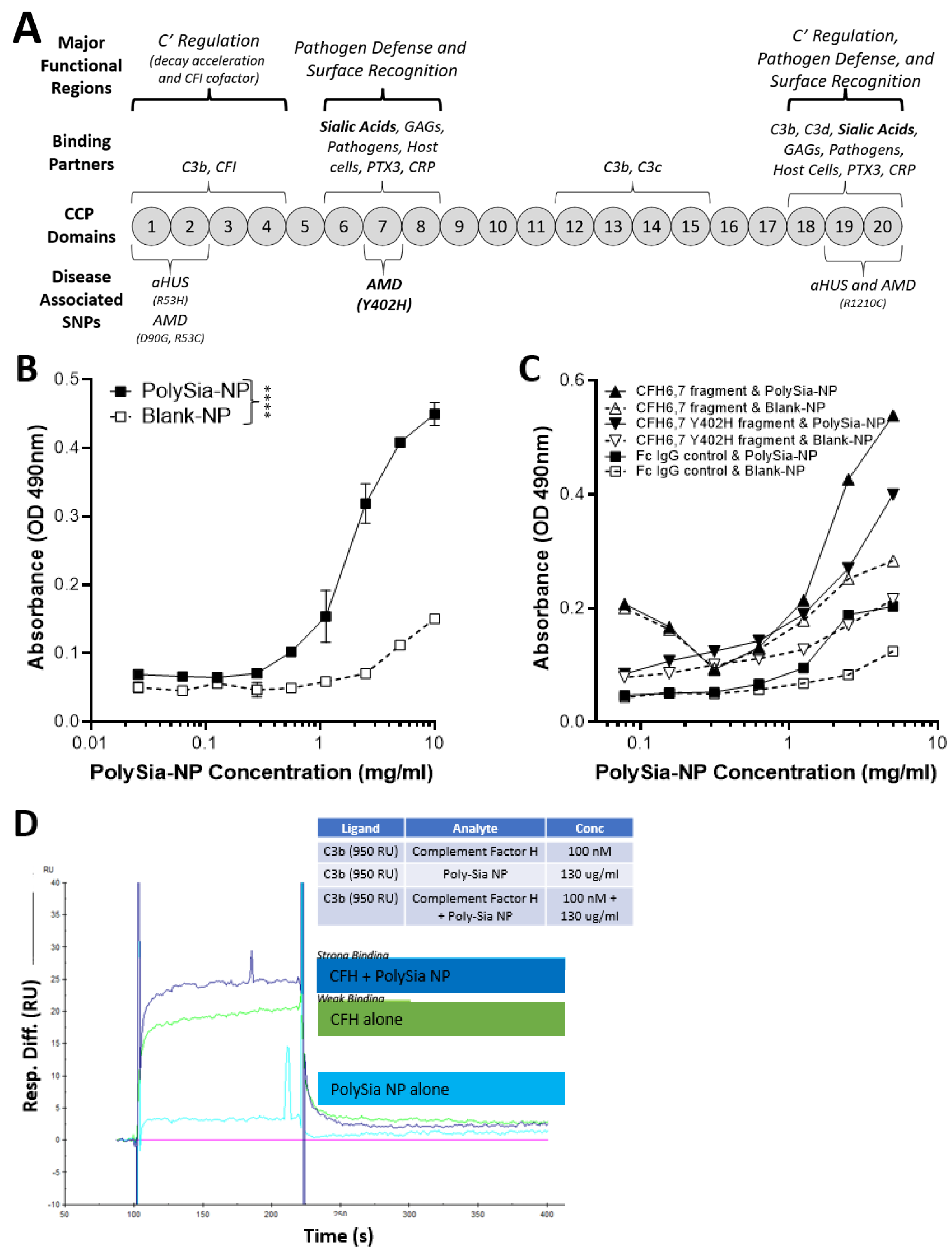
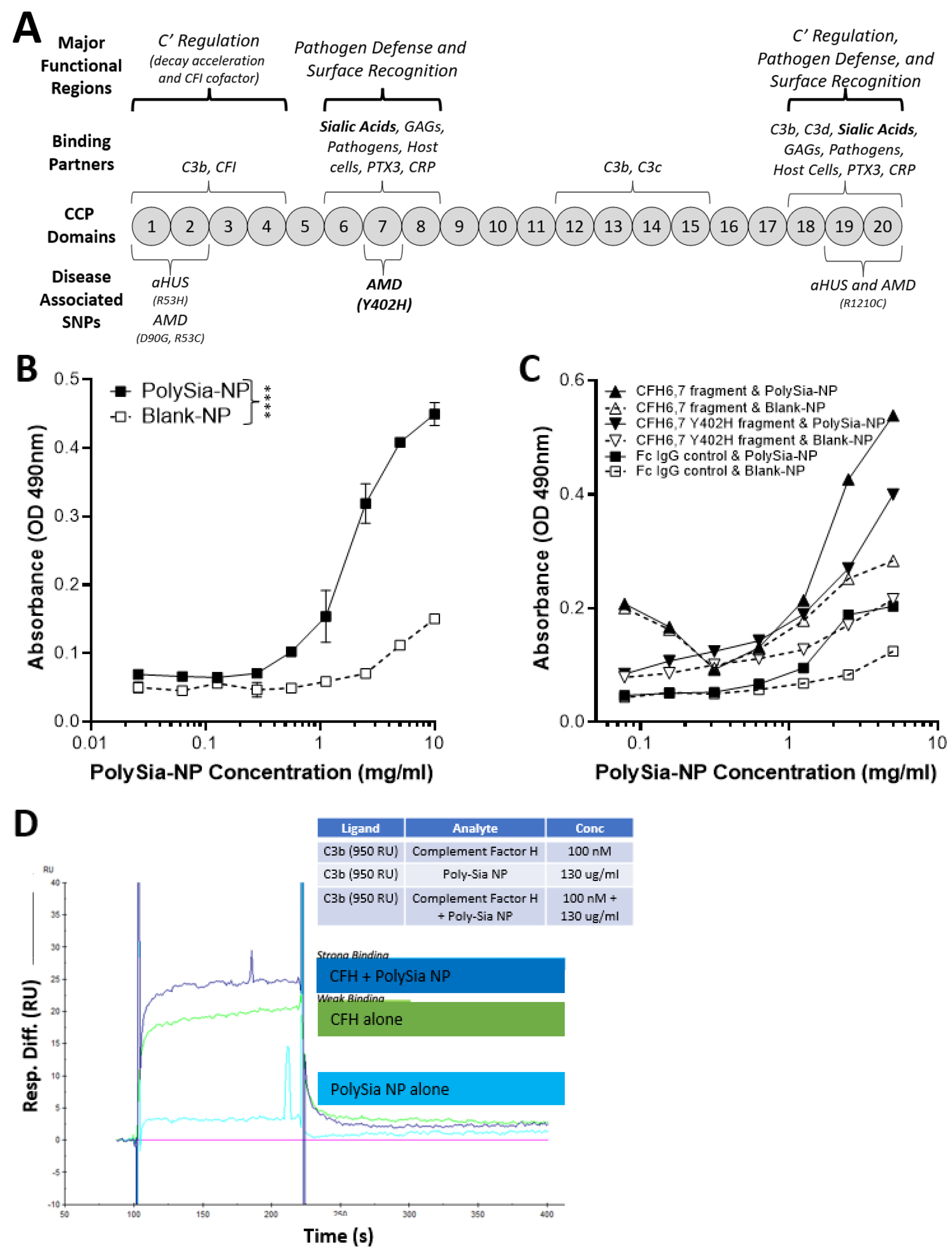
2.1. PolySia-NPs Interact with Alternative Complement Inhibitor CFH
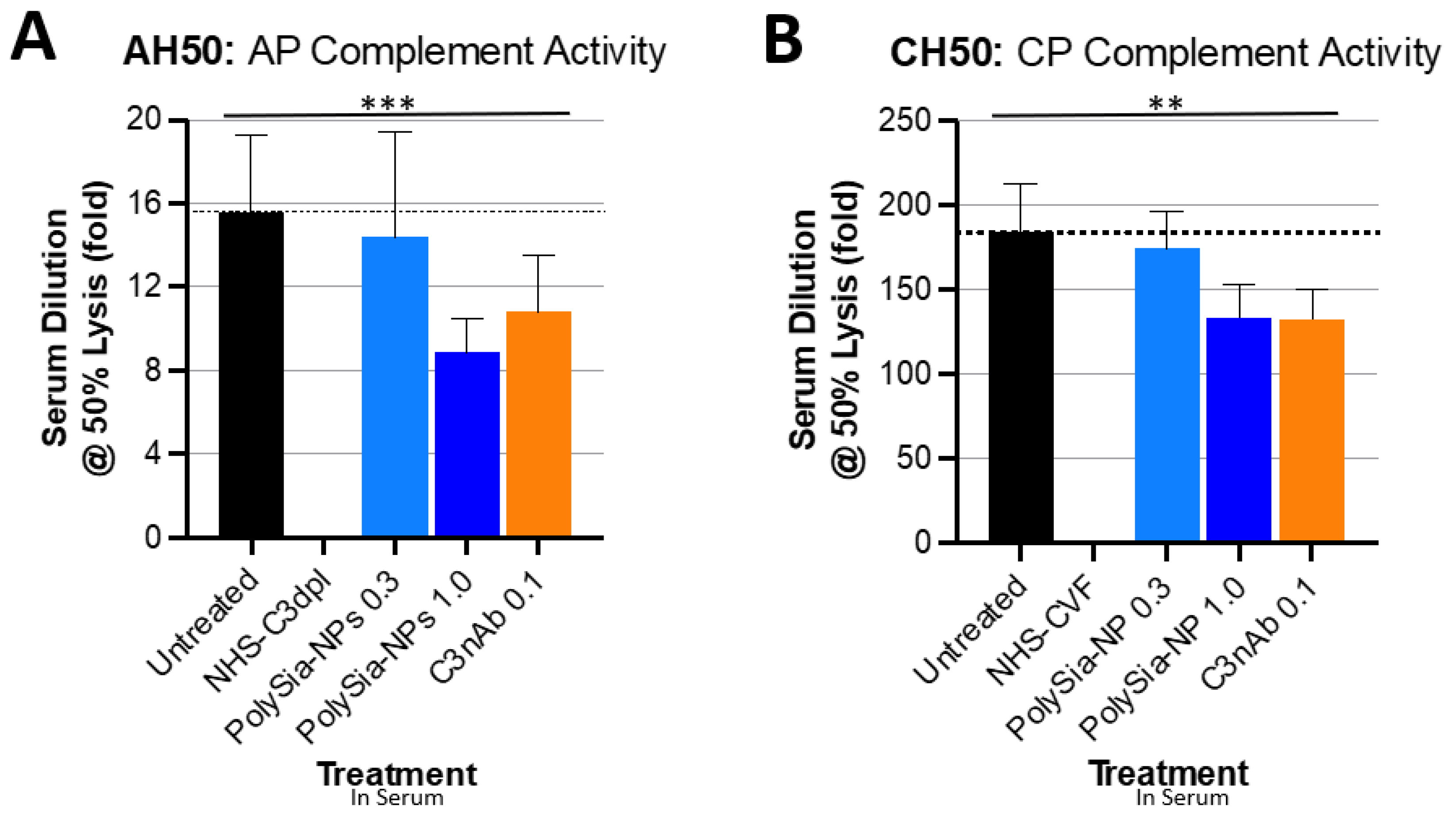
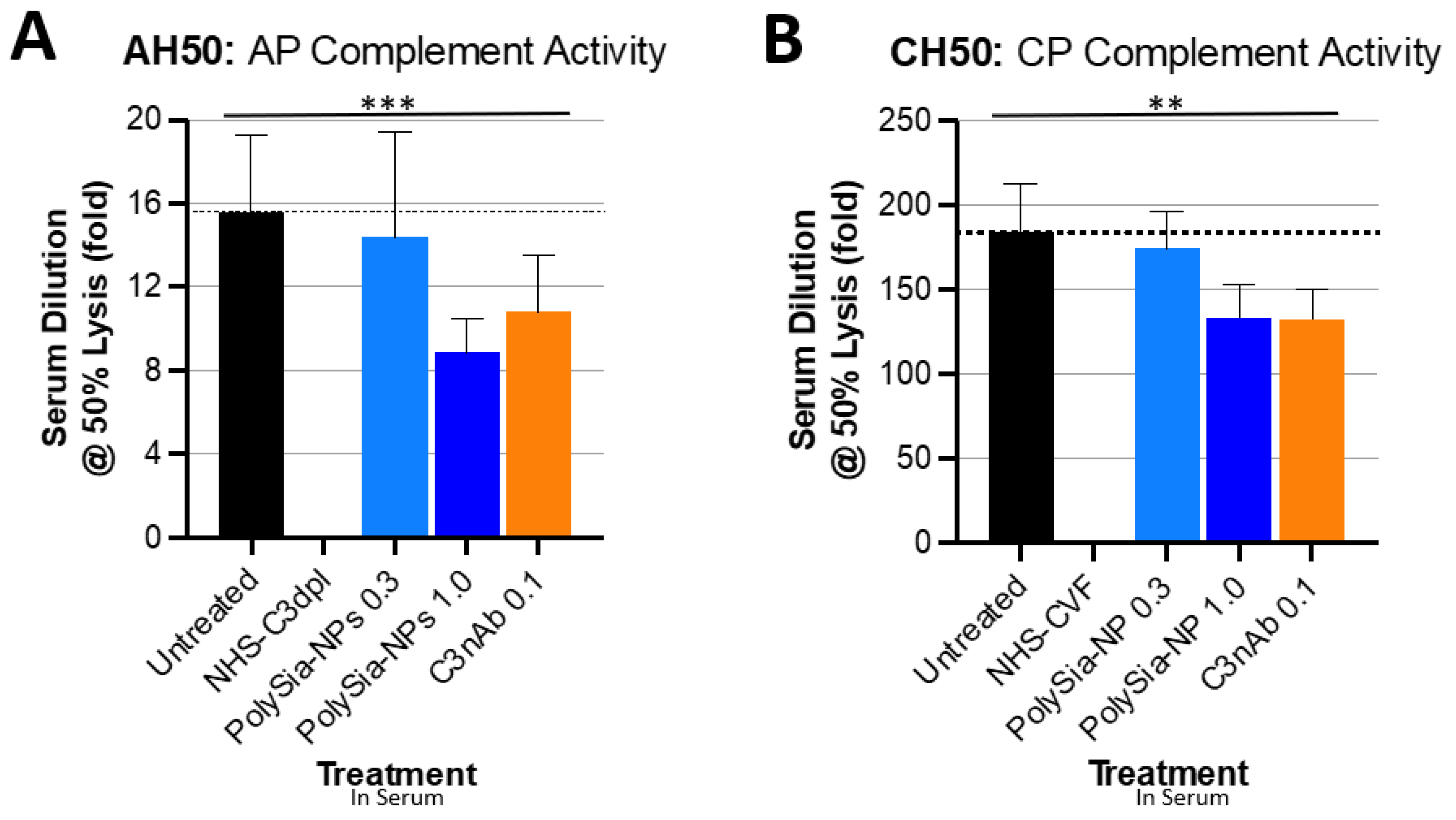
2.2. PolySia-NPs Suppress Complement Activity in Human Serum: Hemolysis
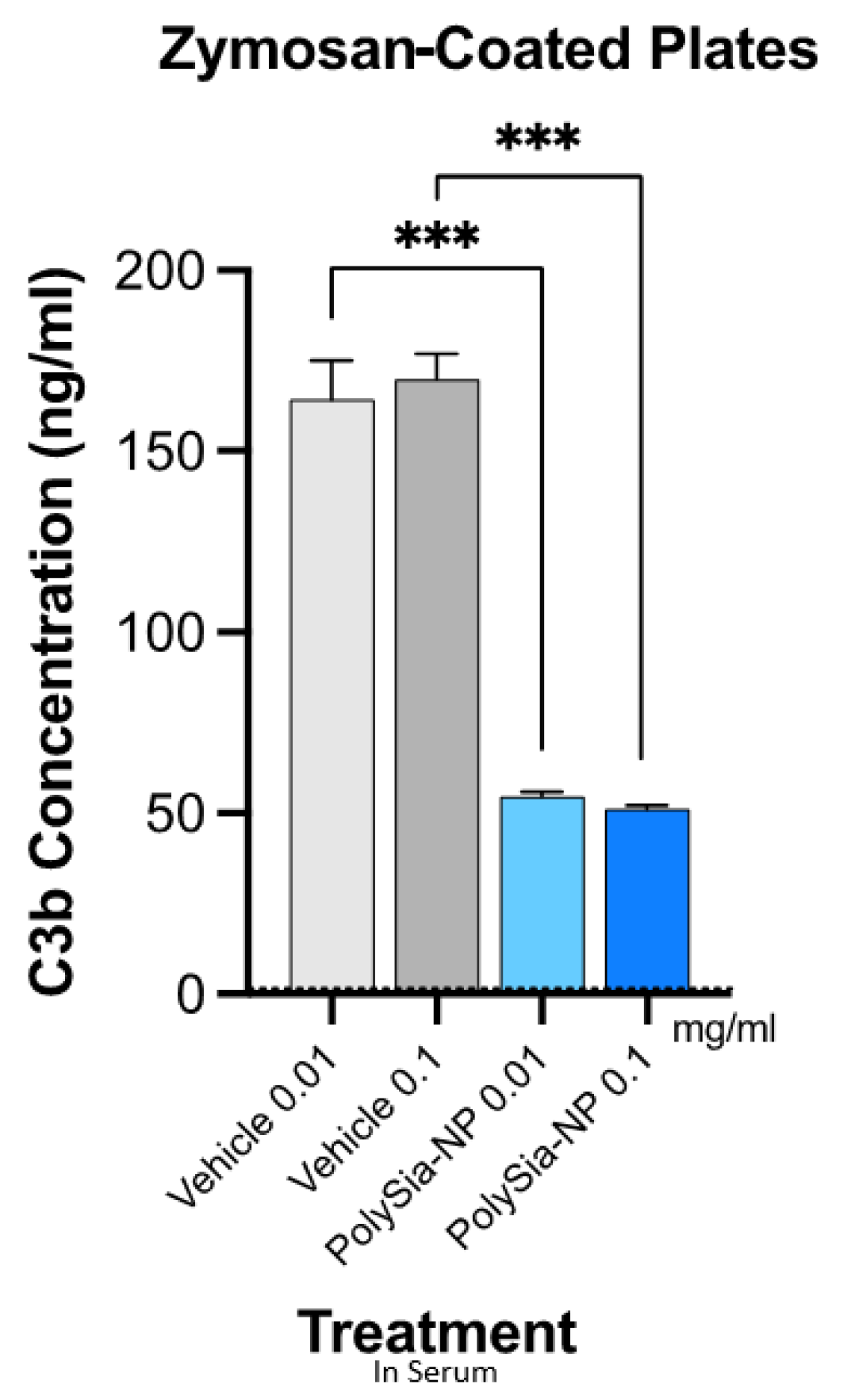
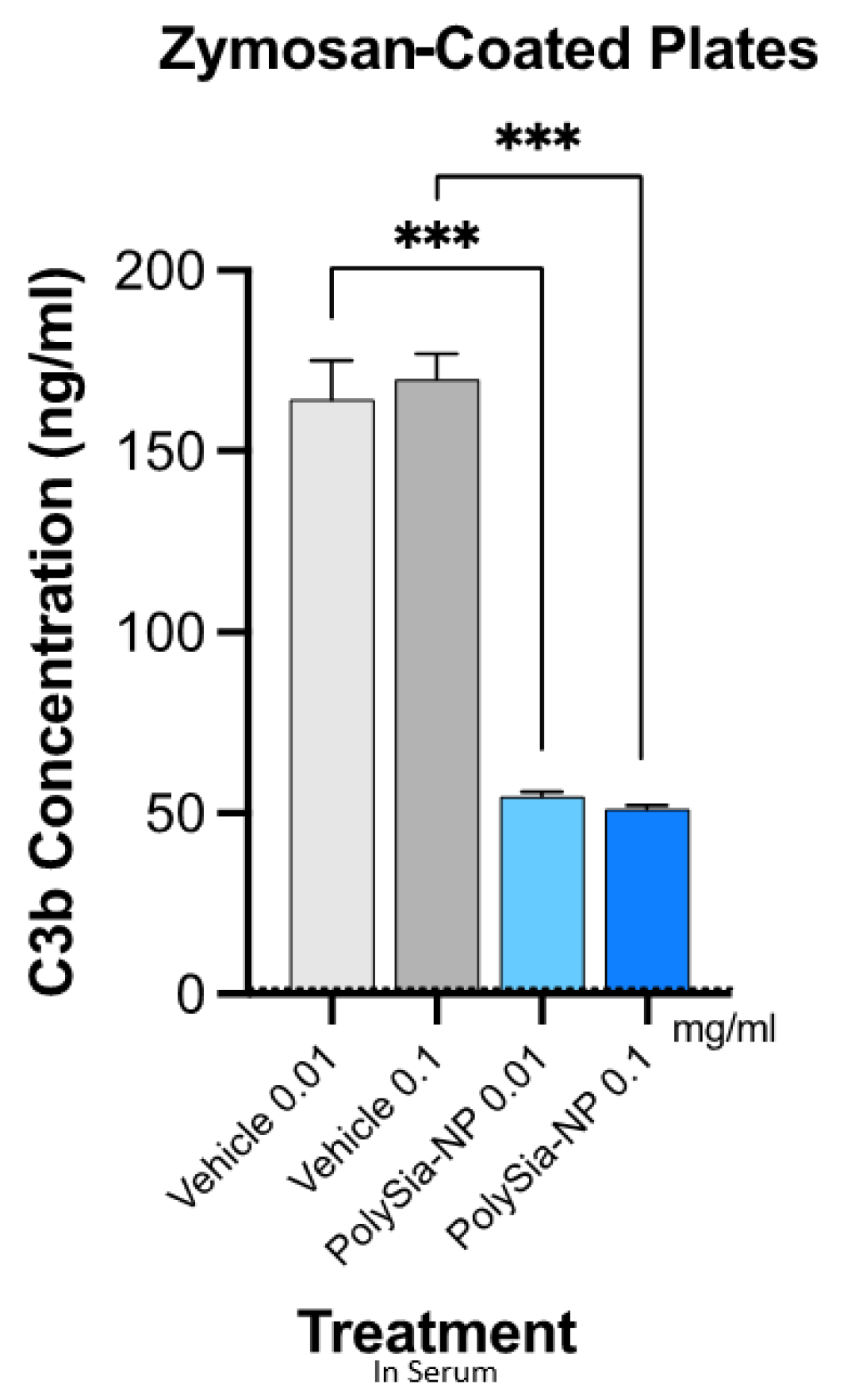
2.3. PolySia-NPs Suppress Complement Activity in Human Serum: Opsonization (C3b Deposition)
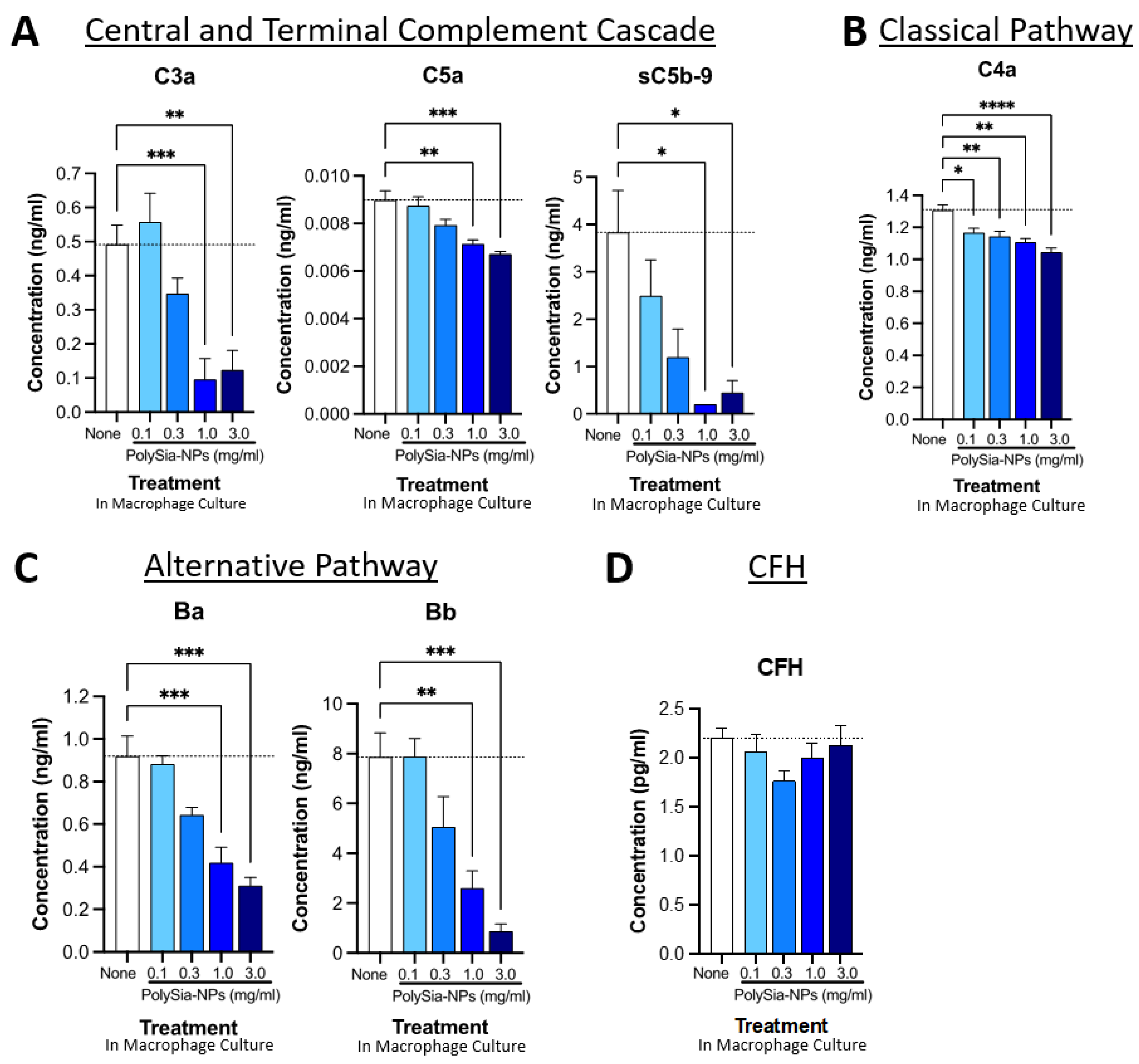
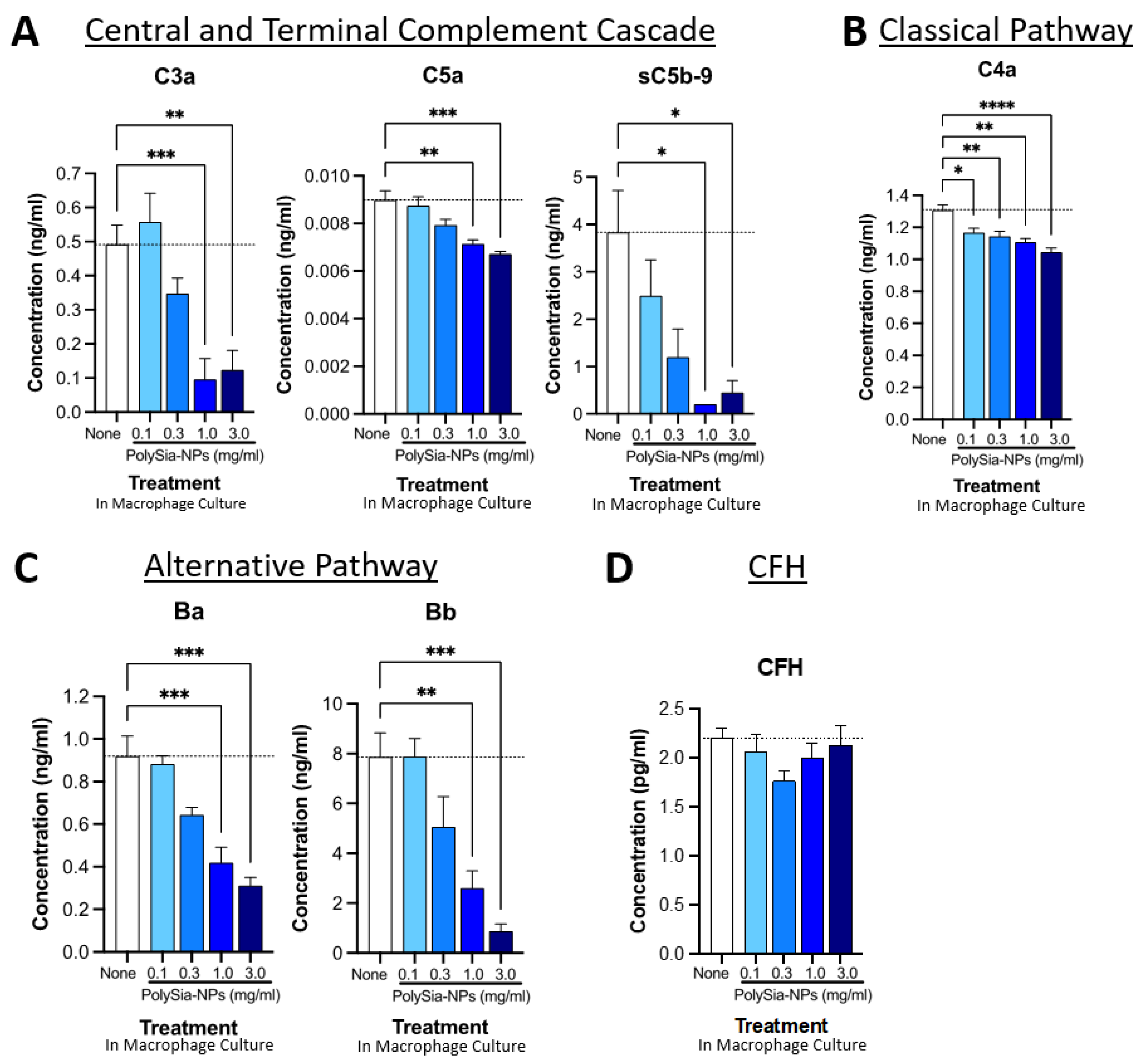
2.4. PolySia-NPs Suppress Complement Activity in Human Macrophages In Vitro
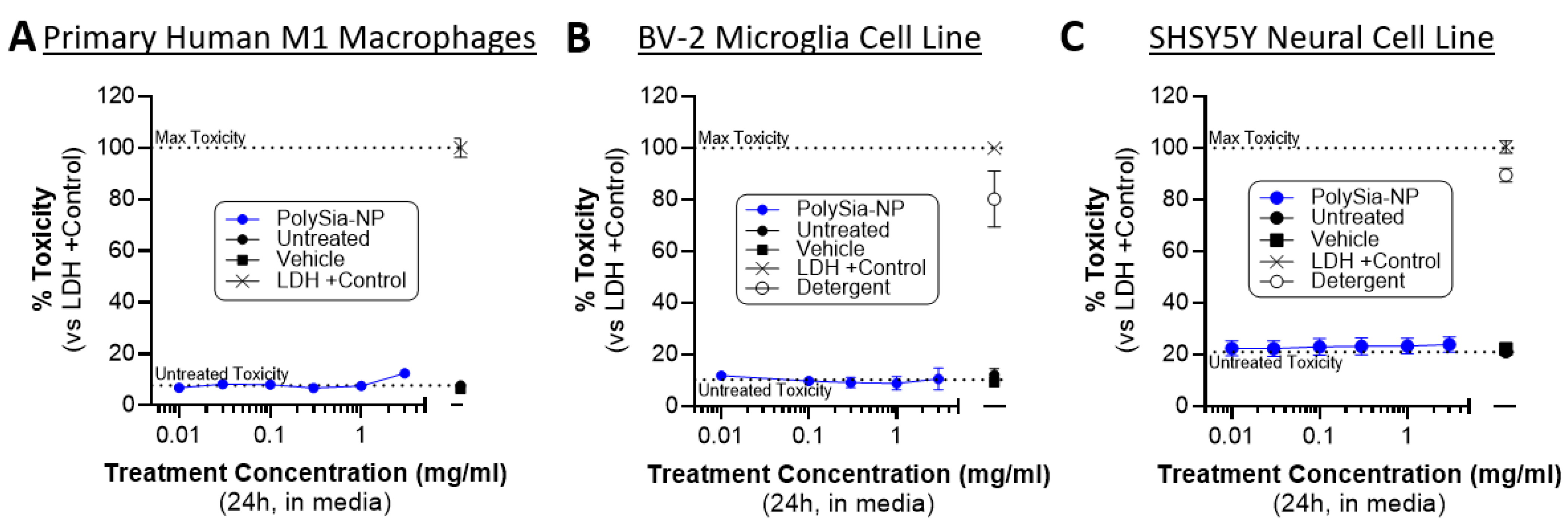
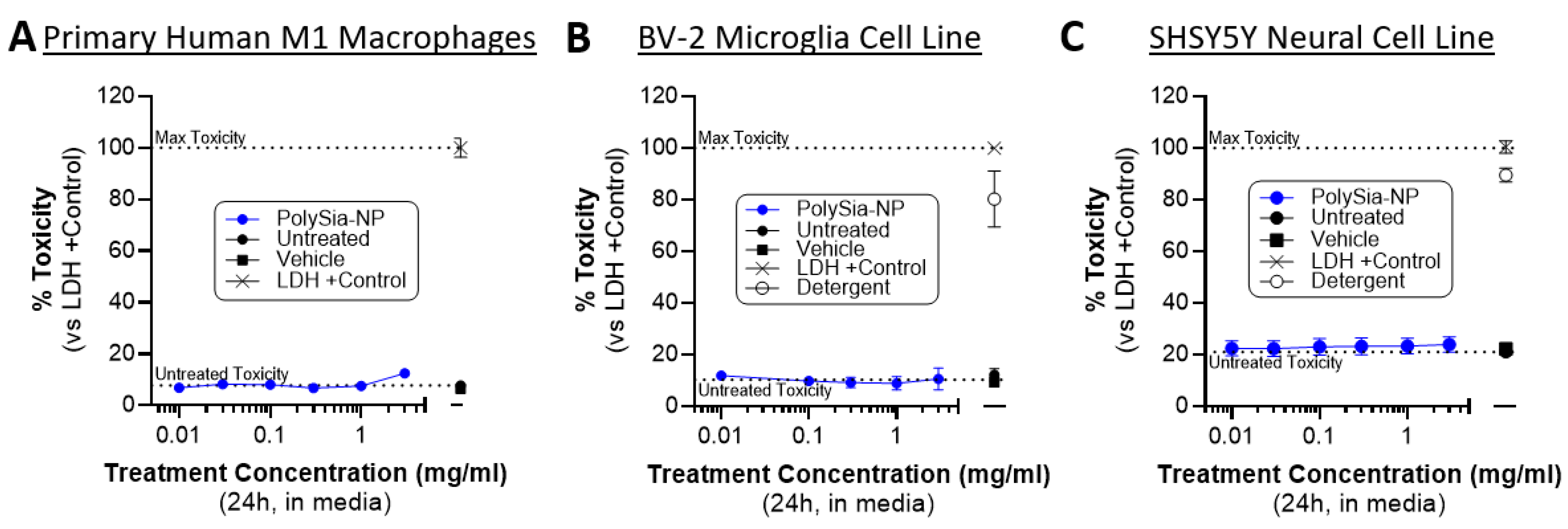
2.5. PolySia-NP Treatment Is Not Cytotoxic to Cultured Macrophages, Microglia, and Neural Cells
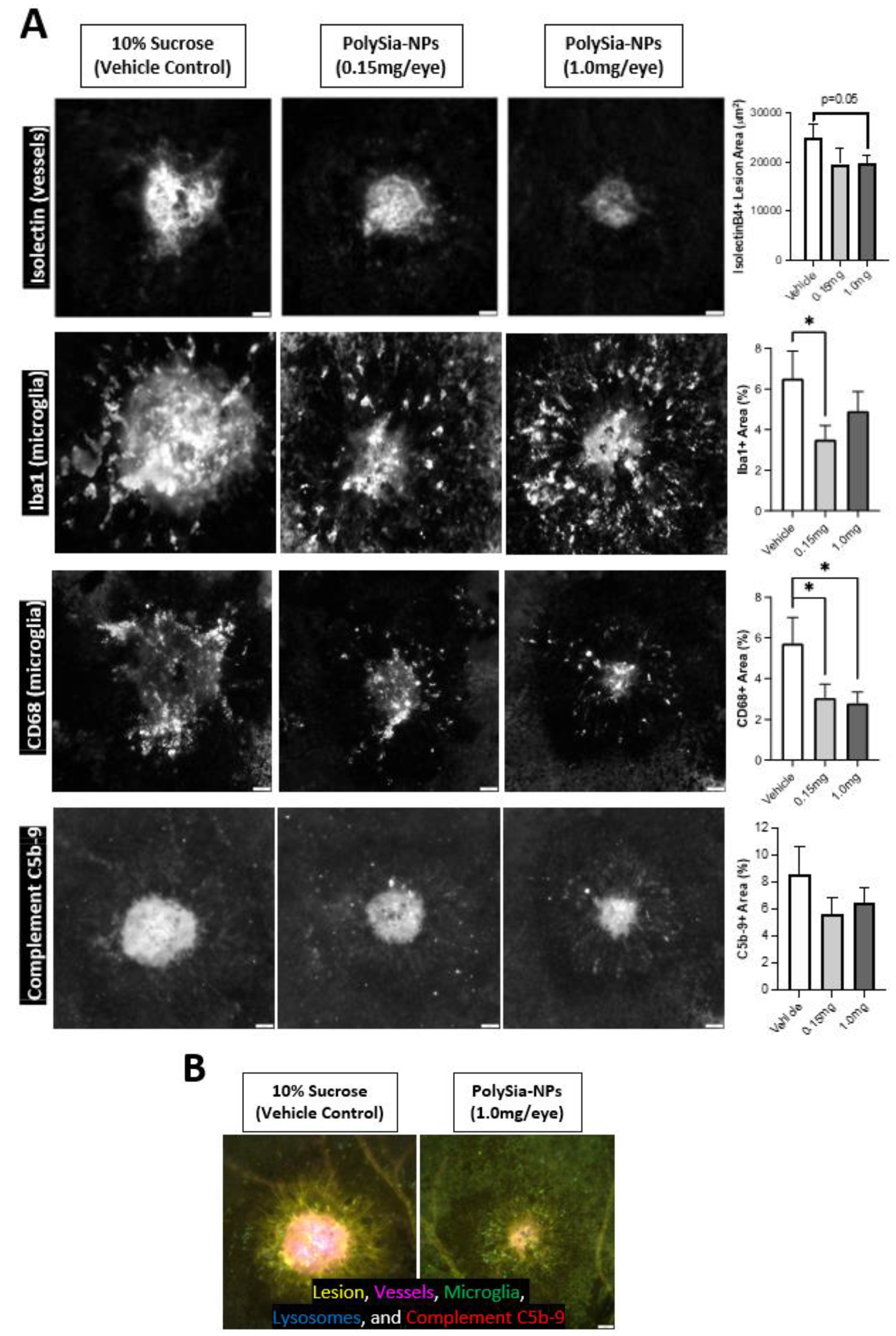
2.6. PolySia-NP Treatment Reduces Microglia and Complement Response to Laser CNV In Vivo
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. PolySia-NP
4.3. Binding Assays
4.3.1. Biacore
4.3.2. ELISA-like
4.4. Complement Hemolytic Assays
4.4.1. CH50
4.4.2. AH50
4.5. Complement Opsonization/Deposition Assay
4.6. Cells
4.6.1. Primary M1 Human Macrophages
4.6.2. THP-1 Cell Line
4.6.3. BV-2 Cell Line
4.6.4. SHSY5Y Cell Line
4.7. Complement ELISAs
4.8. CytoTox96 Assay
4.9. Mouse In Vivo Laser CNV Model
4.10. Immunohistochemistry in Laser CNV Tissues
4.11. Graphs and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: A defense system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef] [PubMed]
- Mohebnasab, M.; Eriksson, O.; Persson, B.; Sandholm, K.; Mohlin, C.; Huber-Lang, M.; Keating, B.J.; Ekdahl, K.N.; Nilsson, B. Current and Future Approaches for Monitoring Responses to Anti-complement Therapeutics. Front. Immunol. 2019, 10, 2539. [Google Scholar] [CrossRef] [PubMed]
