
The Effects of Selected Extraction Methods and Natural Deep Eutectic Solvents on the Recovery of Active Principles from Aralia elata var. mandshurica (Rupr. & Maxim.) J. Wen: A Non-Targeted Metabolomics Approach

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results and Discussion
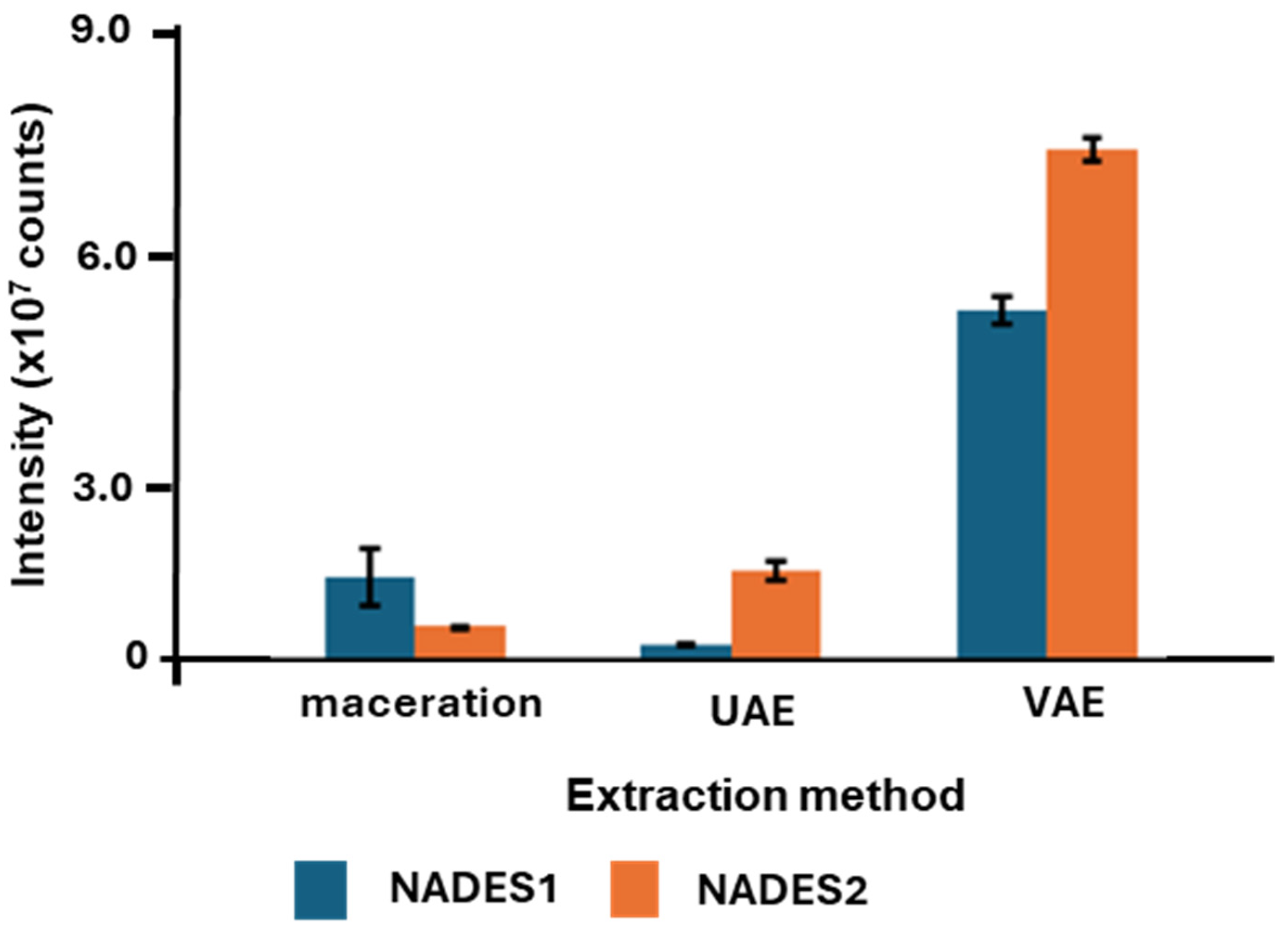
