

Synthesis, Computational Study, and In Vitro α-Glucosidase Inhibitory Action of 1,3,4-Thiadiazole Derivatives of 3-Aminopyridin-2(1H)-ones

 , , , , and
, , , , and 
Abstract

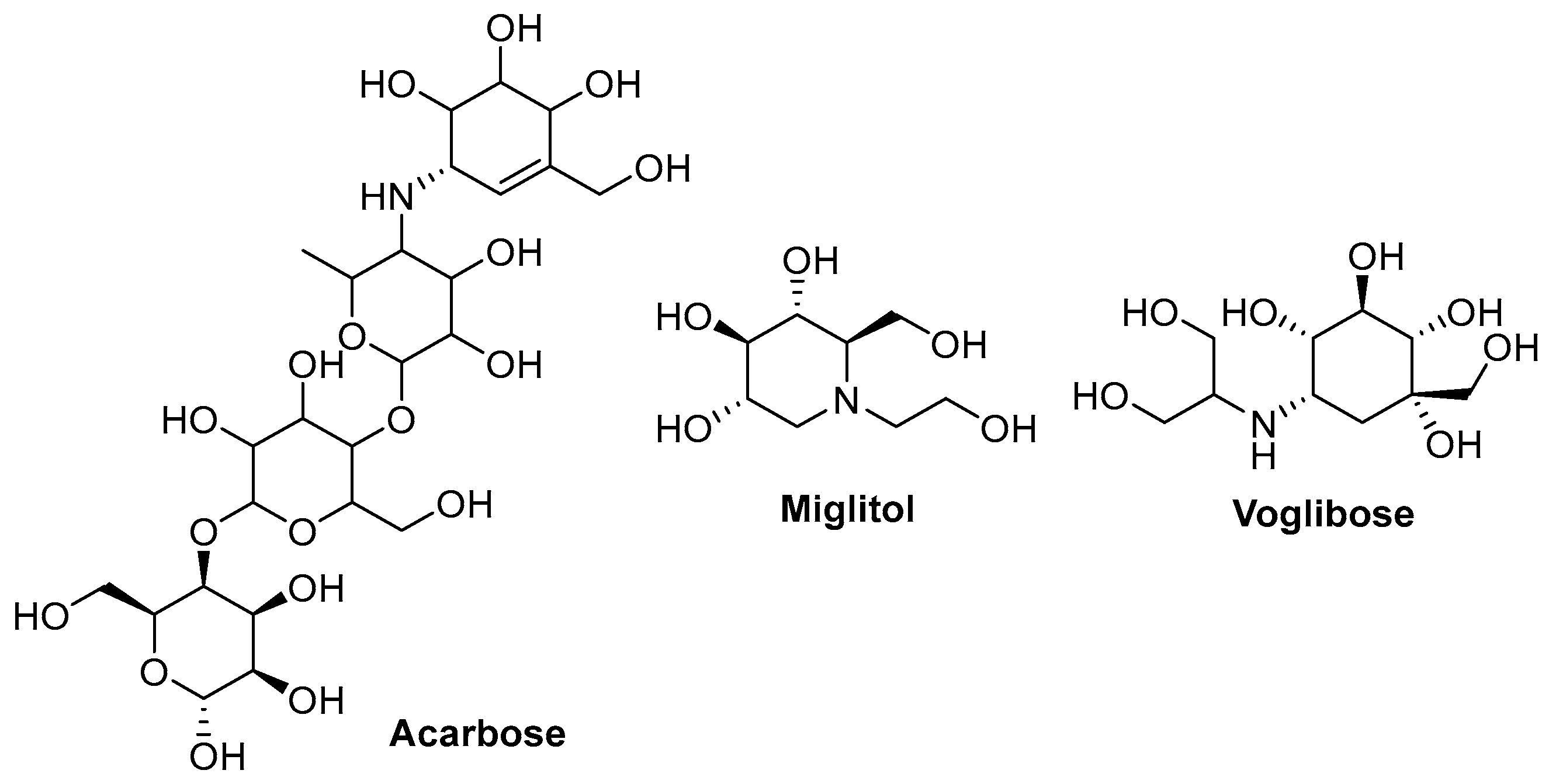
1. Introduction
2. Results and Discussion