- Portilla, D.; Xavier, S. Role of intracellular complement activation in kidney fibrosis. Br. J. Pharmacol. 2021, 178, 2880–2891. [Google Scholar] [CrossRef] [PubMed]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Christophe, B.R.; Khahera, A.; Sim, J.L.; Connolly, E.S., Jr. Therapeutic Modulation of the Complement Cascade in Stroke. Front. Immunol. 2019, 10, 1723. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 2019, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wirta, D.; Murahashi, W.; Mathur, V.; Sankaranarayanan, S.; Taylor, L.K.; Yednock, T.; Fong, D.S.; Goldberg, J.L. Safety and Target Engagement of Complement C1q Inhibitor ANX007 in Neurodegenerative Eye Disease: Results from Phase I Studies in Glaucoma. Ophthalmol. Sci. 2023, 3, 100290. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Kajikawa, T.; Hajishengallis, E.; Maekawa, T.; Reis, E.S.; Mastellos, D.C.; Yancopoulou, D.; Hasturk, H.; Lambris, J.D. Complement-Dependent Mechanisms and Interventions in Periodontal Disease. Front. Immunol. 2019, 10, 406. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Heurich, M.; Martinez-Barricarte, R.; Francis, N.J.; Roberts, D.L.; Rodriguez de Cordoba, S.; Morgan, B.P.; Harris, C.L. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl. Acad. Sci. USA 2011, 108, 8761–8766. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Gudmundsson, E.F.; Jonmundsson, T.; Jonsson, B.G.; Twarog, M.; Gudmundsdottir, V.; Li, Z.; Finkel, N.; Poor, S.; Liu, X.; et al. A proteogenomic signature of age-related macular degeneration in blood. Nat. Commun. 2022, 13, 3401. [Google Scholar] [CrossRef] [PubMed]
- Thakkinstian, A.; Han, P.; McEvoy, M.; Smith, W.; Hoh, J.; Magnusson, K.; Zhang, K.; Attia, J. Systematic review and meta-analysis of the association between complement factor H Y402H polymorphisms and age-related macular degeneration. Hum. Mol. Genet. 2006, 15, 2784–2790. [Google Scholar] [CrossRef]
