
Dose Prediction and Pharmacokinetic Simulation of XZP-5610, a Small Molecule for NASH Therapy, Using Allometric Scaling and Physiologically Based Pharmacokinetic Models

Abstract
1. Introduction
2. Results
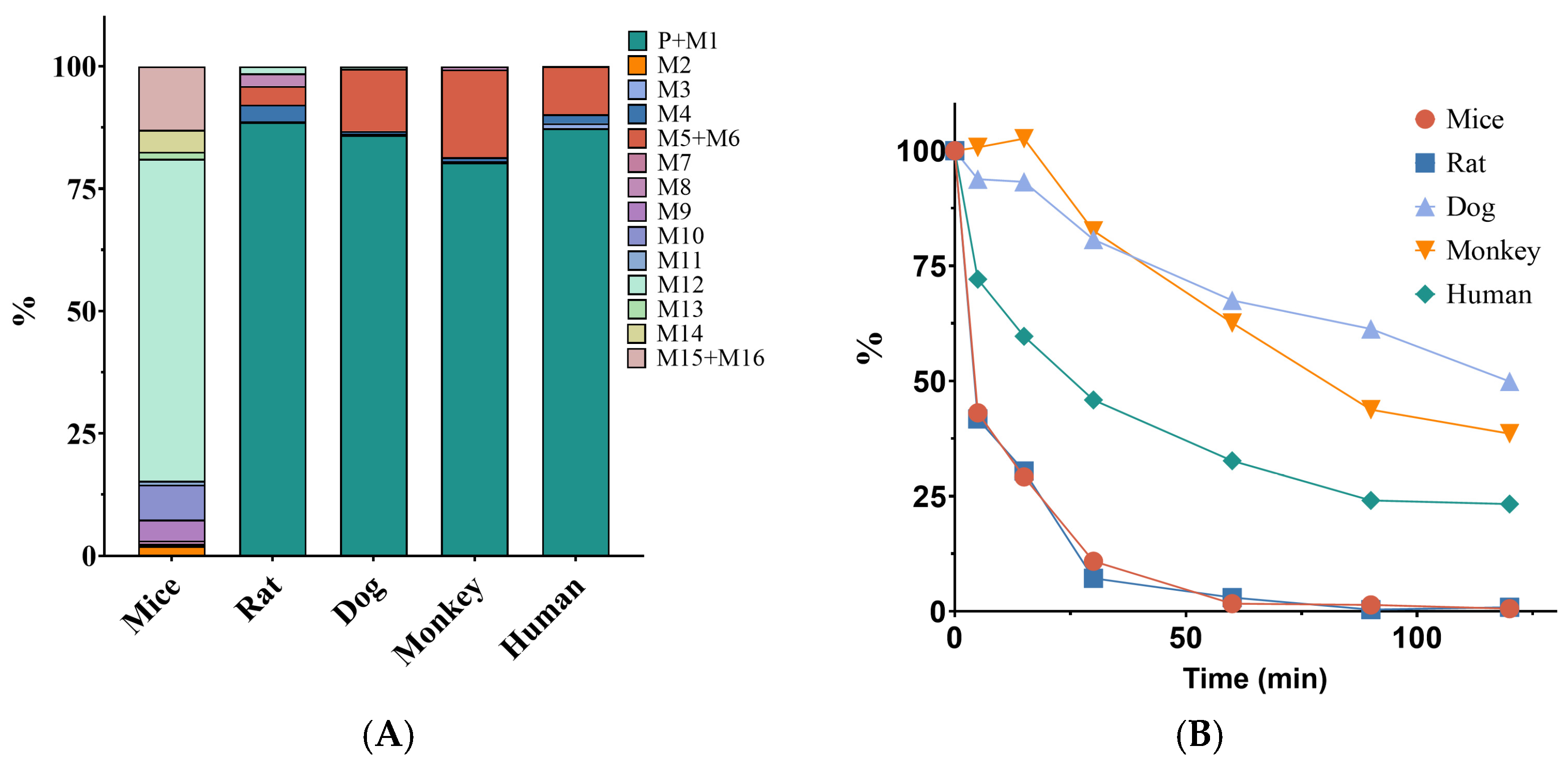
2.1. Selection of Extrapolated Species for XZP-5610
2.2. Parameters of XZP-5610 for Extrapolation and Physiologically Based Pharmacokinetic Model
2.2.1. Apparent Permeability Coefficient of XZP-5610
2.2.2. Plasma Protein Binding of XZP-5610 in Different Species
2.2.3. Blood and Tissue Distribution Coefficients of XZP-5610 in SD Rats
2.2.4. Pharmacokinetic Parameters of XZP-5610 in SD Rats and Beagle Dogs
2.3. Determination of the MABEL and NOAEL Dose of XZP-5610 in Mice, Rats, and Dogs
2.4. Prediction of Human Pharmacokinetic Parameters of XZP-5610
2.5. Prediction of the Doses of XZP-5610 in FIH Trials
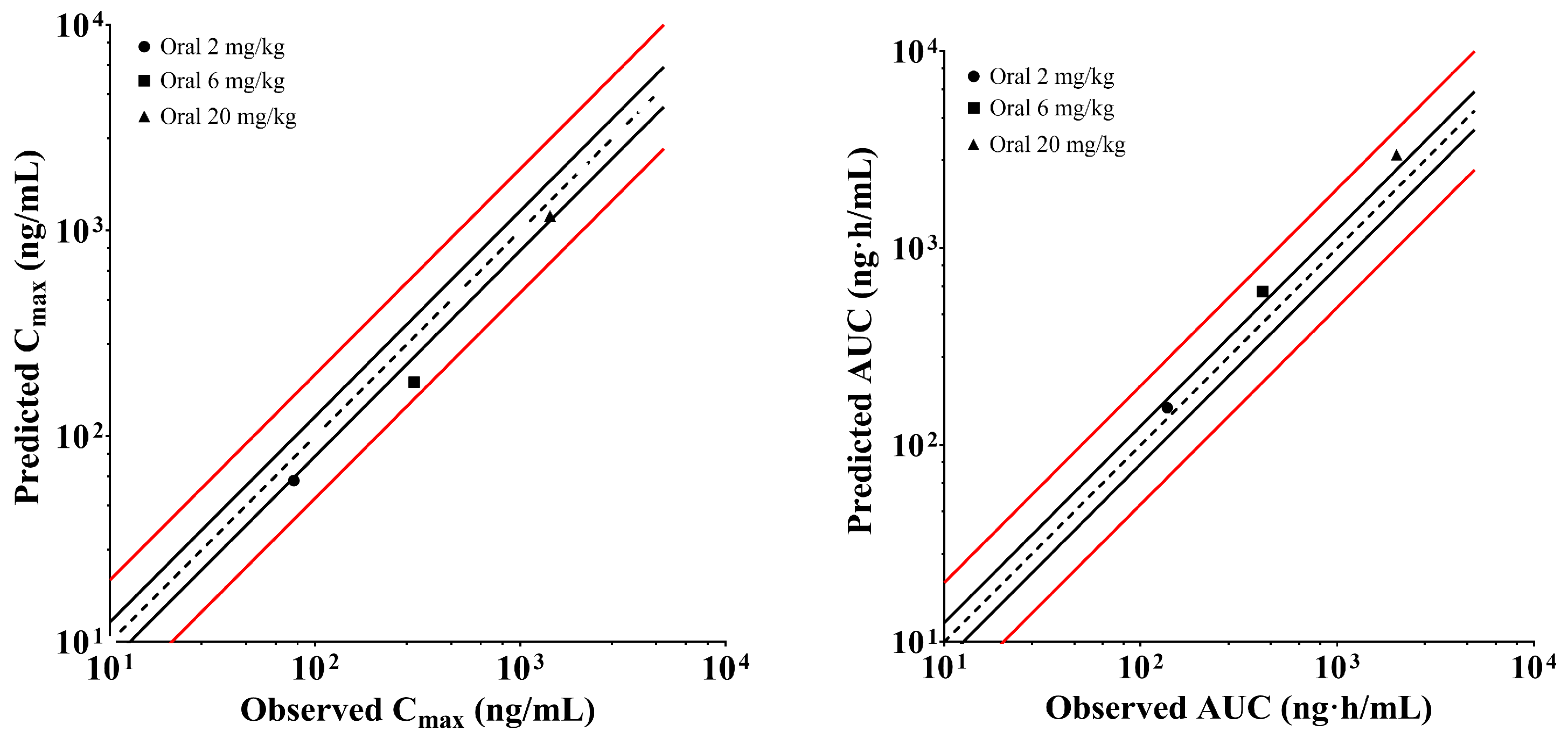
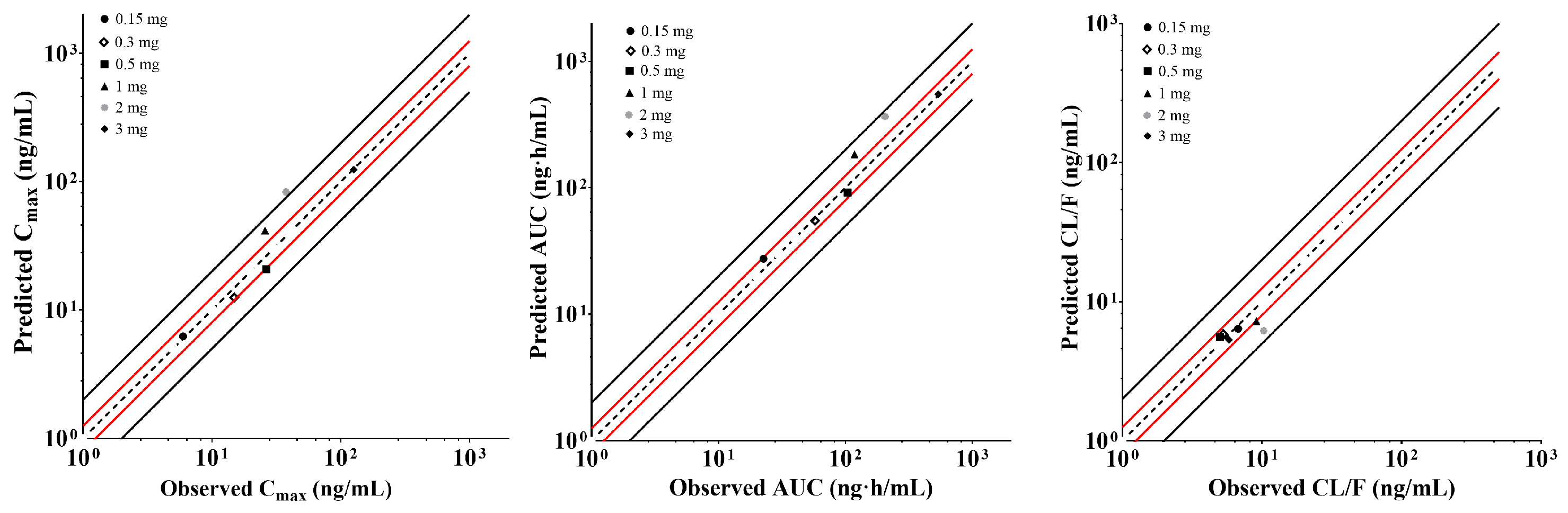
2.6. Establishment and Validation of XZP-5610 PBPK Models in Rats and Healthy Chinese Population
2.7. Prediction of XZP-5610 Liver Concentrations in Healthy Chinese Population Using PBPK Models
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Selection of Extrapolation Species
4.3. Prediction of Human Pharmacokinetic Parameters Using Allometric Scaling Methods
4.3.1. Determination of Pharmacokinetic Parameters for Extrapolation
4.3.2. Extrapolation of Pharmacokinetic Parameters
4.4. Prediction of the Dose Regimen of XZP-5610 in FIH Studies
4.4.1. Determination of the No Observed Adverse Effect Level and Maximum Tolerant Doses in SD Rats and Beagle Dogs
4.4.2. Determination of the Minimum Anticipated Biological Effect Level of XZP-5610 in Mice and Rats
4.4.3. Dose Prediction of XZP-5610 in Healthy Chinese Population
4.5. Establishment and Validation of Physiologically Based Pharmacokinetic Models for XZP-5610 in Rats and Humans
4.5.1. Parameters Employed for the Development of Physiologically Based Pharmacokinetic Model
4.5.2. Establishment and Validation of Physiologically Based Pharmacokinetic Model for XZP-5610 in Rats