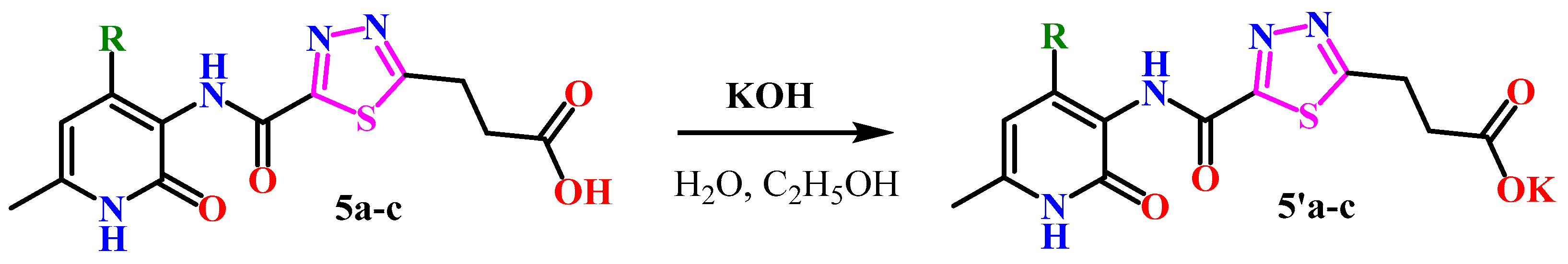
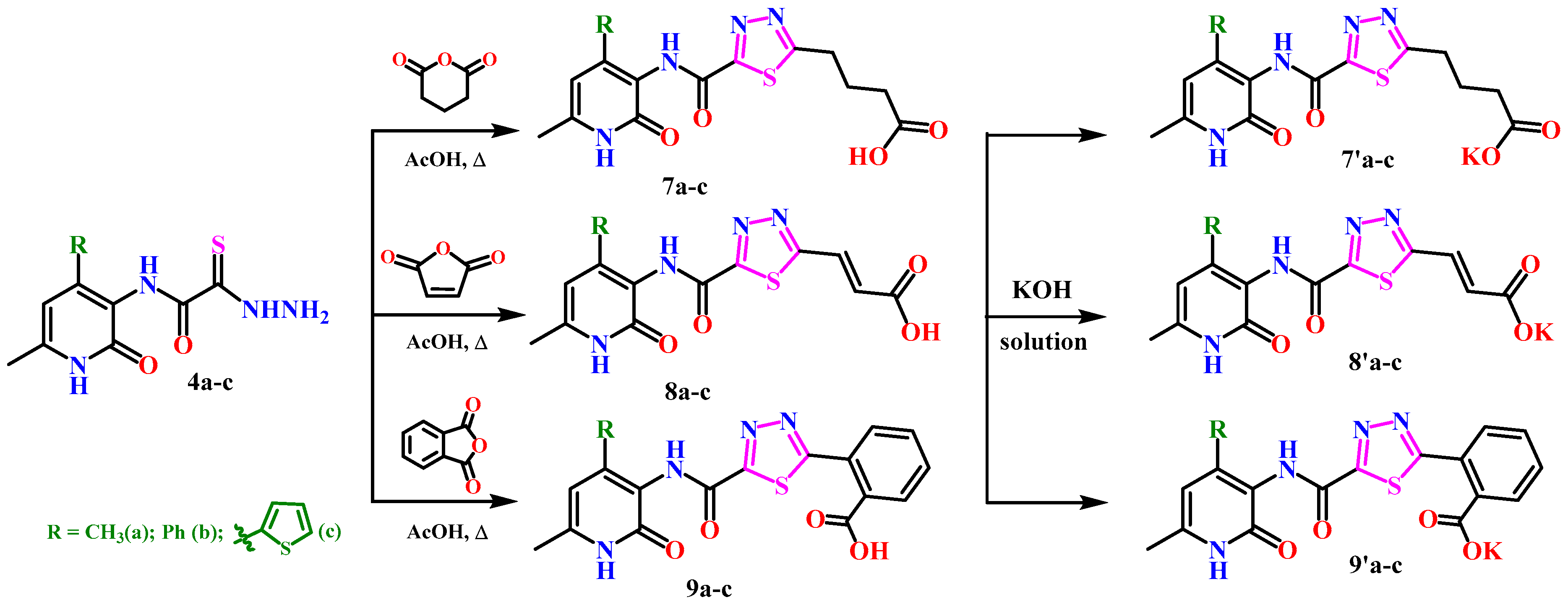
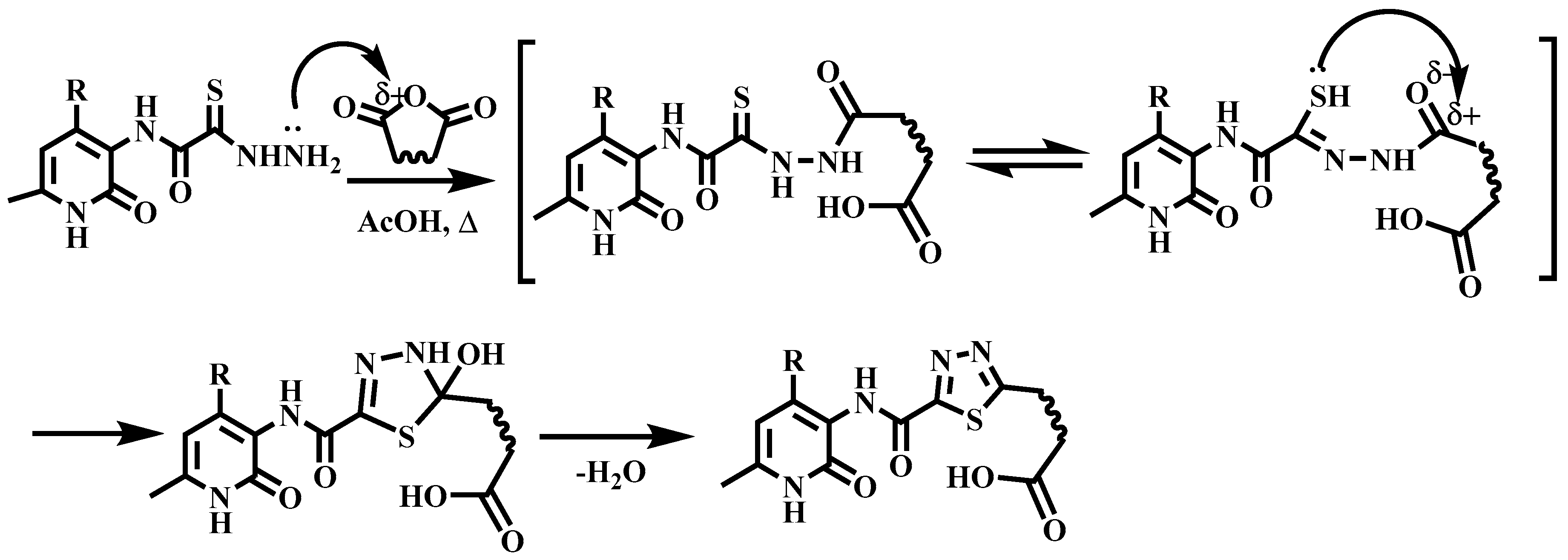
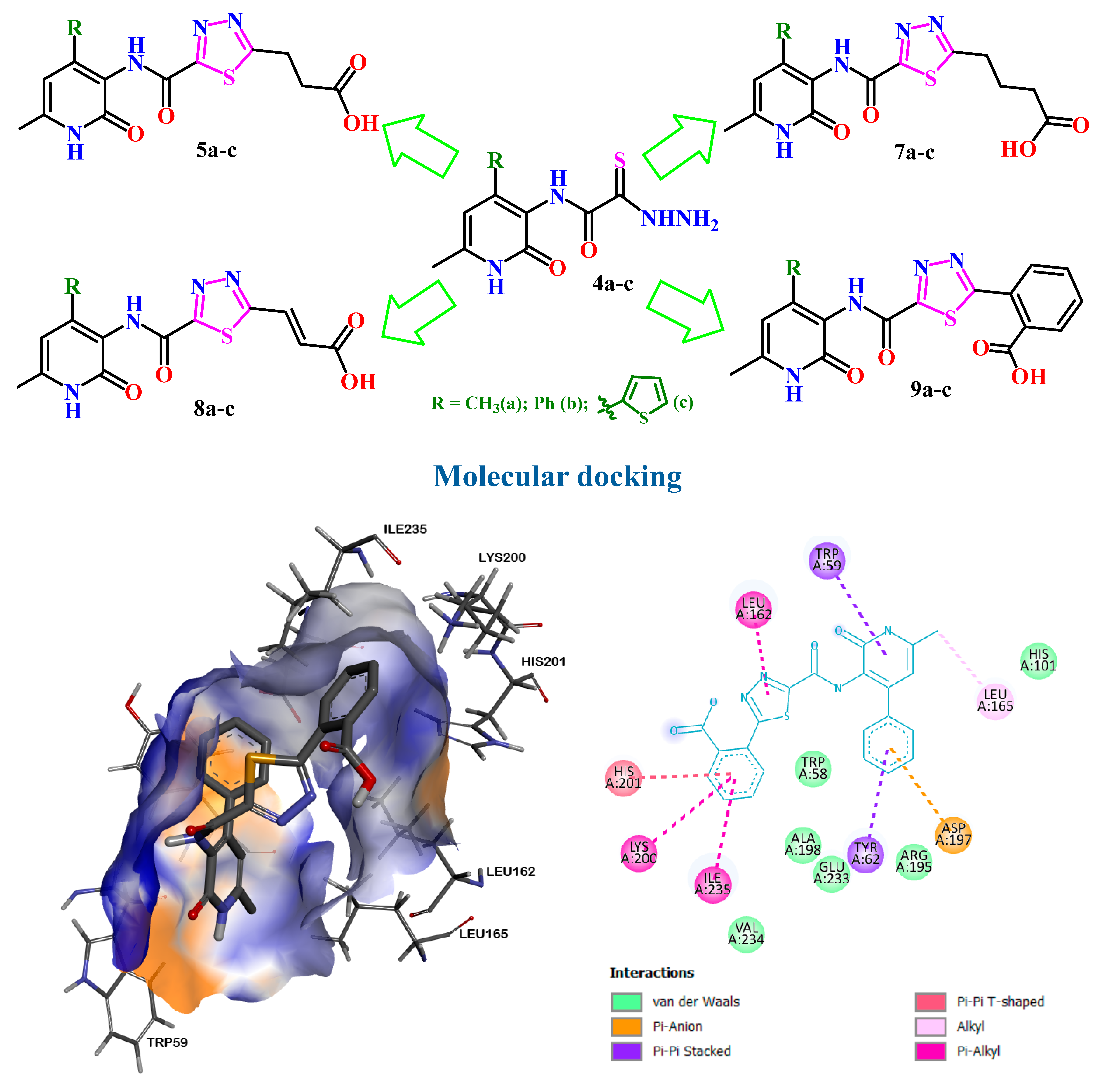
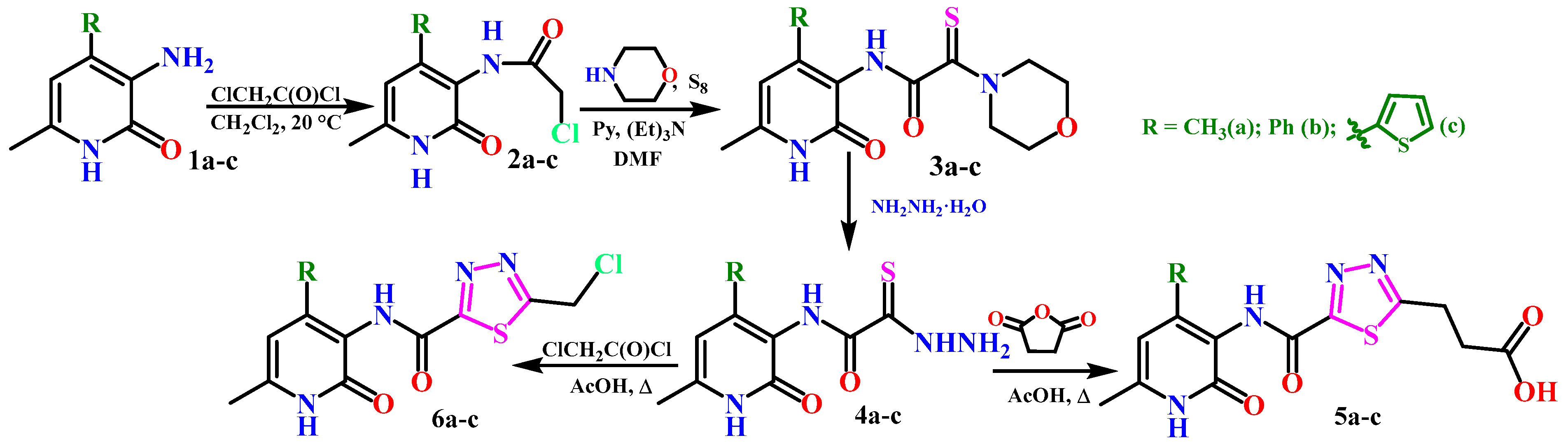
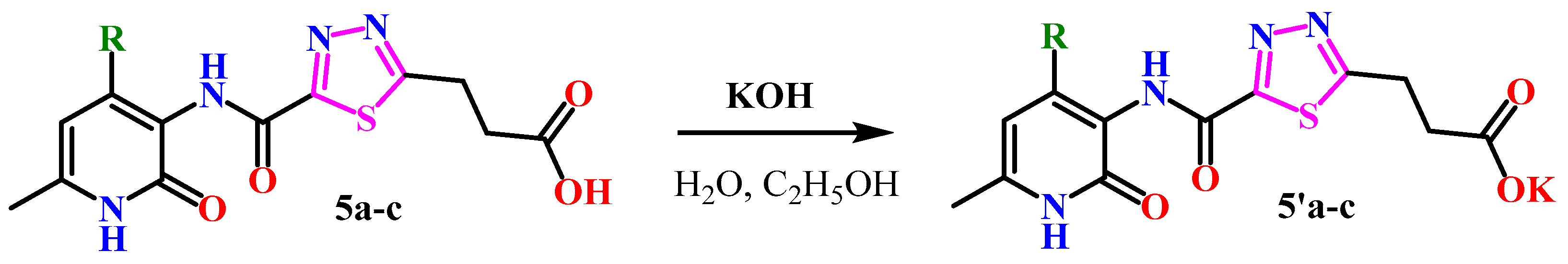
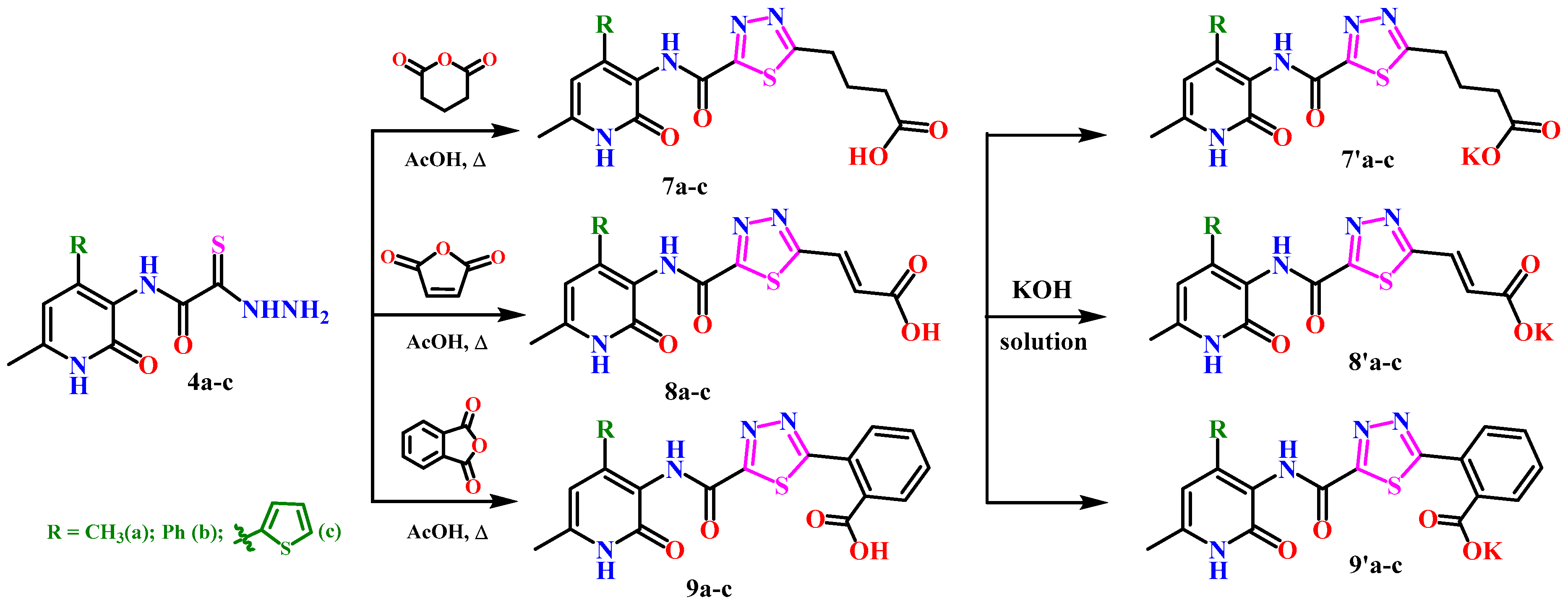