2.1. Comparison of the NADES Extraction Efficiencies and Extraction Method Performance
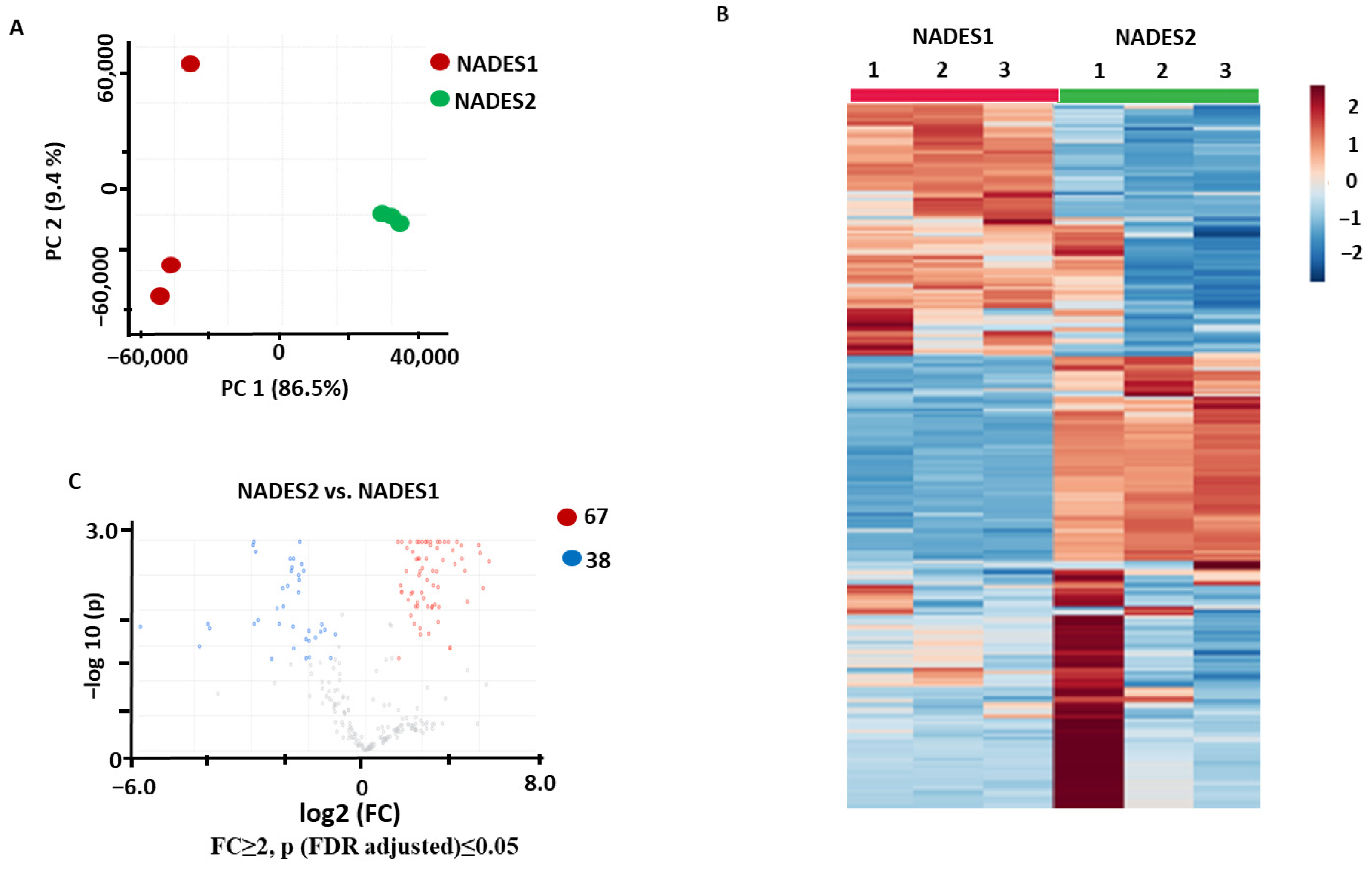