- Ram, S.; Sharma, A.K.; Simpson, S.D.; Gulati, S.; McQuillen, D.P.; Pangburn, M.K.; Rice, P.A. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J. Exp. Med. 1998, 187, 743–752. [Google Scholar] [CrossRef]
- Schmidt, C.Q.; Herbert, A.P.; Hocking, H.G.; Uhrin, D.; Barlow, P.N. Translational mini-review series on complement factor H: Structural and functional correlations for factor H. Clin. Exp. Immunol. 2008, 151, 14–24. [Google Scholar] [CrossRef]
- Ranganathan, S.; Male, D.A.; Ormsby, R.J.; Giannakis, E.; Gordon, D.L. Pinpointing the putative heparin/sialic acid-binding residues in the ‘sushi’ domain 7 of factor H: A molecular modeling study. In Proceedings of the Pacific Symposium on Biocumputing 2000, Honolulu, HI, USA, 5–9 January 2000; pp. 155–167. [Google Scholar] [CrossRef]
- Tolentino, M.; Tolentino, A.J.; Tolentino, E.M.; Krishnan, A.; Genead, M.A. Sialic Acid Mimetic Microglial Sialic Acid-Binding Immunoglobulin-like Lectin Agonism: Potential to Restore Retinal Homeostasis and Regain Visual Function in Age-Related Macular Degeneration. Pharmaceuticals 2023, 16, 1735. [Google Scholar] [CrossRef] [PubMed]
- Khanani, A.M.; Patel, S.S.; Staurenghi, G.; Tadayoni, R.; Danzig, C.J.; Eichenbaum, D.A.; Hsu, J.; Wykoff, C.C.; Heier, J.S.; Lally, D.R.; et al. Efficacy and safety of avacincaptad pegol in patients with geographic atrophy (GATHER2): 12-month results from a randomised, double-masked, phase 3 trial. Lancet 2023, 402, 1449–1458. [Google Scholar] [CrossRef]
- Heier, J.S.; Lad, E.M.; Holz, F.G.; Rosenfeld, P.J.; Guymer, R.H.; Boyer, D.; Grossi, F.; Baumal, C.R.; Korobelnik, J.F.; Slakter, J.S.; et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): Two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet 2023, 402, 1434–1448. [Google Scholar] [CrossRef]