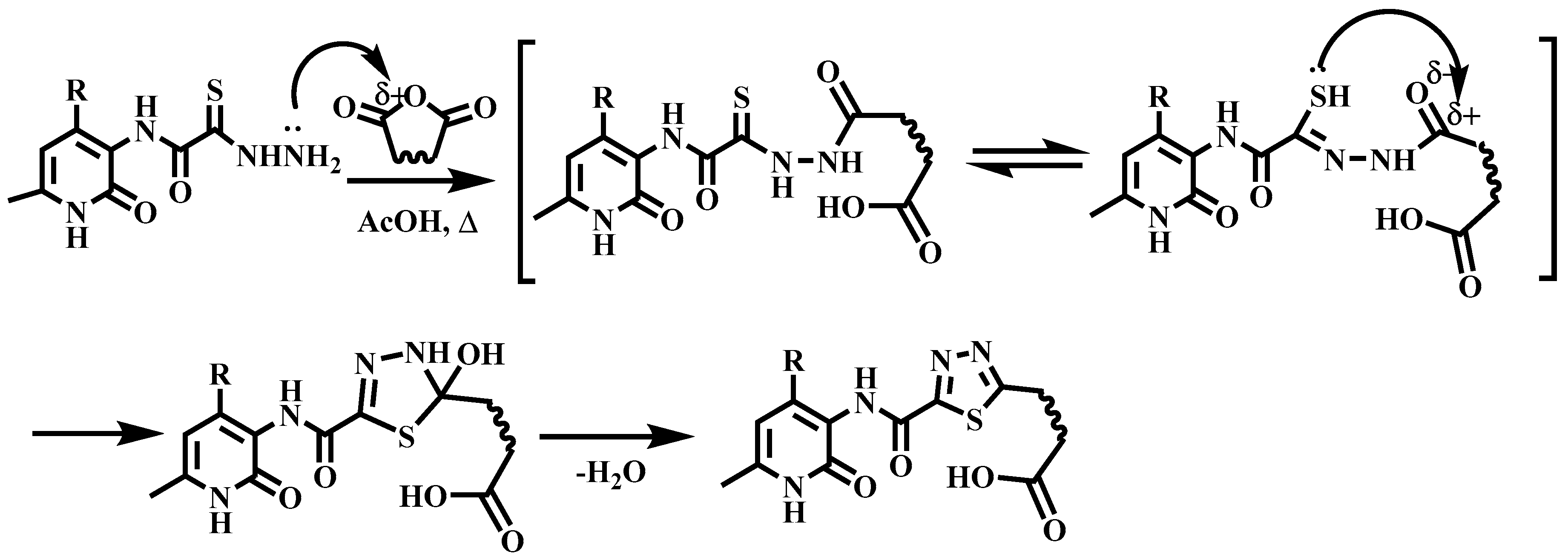
2.1. Chemistry
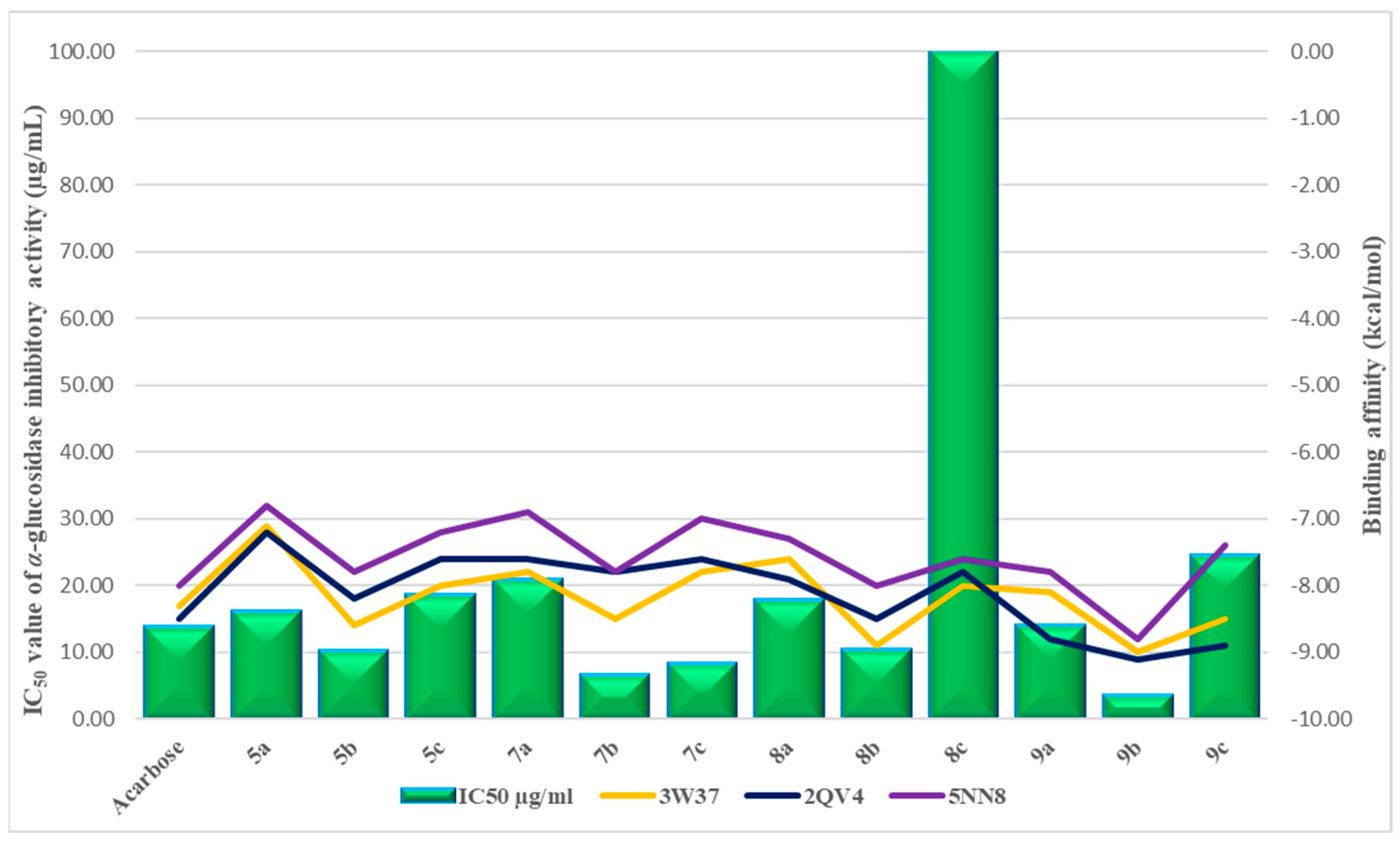
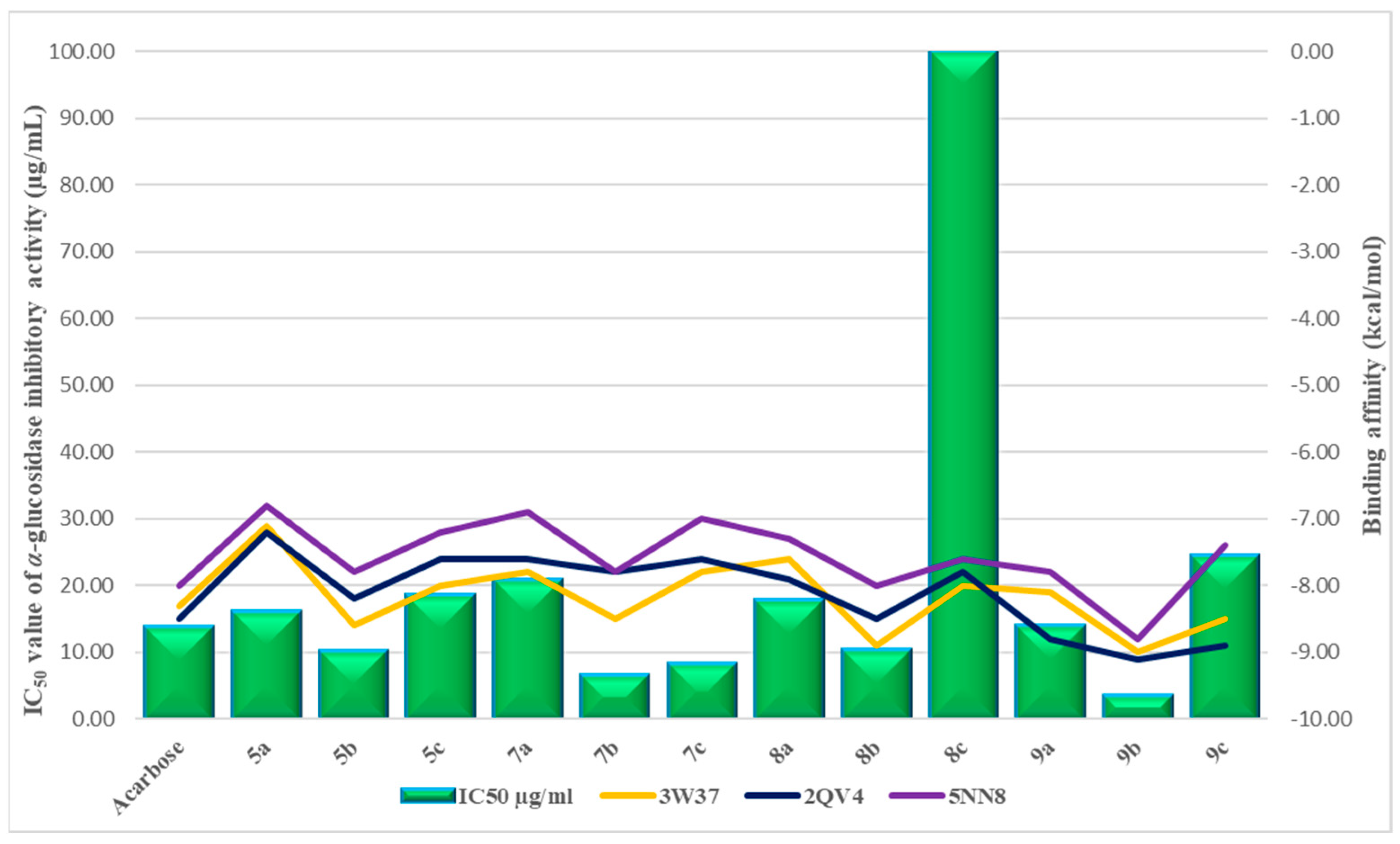
2.2. In Vitro α-Glucosidase Inhibition Assay
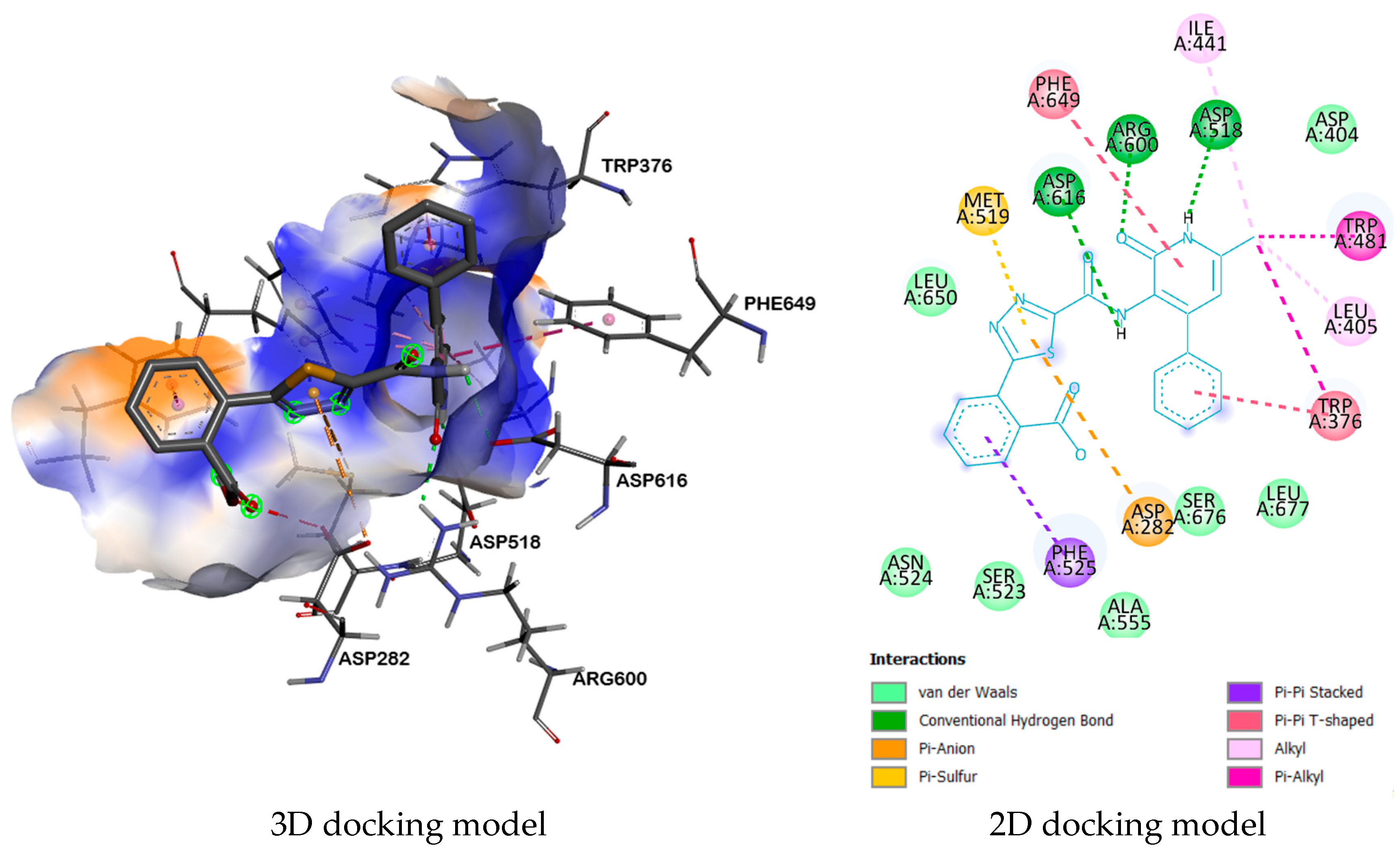
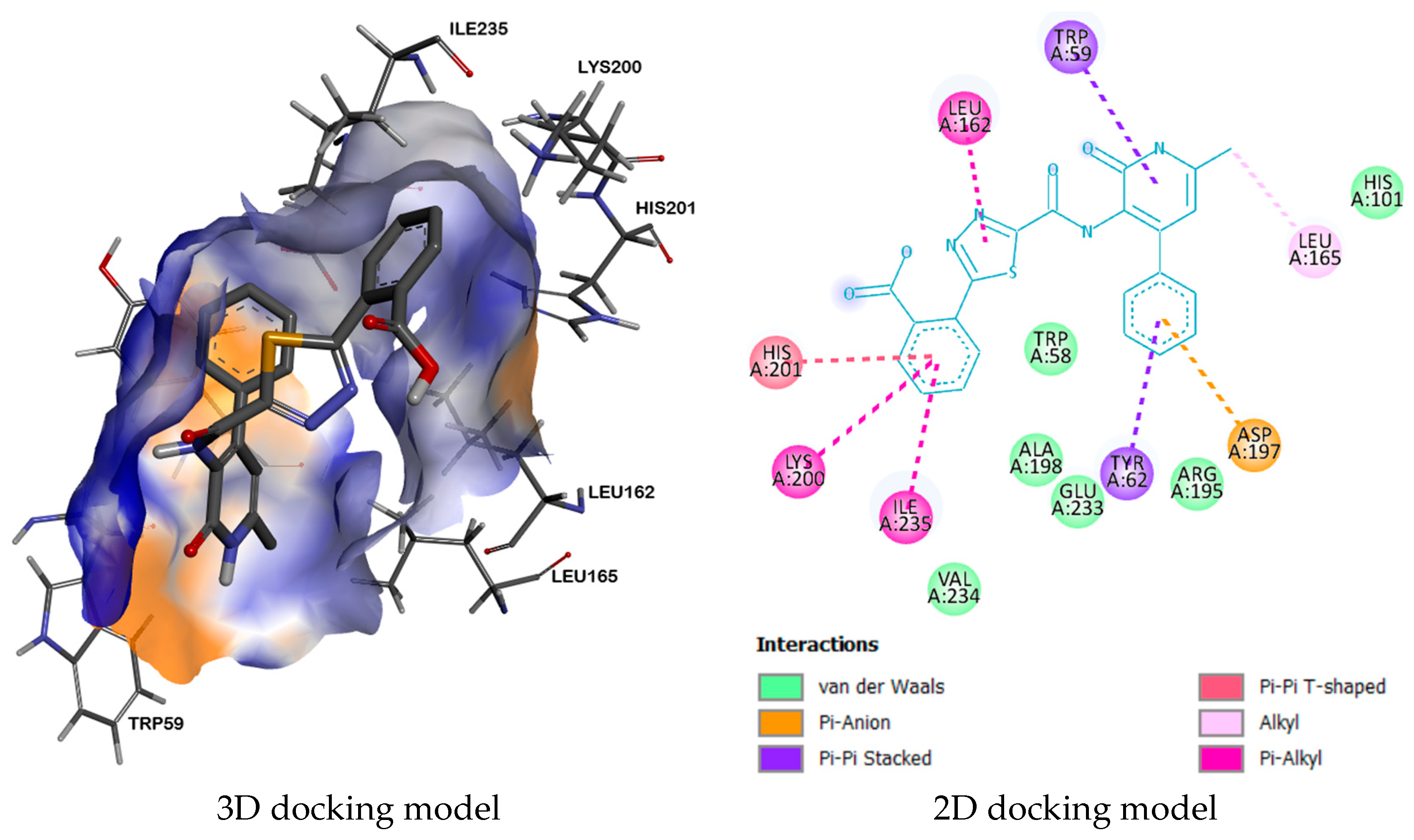