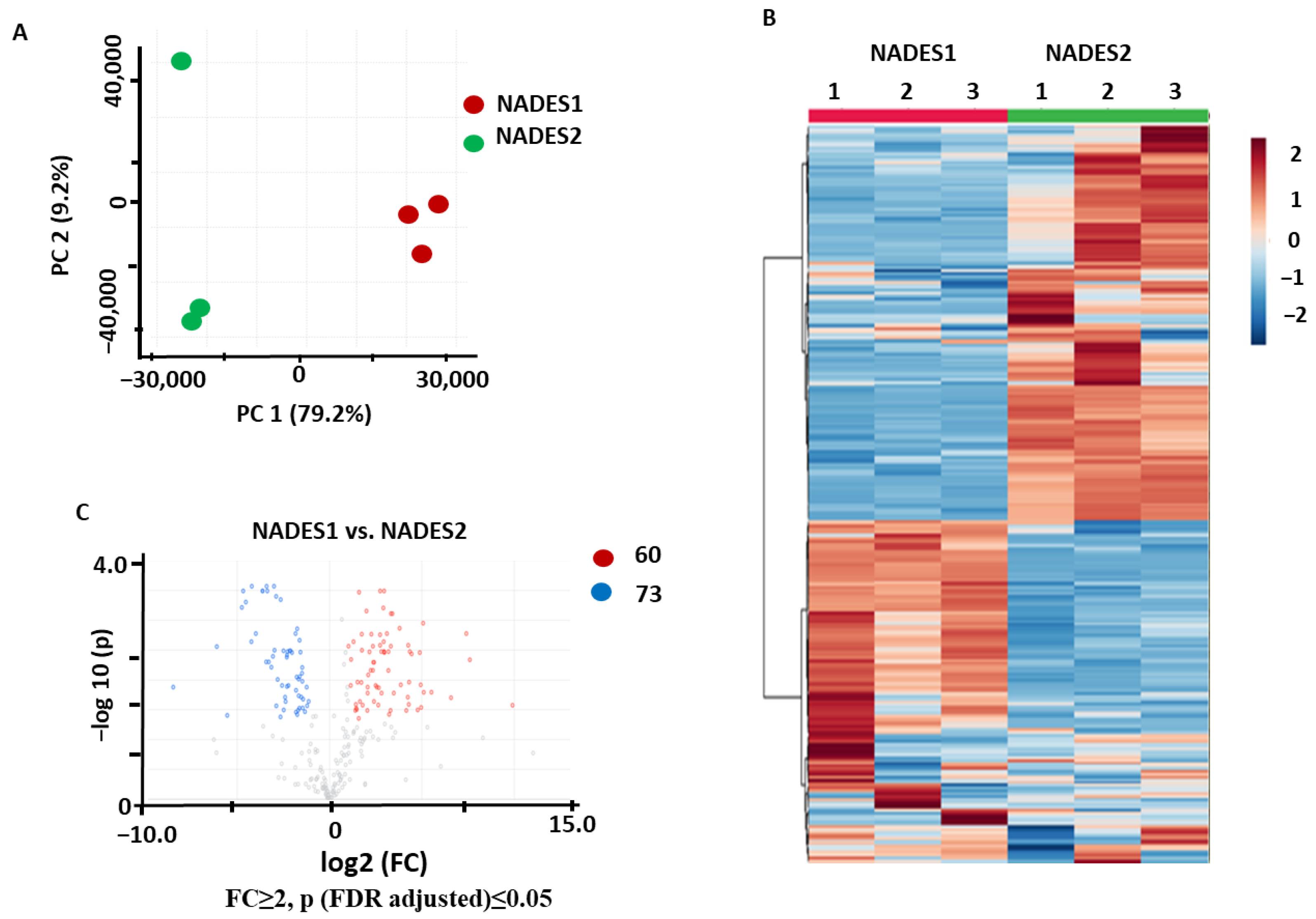
2.1.1. Comparison of Individual NADES in Terms of Their Efficiencies as the Extractants for Maceration