- Cruz-Pimentel, M.; Wu, L. Complement Inhibitors for Advanced Dry Age-Related Macular Degeneration (Geographic Atrophy): Some Light at the End of the Tunnel? J. Clin. Med. 2023, 12, 5131. [Google Scholar] [CrossRef]
- Apellis. FDA Approves SYFOVRE™ (Pegcetacoplan injection) as the First and Only Treatment for Geographic Atrophy (GA), a Leading Cause of Blindness. 2023. Available online: https://investors.apellis.com/news-releases/news-release-details/fda-approves-syfovretm-pegcetacoplan-injection-first-and-only (accessed on 11 November 2023).
- Spaide, R.F.; Vavvas, D.G. Complement Inhibition for Geographic Atrophy: Review of Salient Functional Outcomes and Perspective. Retina 2023, 43, 1064–1069. [Google Scholar] [CrossRef]
- Poor, S.H.; Qiu, Y.; Fassbender, E.S.; Shen, S.; Woolfenden, A.; Delpero, A.; Kim, Y.; Buchanan, N.; Gebuhr, T.C.; Hanks, S.M.; et al. Reliability of the mouse model of choroidal neovascularization induced by laser photocoagulation. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6525–6534. [Google Scholar] [CrossRef] [PubMed]
- Gadjeva, M.G.; Rouseva, M.M.; Zlatarova, A.S.; Reid, K.B.; Kishore, U.; Kojouharova, M.S. Interaction of human C1q with IgG and IgM: Revisited. Biochemistry 2008, 47, 13093–13102. [Google Scholar] [CrossRef] [PubMed]
- McGrath, F.D.; Brouwer, M.C.; Arlaud, G.J.; Daha, M.R.; Hack, C.E.; Roos, A. Evidence that complement protein C1q interacts with C-reactive protein through its globular head region. J. Immunol. 2006, 176, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Nauta, A.J.; Daha, M.R.; van Kooten, C.; Roos, A. Recognition and clearance of apoptotic cells: A role for complement and pentraxins. Trends Immunol. 2003, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Ruseva, M.M.; Zlatarova, A.; Ghai, R.; Kolev, M.; Olova, N.; Gadjeva, M.; Agrawal, A.; Bottazzi, B.; Mantovani, A.; et al. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: Mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry 2006, 45, 4093–4104. [Google Scholar] [CrossRef]