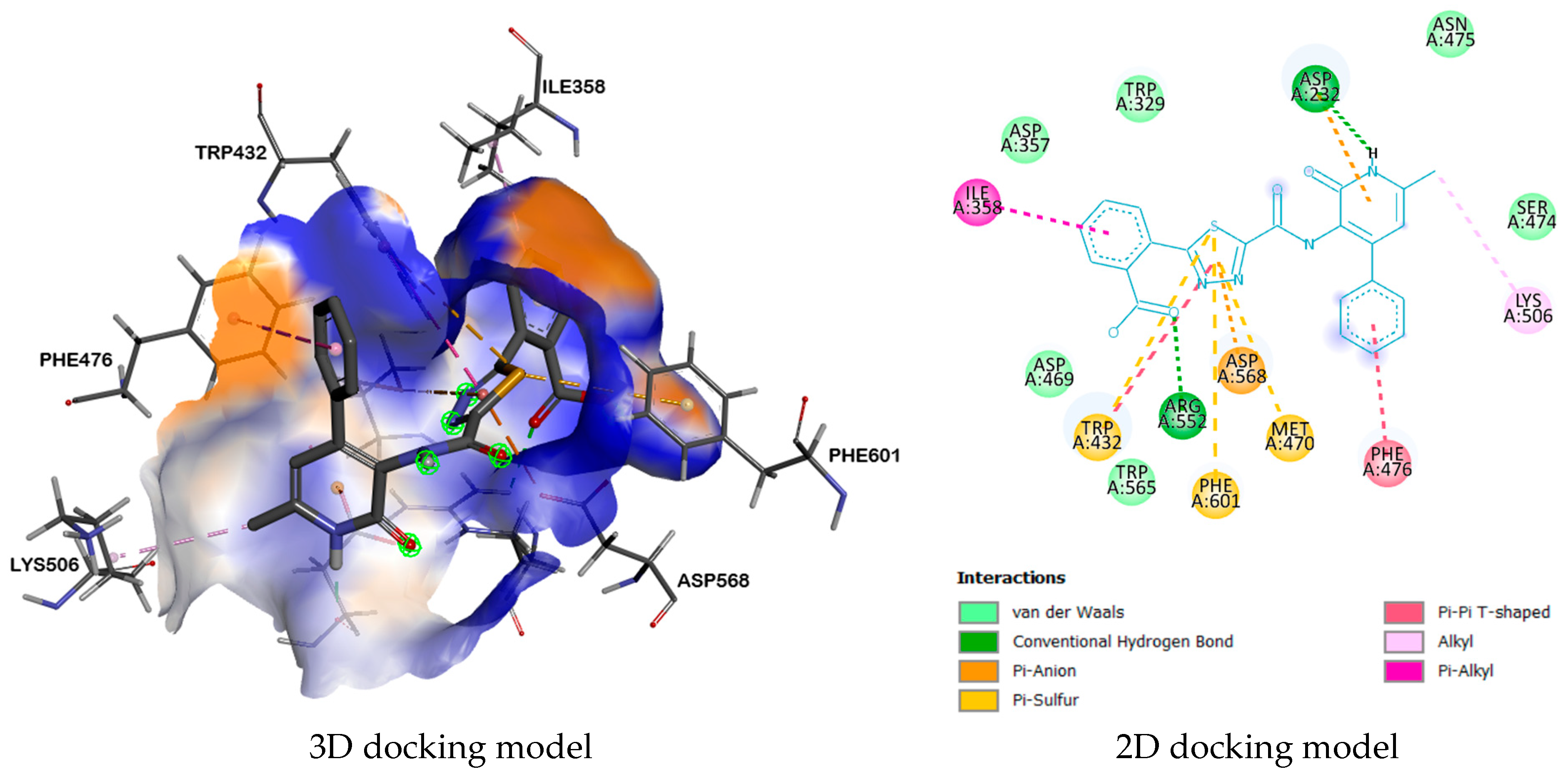
2.3. Molecular Docking
3. Materials and Methods
3.1. Experimental Procedures
3.2. Biological Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banday, M.Z.; Sameer, A.S.; Nissar, S. Pathophysiology of diabetes: An overview. Avicenna J. Med. 2020, 10, 174–188. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/news/item/12-11-2021-new-who-report-maps-barriers-to-insulin-availability-and-suggests-actions-to-promote-universal-access (accessed on 30 November 2023).