4.5.3. Establishment and Validation of Physiologically Based Pharmacokinetic Model for XZP-5610 in Healthy Chinese Adults
4.6. Prediction of XZP-5610 Concentration in Human Livers Using the Physiologically Based Pharmacokinetic Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Cao, H.X.; Li, F.; Cai, X.B.; Ao, Q.H.; Gao, Y.; Fan, J.G. New risk-scoring system including non-alcoholic fatty liver disease for predicting incident type 2 diabetes in East China: Shanghai Baosteel Cohort. J. Diabetes Investig. 2016, 7, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Poulsen, K.L.; Wu, L.; Liu, S.; Miyata, T.; Song, Q.; Wei, Q.; Zhao, C.; Lin, C.; Yang, J. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target. Ther. 2022, 7, 287. [Google Scholar] [CrossRef]
- Adorini, L.; Trauner, M. FXR agonists in NASH treatment. J. Hepatol. 2023, 79, 1317–1331. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Ferrell, J.M.; Pathak, P.; Boehme, S.; Gilliland, T.; Chiang, J.Y.L. Deficiency of Both Farnesoid X Receptor and Takeda G Protein-Coupled Receptor 5 Exacerbated Liver Fibrosis in Mice. Hepatology 2019, 70, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhang, Y.; Ding, H.; Wang, X.; Chen, L.; Jiang, H.; Shen, X. Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cell Physiol. Biochem. 2008, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Wahlström, A.; Marschall, H.U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Kremoser, C. FXR agonists for NASH: How are they different and what difference do they make? J. Hepatol. 2021, 75, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Zollner, G.; Marschall, H.U.; Wagner, M. Recent advances on FXR-targeting therapeutics. Mol. Cell Endocrinol. 2022, 552, 111678. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration; Center for Drug Evaluation and Research. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; US Food and Drug Administration: Silver Spring, ML, USA, 2008.