- Bexborn, F.; Andersson, P.O.; Chen, H.; Nilsson, B.; Ekdahl, K.N. The tick-over theory revisited: Formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol. Immunol. 2008, 45, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Xie, Q.; Xia, M.; Gong, K.; Wang, N.; Chen, Y.; Zhao, M. The importance of sialic acid, pH and ion concentration on the interaction of uromodulin and complement factor H. J. Cell Mol. Med. 2021, 25, 4316–4325. [Google Scholar] [CrossRef] [PubMed]
- Blaum, B.S.; Hannan, J.P.; Herbert, A.P.; Kavanagh, D.; Uhrin, D.; Stehle, T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat. Chem. Biol. 2015, 11, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.Q.; Hipgrave Ederveen, A.L.; Harder, M.J.; Wuhrer, M.; Stehle, T.; Blaum, B.S. Biophysical analysis of sialic acid recognition by the complement regulator Factor H. Glycobiology 2018, 28, 765–773. [Google Scholar] [CrossRef]
- Varki, A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan "self-associated molecular patterns" dampen innate immunity, but pathogens can mimic them. Glycobiology 2011, 21, 1121–1124. [Google Scholar] [CrossRef]
- Krishnan, A.; Sendra, V.G.; Patel, D.; Lad, A.; Greene, M.K.; Smyth, P.; Gallaher, S.A.; Herron, U.M.; Scott, C.J.; Genead, M.; et al. PolySialic acid-nanoparticles inhibit macrophage mediated inflammation through Siglec agonism: A potential treatment for age related macular degeneration. Front. Immunol. 2023, 14, 1237016. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T. Regulation by membrane sialic acid of beta1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc. Natl. Acad. Sci. USA 1978, 75, 1971–1975. [Google Scholar] [CrossRef] [PubMed]