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the international diabetes federation diabetes atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843–107852. [Google Scholar] [CrossRef]
- Available online: https://www.statista.com/topics/1723/diabetes/ (accessed on 30 November 2023).
- Pellegrini, N.; Roberta, R.; Yang, M.; Rice-Evans, C. Screening of dietary carotenoids and carotenoid–rich fruit extracts for antioxidant activities applying 2,2′–azinobis(3-ethyenebenzothiazoline-6-sulfonic acid) radical cation decolorization assay. Methods Enzymol. 1991, 299, 379–389. [Google Scholar] [CrossRef]
- Sindhu, S.; Vaibhavi, K.; Anshu, M. In vitro studies on alpha-amylase and alpha-glucosidase inhibitory activities of selected plant extracts. Eur. J. Exp. Biol. 2013, 3, 128–132. [Google Scholar]
- Kaur, N.; Kumar, V.; Nayak, S.K.; Wadhwa, P.K.; Kaur, P.; Sahu, S.K. Alpha-amylase as molecular target for treatment of diabetes mellitus: A comprehensive review. Chem. Biol. Drug Des. 2021, 98, 539–560. [Google Scholar] [CrossRef]
- Kazmi, M.; Zaib, S.; Ibrar, A.; Amjad, S.T.; Shafique, Z.; Mehsud, S.; Saeed, A.; Iqbal, J.; Khan, I. A new entry into the portfolio of α-glucosidase inhibitors as potent therapeutics for type 2 diabetes: Design, bioevaluation and one-pot multi-component synthesis of diamine-bridged coumarinyl oxadiazole conjugates. Bioorg. Chem. 2018, 77, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, S. Recent advances in synthetic α-glucosidase inhibitors. ChemMedChem 2017, 12, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Pervin, R. Chapter 34—Current Antidiabetic Drugs: Review of Their Efficacy and Safety. In Nutritional and Therapeutic Interventions for Diabetes and Metabolic Syndrome; Academic Press: Cambridge, MA, USA, 2018; pp. 455–473. [Google Scholar] [CrossRef]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Datar, P.A.; Deokule, T.A. Development of thiadiazole as an antidiabetic agent—A review. Mini Rev. Med. Chem. 2014, 14, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Anthwal, T.; Paliwal, S.; Nain, S. Diverse Biological Activities of 1,3,4-Thiadiazole Scaffold. Chemistry 2022, 4, 1654–1671. [Google Scholar] [CrossRef]
- Vaishnav, Y.; Jha, A.K.; Verma, S.; Kashyap, P.; Kaur, C.D. A Review on Antidiabetic Activity of Substituted 1,3,4-thiadiazole Derivatives. Res. J. Pharm. Tech. 2017, 10, 4467–4470. [Google Scholar] [CrossRef]
- Gowda, K.; Swarup, H.A.; Nagarakere, S.C.; Rangappa, S.; Kanchugarkoppal, R.S.; Kempegowda, M. Structural Studies of 2,5-disubstituted 1,3,4-thiadiazole Derivatives from Dithioesters under the Mild Condition: Studies on Antioxidant, Antimicrobial Activities, and Molecular Docking. Synth. Commun. 2020, 50, 1528–1544. [Google Scholar] [CrossRef]
- Janowska, S.; Paneth, A.; Wujec, M. Cytotoxic properties of 1,3,4-thiadiazole derivatives—A review. Molecules 2020, 25, 4309. [Google Scholar] [CrossRef]
- Kulakov, I.V.; Matsukevich, M.V.; Shulgau, Z.T.; Sergazy, S.; Seilkhanov, T.M.; Puzari, A.; Fisyuk, A.S. Synthesis and antiradical activity of 4-aryl(hetaryl)-substituted 3-aminopyridin-2(1H)-ones. Chem. Heterocycl. Compd. 2015, 51, 991–996. [Google Scholar] [CrossRef]
- Sergazy, S.; Shulgau, Z.; Zhulikeyeva, A.; Ramankulov, Y.; Palamarchuk, I.V.; Kulakov, I.V. Cytoprotective Activity of Newly Synthesized 3-(Arylmethylamino)-6-Methyl-4-Phenylpyridin-2(1H)-ones Derivatives. Molecules 2022, 27, 5362. [Google Scholar] [CrossRef]
- Palamarchuk, I.V.; Shulgau, Z.T.; Kharitonova, M.A.; Kulakov, I.V. Synthesis and neurotropic activity of new 3-(arylmethyl)aminopyridine-2(1H)-one. Chem. Pap. 2021, 75, 4729–4739. [Google Scholar] [CrossRef]
- Palamarchuk, I.V.; Shulgau, Z.T.; Sergazy, S.D.; Zhulikeeva, A.M.; Seilkhanov, T.M.; Kulakov, I.V. Synthesis, molecular docking, and hemorheological activity of new 4-(thien-2-yl)-3-aminopyridine-2(1H)-one derivatives. Russ. J. Gen. Chem. 2022, 92, 1692–1705. [Google Scholar] [CrossRef]
- Palamarchuk, I.V.; Shulgau, Z.T.; Dautov, A.Y.; Sergazy, S.D.; Kulakov, I.V. Design, synthesis, spectroscopic characterization, computational analysis, and in vitro α-amylase and α-glucosidase evaluation of 3-aminopyridin-2(1H)-one based novel monothiooxamides and 1,3,4-thiadiazoles. Org. Biomol. Chem. 2022, 20, 8962–8976. [Google Scholar] [CrossRef] [PubMed]
- Shai, L.J.; Masoko, P.; Mokgotho, M.P.; Magano, S.R.; Mogale, A.M.; Boaduo, N.; Eloff, J.N. Yeast alpha glucosidase inhibitory and antioxidant activities of six medicinal plants collected in Phalaborwa. S. Afr. J. Bot. 2010, 76, 465–470. [Google Scholar] [CrossRef]
- Hassan, L.R.; Anouar, E.H.; Bahron, H.; Abdullah, F.; Tajuddin, A.M. Cytotoxicity, alpha-glucosidase inhibition and molecular docking studies of hydroxamic acid chromium(III) complexes. J. Biol. Inorg. Chem. 2020, 25, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Yamashita, K.; Okuyama, M.; Mori, H.; Yao, M.; Kimura, A. Molecular basis for the recognition of long-chain substrates by plant α-glucosidases. J. Biol. Chem. 2013, 288, 19296–19303. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Muhammad, I.; Khan, H.; Aschner, M.; Filosa, R.; Daglia, M. Molecular docking of isolated alkaloids for possible a-glucosidase inhibition. Biomolecules 2019, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh-Pillay, A.; Awolade, P.; Singh, P. Current antidiabetic agents and their molecular targets: A review. Eur. J. Med. Chem. 2018, 152, 436–488. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Lacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase—A guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef]
- Maurus, R.; Begum, A.; Williams, L.K.; Fredriksen, J.R.; Zhang, R.; Withers, S.G.; Brayer, G.D. Alternative catalytic anions differentially modulate human α-amylase activity and specificity. Biochemistry 2008, 47, 3332–3344. [Google Scholar] [CrossRef]
- Available online: https://www.rcsb.org/ (accessed on 10 November 2023).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comp. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Discovery Studio 2015: Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Dassault Systemes: San Diego, CA, USA, 2015.