- Shen, J.; Swift, B.; Mamelok, R.; Pine, S.; Sinclair, J.; Attar, M. Design and Conduct Considerations for First-in-Human Trials. Clin. Transl. Sci. 2019, 12, 6–19. [Google Scholar] [CrossRef]
- Tian, S.Y.; Chen, S.M.; Pan, C.X.; Li, Y. FXR: Structures, biology, and drug development for NASH and fibrosis diseases. Acta Pharmacol. Sin. 2022, 43, 1120–1132. [Google Scholar] [CrossRef]
- Hambruch, E.; Miyazaki-Anzai, S.; Hahn, U.; Matysik, S.; Boettcher, A.; Perović-Ottstadt, S.; Schlüter, T.; Kinzel, O.; Krol, H.D.; Deuschle, U.; et al. Synthetic farnesoid X receptor agonists induce high-density lipoprotein-mediated transhepatic cholesterol efflux in mice and monkeys and prevent atherosclerosis in cholesteryl ester transfer protein transgenic low-density lipoprotein receptor (−/−) mice. J. Pharmacol. Exp. Ther. 2012, 343, 556–567. [Google Scholar] [CrossRef]
- Rasmussen, D.G.K.; Anstee, Q.M.; Torstenson, R.; Golding, B.; Patterson, S.D.; Brass, C.; Thakker, P.; Harrison, S.; Billin, A.N.; Schuppan, D.; et al. NAFLD and NASH biomarker qualification in the LITMUS consortium—Lessons learned. J. Hepatol. 2023, 78, 852–865. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Walsh, D.T.; Warner, T.D. Expression and activation of the farnesoid X receptor in the vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 3668–3673. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, E.; Panzitt, K.; Marschall, H.U.; Thallinger, G.G.; Wagner, M. Meta-analysis and Consolidation of Farnesoid X Receptor Chromatin Immunoprecipitation Sequencing Data Across Different Species and Conditions. Hepatol. Commun. 2021, 5, 1721–1736. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, F.; Waters, N.J.; Argikar, U.A.; Dennehy, M.K.; Zhan, J.; Gunduz, M.; Harriman, S.P.; Berellini, G.; Rajlic, I.L.; Obach, R.S. Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 1: Volume of distribution at steady state. J. Clin. Pharmacol. 2013, 53, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, F.; Waters, N.J.; Argikar, U.A.; Dennehy, M.K.; Zhan, J.; Gunduz, M.; Harriman, S.P.; Berellini, G.; Liric Rajlic, I.; Obach, R.S. Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 2: Clearance. J. Clin. Pharmacol. 2013, 53, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Anakk, S.; Ku, C.Y.; Vore, M.; Strobel, H.W. Insights into gender bias: Rat cytochrome P450 3A9. J. Pharmacol. Exp. Ther. 2003, 305, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.W.; Choi, M.R.; Kwon, Y.S.; Jeong, J.S.; Son, M.; Kang, H.E. Gender differences in corydaline pharmacokinetics in rats. Xenobiotica 2015, 45, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.M.; Kuffel, M.J.; Ruben, S.L.; Morales, J.J.; Rinehart, K.L.; Squillace, D.P.; Ames, M.M. Rat and human liver cytochrome P-450 isoform metabolism of ecteinascidin 743 does not predict gender-dependent toxicity in humans. Clin. Cancer Res. 2002, 8, 2952–2962. [Google Scholar]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar]
- Sohlenius-Sternbeck, A.K. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol. In Vitro 2006, 20, 1582–1586. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Route | Dose (mg/kg) | Gender | Tmax (h) | T1/2 (h) | Cmax (ng/mL) | AUC(0–t) (h·ng/mL) | Cl (L/h/kg) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| I.V. | 1 | male | NA | 2.9 ± 2.3 | NA | 488.3 ± 31.1 | 2.0 ± 0.1 | 0.9 ± 0.5 | NA |
| I.V. | 1 | female | NA | 2.2 ± 1.5 | NA | 569.8 ± 151.4 | 1.8 ± 0.4 | 0.6 ± 0.1 | NA |
| PO | 2 | male | 0.5 ± 0.0 | 1.3 ± 0.2 | 56.6 ± 11.6 | 94.9 ± 7.8 | NA | NA | 9.6 ± 0.8 |
| PO | 1 | female | 0.5 ± 0.0 | 1.3 ± 0.2 | 50.4 ± 15.0 | 89.0 ± 25.5 | NA | NA | 15.6 ± 4.5 |
| PO | 6 | male | 0.5 ± 0.0 | 1.0 ± 0.2 | 255.8 ± 47.7 | 293.8 ± 43.4 | NA | NA | 9.9 ± 1.5 |
| PO | 3 | female | 0.8 ± 0.3 | 1.2 ± 0.4 | 175.6 ± 17.9 | 271.1 ± 45.8 | NA | NA | 15.9 ± 2.7 |
| PO | 20 | male | 1.0 ± 0.0 | 1.4 ± 0.5 | 762.9 ± 283.0 | 1276.6 ± 447.0 | NA | NA | 12.9 ± 4.5 |
| PO | 10 | female | 0.5 ± 0.0 | 1.9 ± 0.9 | 1014.0 ± 544.6 | 1367.8 ± 341.4 | NA | NA | 24.1 ± 6.0 |
| Route | Dose (mg/kg) | Gender | Tmax (h) | T1/2 (h) | Cmax (ng/mL) | AUC(0–t) (h·ng/mL) | Cl (L/h/kg) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| I.V. | 0.2 | Male | NA | 7.9 ± 6.7 | NA | 864.4 ± 56.8 | 0.2 ± 0.03 | 1.1 ± 0.6 | NA |