- Meri, S.; Pangburn, M.K. Discrimination between activators and nonactivators of the alternative pathway of complement: Regulation via a sialic acid/polyanion binding site on factor H. Proc. Natl. Acad. Sci. USA 1990, 87, 3982–3986. [Google Scholar] [CrossRef] [PubMed]
- Hyvarinen, S.; Meri, S.; Jokiranta, T.S. Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood 2016, 22, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Biggs, R.M.; Makou, E.; Lauder, S.; Herbert, A.P.; Barlow, P.N.; Katti, S.K. A Novel Full-Length Recombinant Human Complement Factor H (CFH; GEM103) for the Treatment of Age-Related Macular Degeneration Shows Similar In Vitro Functional Activity to Native CFH. Curr. Eye Res. 2022, 47, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Karlstetter, M.; Kopatz, J.; Aslanidis, A.; Shahraz, A.; Caramoy, A.; Linnartz-Gerlach, B.; Lin, Y.; Luckoff, A.; Fauser, S.; Duker, K.; et al. Polysialic acid blocks mononuclear phagocyte reactivity, inhibits complement activation, and protects from vascular damage in the retina. EMBO Mol. Med. 2017, 9, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Hartung, H.P.; Hadding, U. Complement components in relation to macrophage function. Agents Actions 1983, 13, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Meri, S.; Pangburn, M.K. Regulation of alternative pathway complement activation by glycosaminoglycans: Specificity of the polyanion binding site on factor H. Biochem. Biophys. Res. Commun. 1994, 198, 52–59. [Google Scholar] [CrossRef]