- Available online: http://admet.scbdd.com/ (accessed on 10 November 2023).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Compounds | The Degree of Inhibition of α-Glucosidase Activity, % | IC50 Value of α-Glucosidase Inhibitory Activity (µg/mL) |
|---|---|---|---|
| 1. | 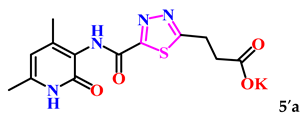 | 36.3 ± 4.8 | 16.22 ± 0.76 |
| 2. | 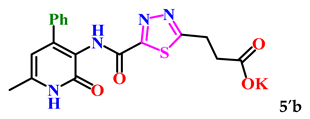 | 72.4 ± 2.3 | 10.22 ± 0.82 |
| 3. | 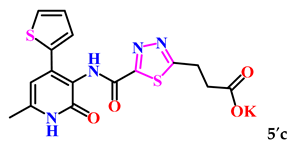 | 38.3 ± 2.4 | 18.58 ± 1.20 |
| 4. | 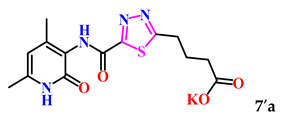 | 34.8 ± 4.2 | 20.83 ± 1.06 |
| 5. | 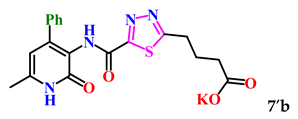 | 95.0 ± 0.2 | 6.70 ± 0.78 |
| 6. | 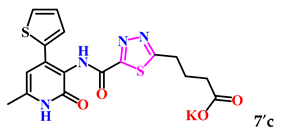 | 93.9 ± 0.4 | 8.42 ± 0.29 |
| 7. | 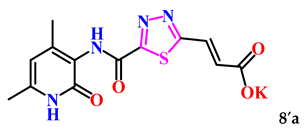 | 14.0 ± 3.0 | 17.93 ± 0.28 |
| 8. | 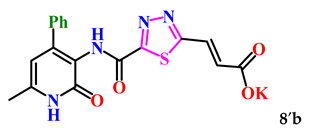 | 68.2 ± 4.7 | 10.36 ± 0.60 |
| 9. | 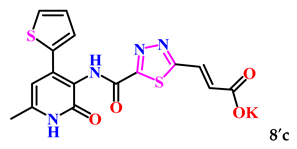 | 8.0 ± 3.0 | 48.00±1.67 |
| 10. | 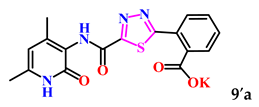 | 33.6 ± 6.1 | 14.02 ± 0.49 |
| 11. | 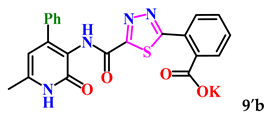 | 95.2 ± 0.1 | 3.66 ± 0.05 |
| 12. | 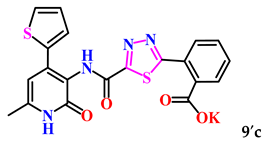 | 21.0 ± 2.2 | 24.51 ± 1.54 |
| 13. | Acarbose | 49.5 ± 1.8 | 13.88 ± 0.81 |
| Receptor | 3W37 | 2QV4 | 5NN8 | |
|---|---|---|---|---|
| Ligand | ||||
| Acarbose | −8.3 | −8.5 | −8.0 | |
| 5a | −7.1 | −7.2 | −6.8 | |
| 5b | −8.6 | −8.2 | −7.8 | |
| 5c | −8.0 | −7.6 | −7.2 | |
| 7a | −7.8 | −7.6 | −6.9 | |
| 7b | −8.5 | −7.8 | −7.8 | |
| 7c | −7.8 | −7.6 | −7.0 | |
| 8a | −7.6 | −7.9 | −7.3 | |
| 8b | −8.9 | −8.5 | −8.0 | |
| 8c | −8.0 | −7.8 | −7.6 | |
| 9a | −8.1 | −8.8 | −7.8 | |
| 9b | −9.0 | −9.1 | −8.8 | |
| 9c | −8.5 | −8.9 | −7.4 | |
| Compound | Receptor | H-Bond | Residual Amino Acid Interactions | |
|---|---|---|---|---|
| Pi-Sulfur/Pi-Anion/Pi-Pi Stacked/Pi-Pi T-Shaped/Pi-Alkyl/Amide-Pi Stacked/Pis Interactions | Van-der Walls Interactions | |||
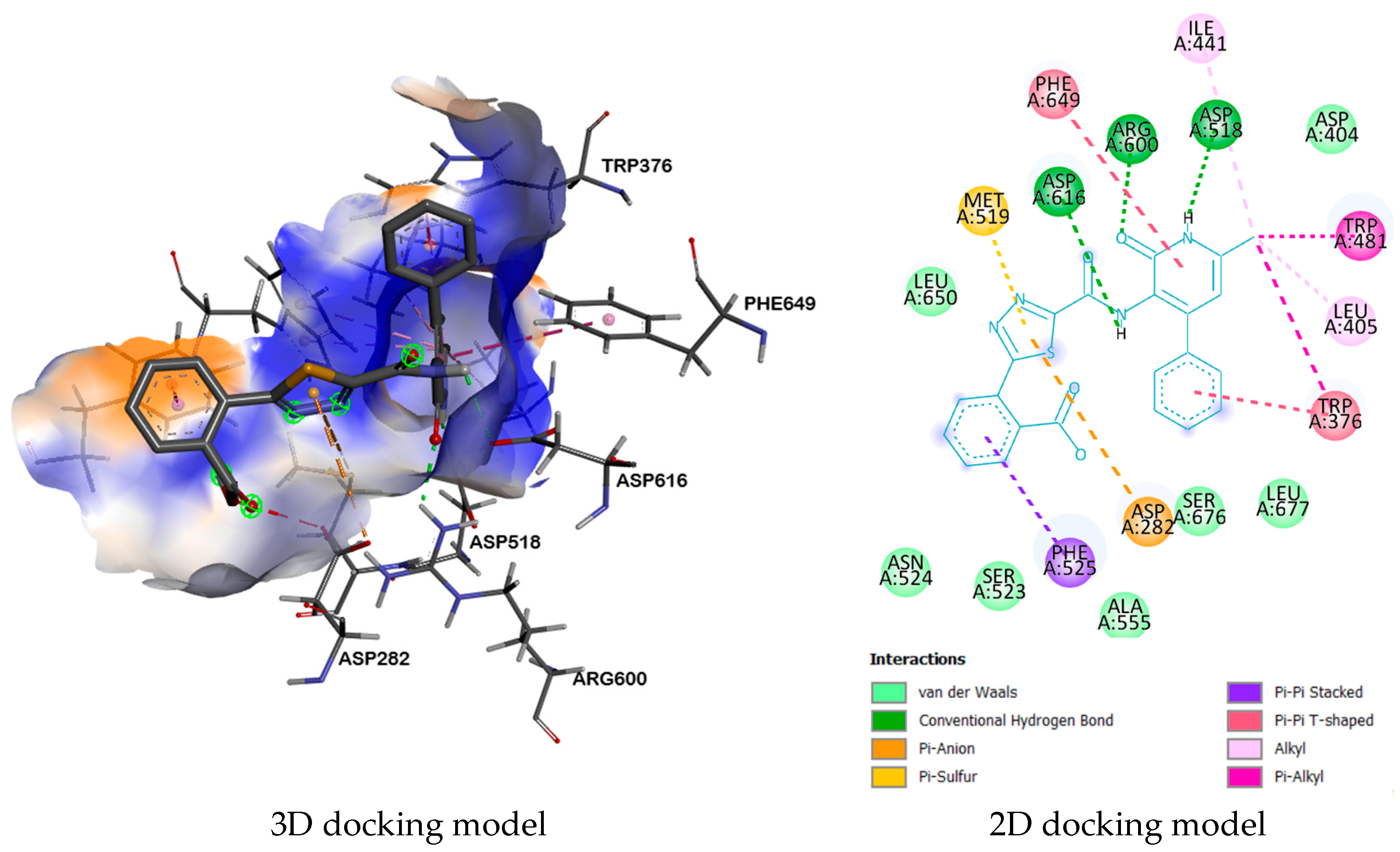