| Female | 5.5 ± 2.5 | 871.4 ± 375.8 | 0.2 ± 0.1 | 1.0 ± 0.5 | |||||
| PO | 0.05 | Male | 1.7 ± 0.6 | 10.1 ± 3.4 | 15.8 ± 5.5 | 88.2 ± 10.1 | NA | NA | 43.4 ± 4.0 |
| Female | 1.7 ± 0.6 | 13.1 ± 5.0 | 21.1 ± 9.6 | 125.9 ± 63.1 | 82.9 ± 59.8 | ||||
| PO | 0.2 | Male | 2.0 ± 1.7 | 6.1 ± 1.7 | 126.0 ± 77.5 | 508.3 ± 155.0 | NA | NA | 51.2 ± 21.1 |
| Female | 1.3 ± 0.6 | 6.5 ± 1.8 | 67.3 ± 12.9 | 319.4 ± 89.6 | 38.2 ± 12.2 | ||||
| PO | 0.8 | Male | 1.0 ± 0.0 | 5.8 ± 1.1 | 533.7 ± 277.7 | 2140.0 ± 1069.5 | NA | NA | 61.4 ± 28.9 |
| Female | 1.3 ± 0.6 | 7.0 ± 2.4 | 626.7 ± 144.8 | 2279.2 ± 383.2 | 67.1 ± 11.1 |
| Predicted Parameter | Predicted Method | Value |
|---|---|---|
| Human CLi.v (mL/min) | Single-species scaling (Rat) | 293 |
| Single-species scaling (Dog) | 96.4 | |
| Singl-species allometric scaling (Rat) | 75.4 | |
| Single-species allometric scaling (Dog) | 92.3 | |
| Two-species allometric scaling | 177 | |
| Fu-corrected intercept (Rat) | 58.6 | |
| Fu-corrected intercept (Dog) | 181 | |
| Hepatic blood flow (Rat) | 845 | |
| Hepatic blood flow (Dog) | 184 | |
| Human Vss (L) | Øie–Tozer | 17.1 |
| Single-species allometric scaling (Rat) | 45.3 | |
| Single-species allometric scaling (Dog) | 63.0 |
| Species | NOAEL Dose (mg/kg) | HED (mg) | Safety Factor | MRSD (mg) |
|---|---|---|---|---|
| SD rat (male) | 1.5 | 14.4 | 10 | 1.44 |
| SD rat (female) | 1 | 9.60 | 10 | 0.960 |
| Beagle dog | 0.05 | 1.62 | 10 | 0.162 |
| Species | Gender | NOAEL Dose (mg/kg) | Steady AUC0–24 (ng·h/mL) | Human Equivalent AUC0–24 (ng·h/mL) | HED (mg) | SF | MRSD (mg) |
|---|---|---|---|---|---|---|---|
| SD rat | Male | 1.5 | 75.2 | 489 | 7.07 | 10 | 0.71 |
| Female | 1 | 46.4 | 302 | 4.36 | 10 | 0.44 | |
| Beagle dog | Male | 0.05 | 127.6 | 191 | 2.77 | 10 | 0.28 |
| Female | 0.05 | 152.1 | 228 | 3.30 | 10 | 0.33 |
| Property (Units) | Values Used in the Model | Data Source | Descriptions |
|---|---|---|---|
| MW(g/mol) | 556.46 | - | Molecular weight |