- Tzoumas, N.; Riding, G.; Williams, M.A.; Steel, D.H. Complement inhibitors for age-related macular degeneration. Cochrane Database Syst. Rev. 2023, 6, CD009300. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, J.; Lee, J.; Cho, S.Y.; Kang, H.J.; Kim, K.Y.; Jin, D.K. Intravitreal human complement factor H in a rat model of laser-induced choroidal neovascularisation. Br. J. Ophthalmol. 2013, 97, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Khanani, A.M.; Maturi, R.K.; Bagheri, N.; Bakall, B.; Boyer, D.S.; Couvillion, S.S.; Dhoot, D.S.; Holekamp, N.M.; Jamal, K.N.; Marcus, D.M.; et al. A Phase I, Single Ascending Dose Study of GEM103 (Recombinant Human Complement Factor H) in Patients with Geographic Atrophy. Ophthalmol. Sci. 2022, 2, 100154. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef]
- Aviceda Therapeutics Inc. A Multiple Dose Study of AVD-104 for Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration (AMD) (SIGLEC). 2023. Available online: https://clinicaltrials.gov/study/NCT05839041 (accessed on 11 November 2023).
- Hoh Kam, J.; Lenassi, E.; Malik, T.H.; Pickering, M.C.; Jeffery, G. Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degeneration. Am. J. Pathol. 2013, 183, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Tolentino, M.J.; Tolentino, A.J. Investigational drugs in clinical trials for macular degeneration. Expert. Opin. Investig. Drugs 2022, 31, 1067–1085. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.; Gulati, S.; Agarwal, S.; Unemo, M.; Ohnishi, M.; Su, X.H.; Monks, B.G.; Visintin, A.; Madico, G.; Lewis, L.A.; et al. A Novel Factor H-Fc Chimeric Immunotherapeutic Molecule against Neisseria gonorrhoeae. J. Immunol. 2016, 196, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.; Lewis, L.A.; Zheng, B.; Carr, C.; Bass, I.; Gulati, S.; DeOliveira, R.B.; Gose, S.; Reed, G.W.; Botto, M.; et al. Human Factor H Domains 6 and 7 Fused to IgG1 Fc Are Immunotherapeutic against Neisseria gonorrhoeae. J. Immunol. 2018, 201, 2700–2709. [Google Scholar] [CrossRef]