| 9b | 5NN8 | ARG600, ASP518, ASP616 | ASP282, PHE525, TRP376, LEU405, TRP481, ILE441, MET519, PHE649, | LEU677, SER676, ASP404, ALA555, SER523, ASN524, LEU650 |
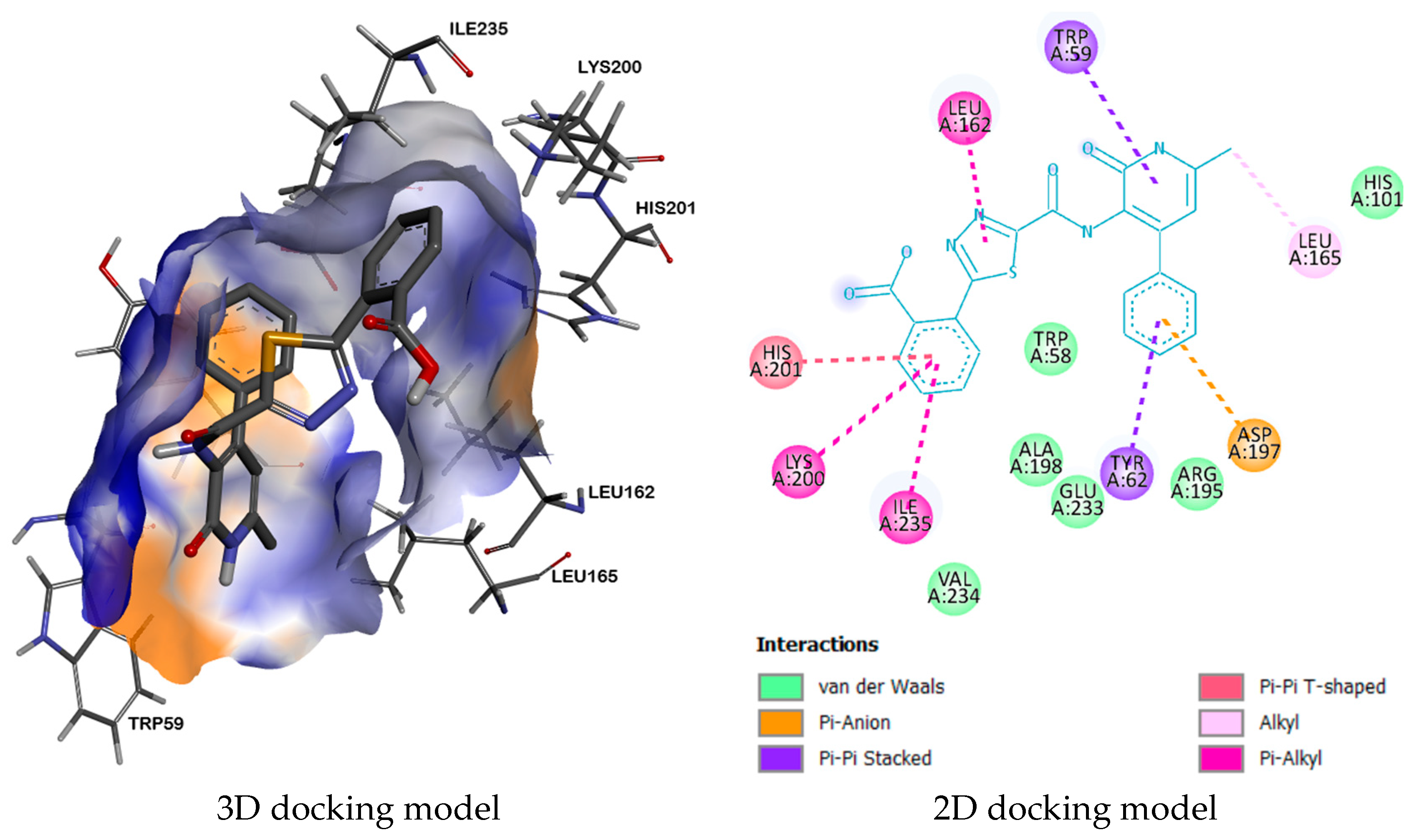
| 2QV4 | - | LEU162, TYR62, LEU165, TRP59, HIS201, LYS200, ILE235, ASP197 | VAL234, ALA198, TRP58, GLU233, ARG195, HIS101 | |
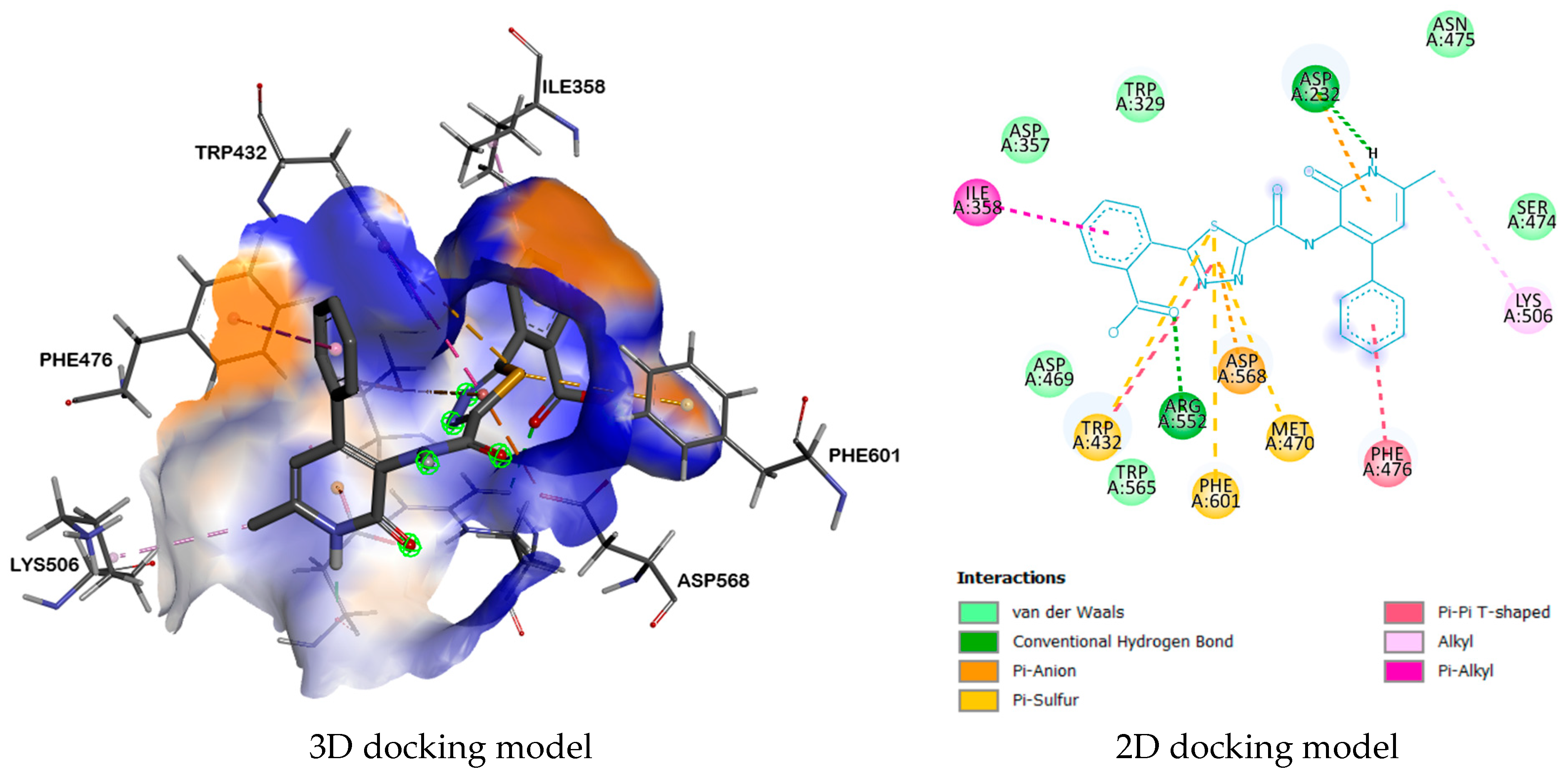
| 3W37 | ASP232, ARG552 | TRP432, PHE601, PHE476, LYS506, MET470, ASP568, ASP232, ILE358 | ASP469, TRP565, SER474, ASN475, TRP329, ASP357. | |
| Ligand | Molecular Weight | miLogP | nHBA | nHBD | nViolation |
|---|---|---|---|---|---|
| 7b | 398.44 | 0.130 | 6 | 3 | 0 |
| 7c | 404.46 | 1.646 | 8 | 3 | 0 |
| 9b | 432.45 | 3.150 | 6 | 3 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shulgau, Z.; Palamarchuk, I.V.; Sergazy, S.; Urazbayeva, A.; Ramankulov, Y.; Kulakov, I.V. Synthesis, Computational Study, and In Vitro α-Glucosidase Inhibitory Action of 1,3,4-Thiadiazole Derivatives of 3-Aminopyridin-2(1H)-ones. Pharmaceuticals 2024, 17, 377. https://doi.org/10.3390/ph17030377
Shulgau Z, Palamarchuk IV, Sergazy S, Urazbayeva A, Ramankulov Y, Kulakov IV. Synthesis, Computational Study, and In Vitro α-Glucosidase Inhibitory Action of 1,3,4-Thiadiazole Derivatives of 3-Aminopyridin-2(1H)-ones. Pharmaceuticals. 2024; 17(3):377. https://doi.org/10.3390/ph17030377
Chicago/Turabian StyleShulgau, Zarina, Irina V. Palamarchuk, Shynggys Sergazy, Assel Urazbayeva, Yerlan Ramankulov, and Ivan V. Kulakov. 2024. "Synthesis, Computational Study, and In Vitro α-Glucosidase Inhibitory Action of 1,3,4-Thiadiazole Derivatives of 3-Aminopyridin-2(1H)-ones" Pharmaceuticals 17, no. 3: 377. https://doi.org/10.3390/ph17030377
APA StyleShulgau, Z., Palamarchuk, I. V., Sergazy, S., Urazbayeva, A., Ramankulov, Y., & Kulakov, I. V. (2024). Synthesis, Computational Study, and In Vitro α-Glucosidase Inhibitory Action of 1,3,4-Thiadiazole Derivatives of 3-Aminopyridin-2(1H)-ones. Pharmaceuticals, 17(3), 377. https://doi.org/10.3390/ph17030377