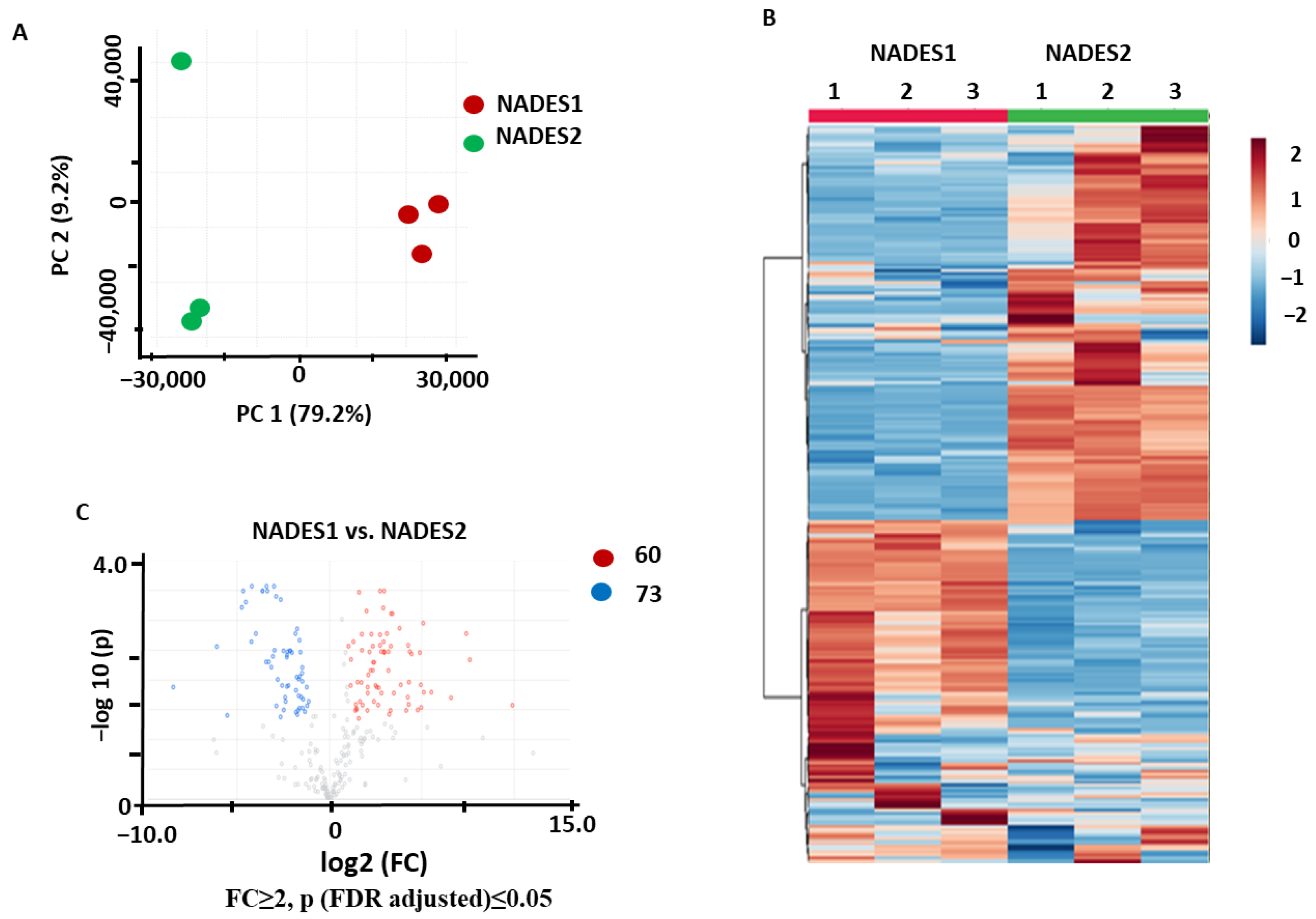
2.1.2. Comparison of the NADES Efficiencies Observed with UAE
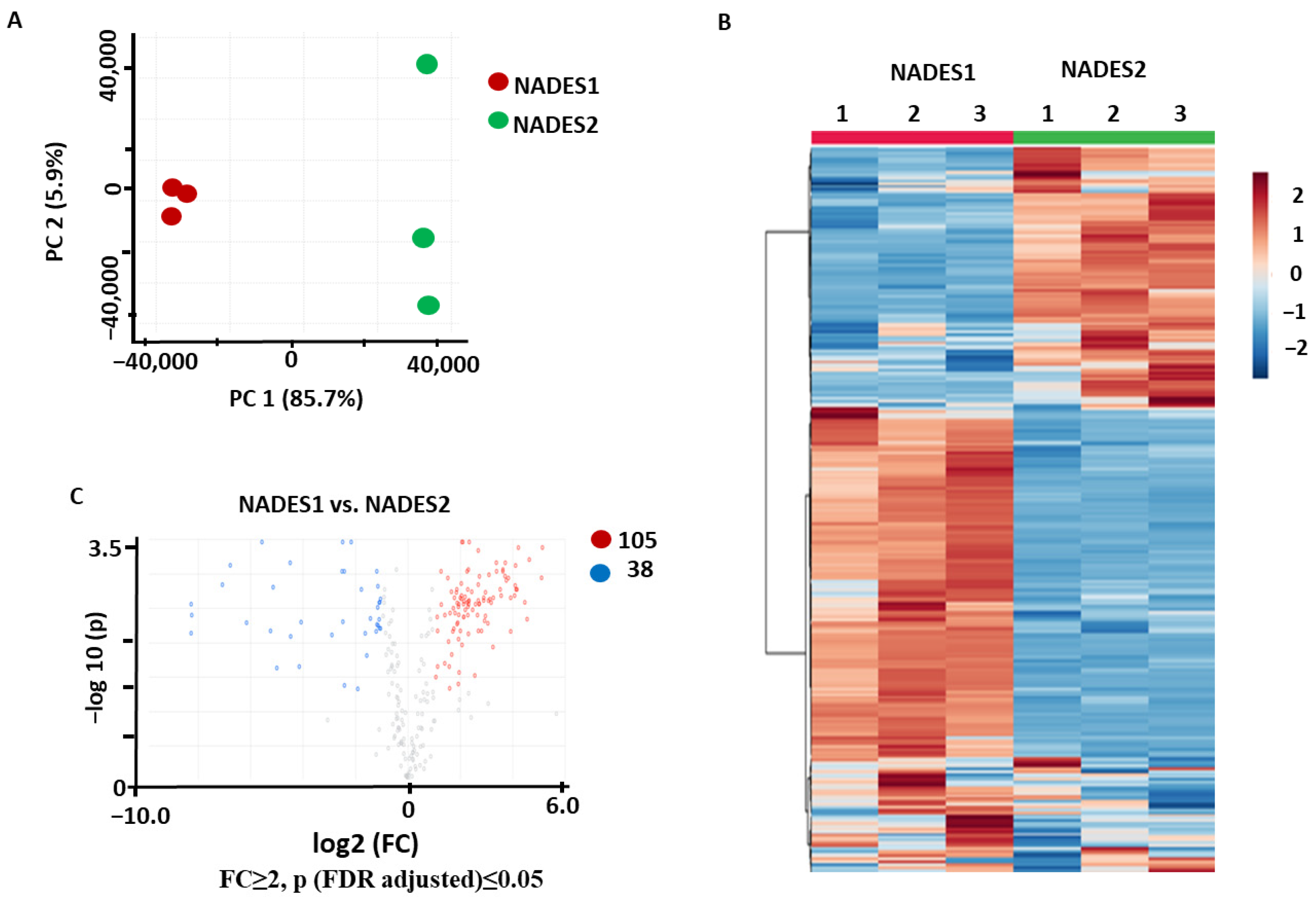
2.1.3. Comparison of the NADES