- Schmitt, M.; Hippelainen, E.; Ravina, M.; Arango-Gonzalez, B.; Antopolsky, M.; Vellonen, K.S.; Airaksinen, A.J.; Urtti, A. Intravitreal Pharmacokinetics in Mice: SPECT/CT Imaging and Scaling to Rabbits and Humans. Mol. Pharm. 2019, 16, 4399–4404. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peterson, S.L.; Krishnan, A.; Patel, D.; Khanehzar, A.; Lad, A.; Shaughnessy, J.; Ram, S.; Callanan, D.; Kunimoto, D.; Genead, M.A.; et al. PolySialic Acid Nanoparticles Actuate Complement-Factor-H-Mediated Inhibition of the Alternative Complement Pathway: A Safer Potential Therapy for Age-Related Macular Degeneration. Pharmaceuticals 2024, 17, 517. https://doi.org/10.3390/ph17040517
Peterson SL, Krishnan A, Patel D, Khanehzar A, Lad A, Shaughnessy J, Ram S, Callanan D, Kunimoto D, Genead MA, et al. PolySialic Acid Nanoparticles Actuate Complement-Factor-H-Mediated Inhibition of the Alternative Complement Pathway: A Safer Potential Therapy for Age-Related Macular Degeneration. Pharmaceuticals. 2024; 17(4):517. https://doi.org/10.3390/ph17040517
Chicago/Turabian StylePeterson, Sheri L., Anitha Krishnan, Diyan Patel, Ali Khanehzar, Amit Lad, Jutamas Shaughnessy, Sanjay Ram, David Callanan, Derek Kunimoto, Mohamed A. Genead, and et al. 2024. "PolySialic Acid Nanoparticles Actuate Complement-Factor-H-Mediated Inhibition of the Alternative Complement Pathway: A Safer Potential Therapy for Age-Related Macular Degeneration" Pharmaceuticals 17, no. 4: 517. https://doi.org/10.3390/ph17040517
APA StylePeterson, S. L., Krishnan, A., Patel, D., Khanehzar, A., Lad, A., Shaughnessy, J., Ram, S., Callanan, D., Kunimoto, D., Genead, M. A., & Tolentino, M. J. (2024). PolySialic Acid Nanoparticles Actuate Complement-Factor-H-Mediated Inhibition of the Alternative Complement Pathway: A Safer Potential Therapy for Age-Related Macular Degeneration. Pharmaceuticals, 17(4), 517. https://doi.org/10.3390/ph17040517