| pKa (Acid) | 10.13, 3.39 | Predicted | Acid dissociation constant |
| LogP | 3.2 | Optimized | Lipophilicity |
| Solubility (μg/mL) | 25 | Optimized | Solubility at pH 6.0 |
| Papp (×10−6 cm/s) | 10.1 | Determined | Caco-2 apparent permeability |
| fup | 0.013 a, 0.002 b | Determined | Fraction of free drug in plasma |
| BP | 0.58 | Determined | Blood-to-plasma concentration ratio |
| CL (L/h/kg) | 1.93 a, 0.138 b | Determined | Clearance |
| CLB CLint,u (L/h/kg) | 0.18 | Optimized | Biliary clearance |
| KP,L | 37.0 | Optimized | Liver-to-plasma partition coefficient |
| Partition coefficients | Rodgers and Rowland | Optimized | Calculation method from cell to plasma coefficients |
| Cellular permeabilities | PK-Sim Standard | Optimized | Permeability calculation method across cell |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Feng, F.; Wang, X.; Liang, H.; Yao, X.; Liu, D. Dose Prediction and Pharmacokinetic Simulation of XZP-5610, a Small Molecule for NASH Therapy, Using Allometric Scaling and Physiologically Based Pharmacokinetic Models. Pharmaceuticals 2024, 17, 369. https://doi.org/10.3390/ph17030369
Zhang L, Feng F, Wang X, Liang H, Yao X, Liu D. Dose Prediction and Pharmacokinetic Simulation of XZP-5610, a Small Molecule for NASH Therapy, Using Allometric Scaling and Physiologically Based Pharmacokinetic Models. Pharmaceuticals. 2024; 17(3):369. https://doi.org/10.3390/ph17030369
Chicago/Turabian StyleZhang, Lei, Feifei Feng, Xiaohan Wang, Hao Liang, Xueting Yao, and Dongyang Liu. 2024. "Dose Prediction and Pharmacokinetic Simulation of XZP-5610, a Small Molecule for NASH Therapy, Using Allometric Scaling and Physiologically Based Pharmacokinetic Models" Pharmaceuticals 17, no. 3: 369. https://doi.org/10.3390/ph17030369
APA StyleZhang, L., Feng, F., Wang, X., Liang, H., Yao, X., & Liu, D. (2024). Dose Prediction and Pharmacokinetic Simulation of XZP-5610, a Small Molecule for NASH Therapy, Using Allometric Scaling and Physiologically Based Pharmacokinetic Models. Pharmaceuticals, 17(3), 369. https://doi.org/10.3390/ph17030369