Efficiencies Observed with VAE
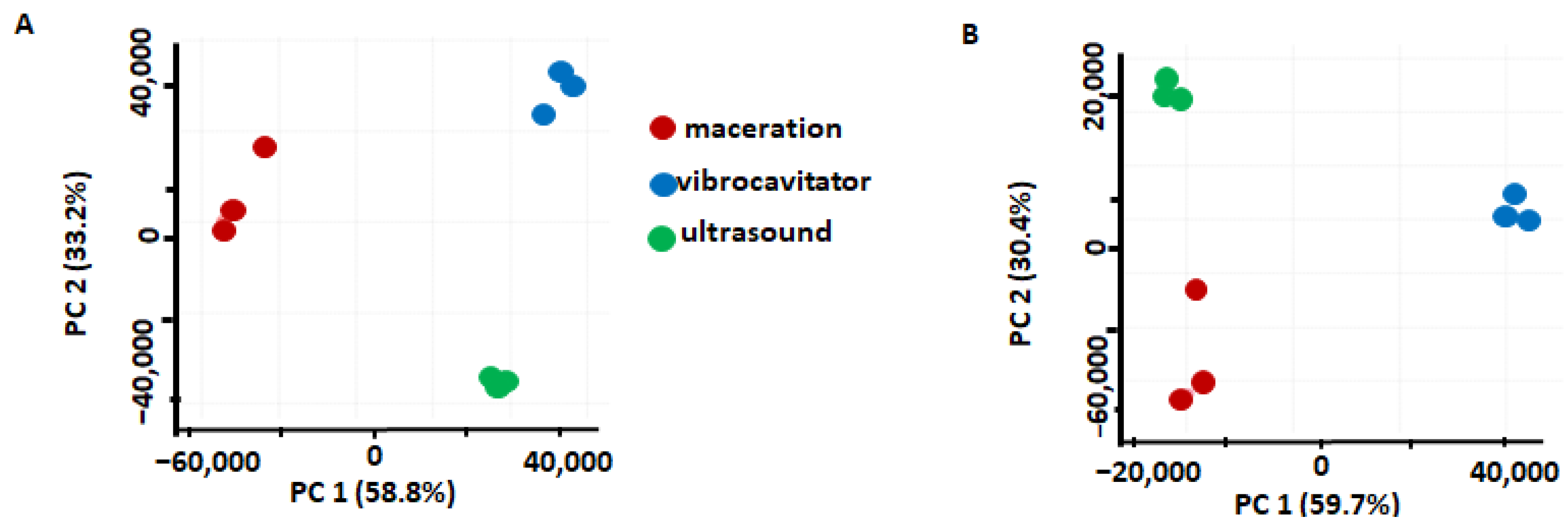
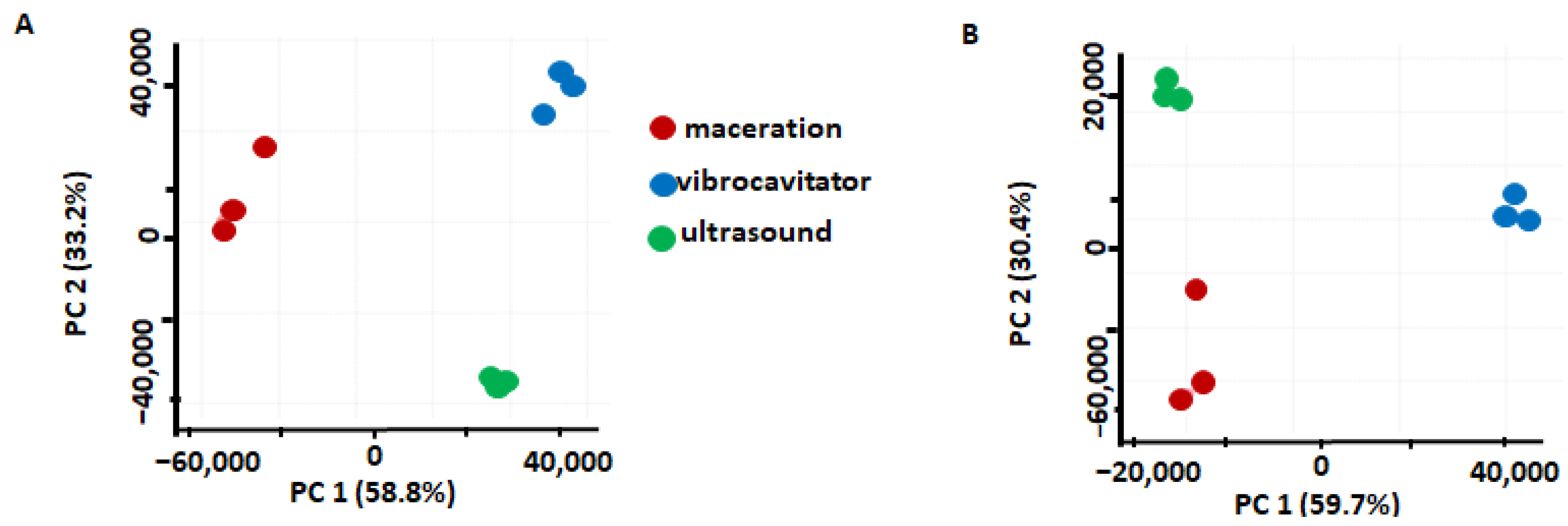
2.2. Comparison of the Extraction Methods
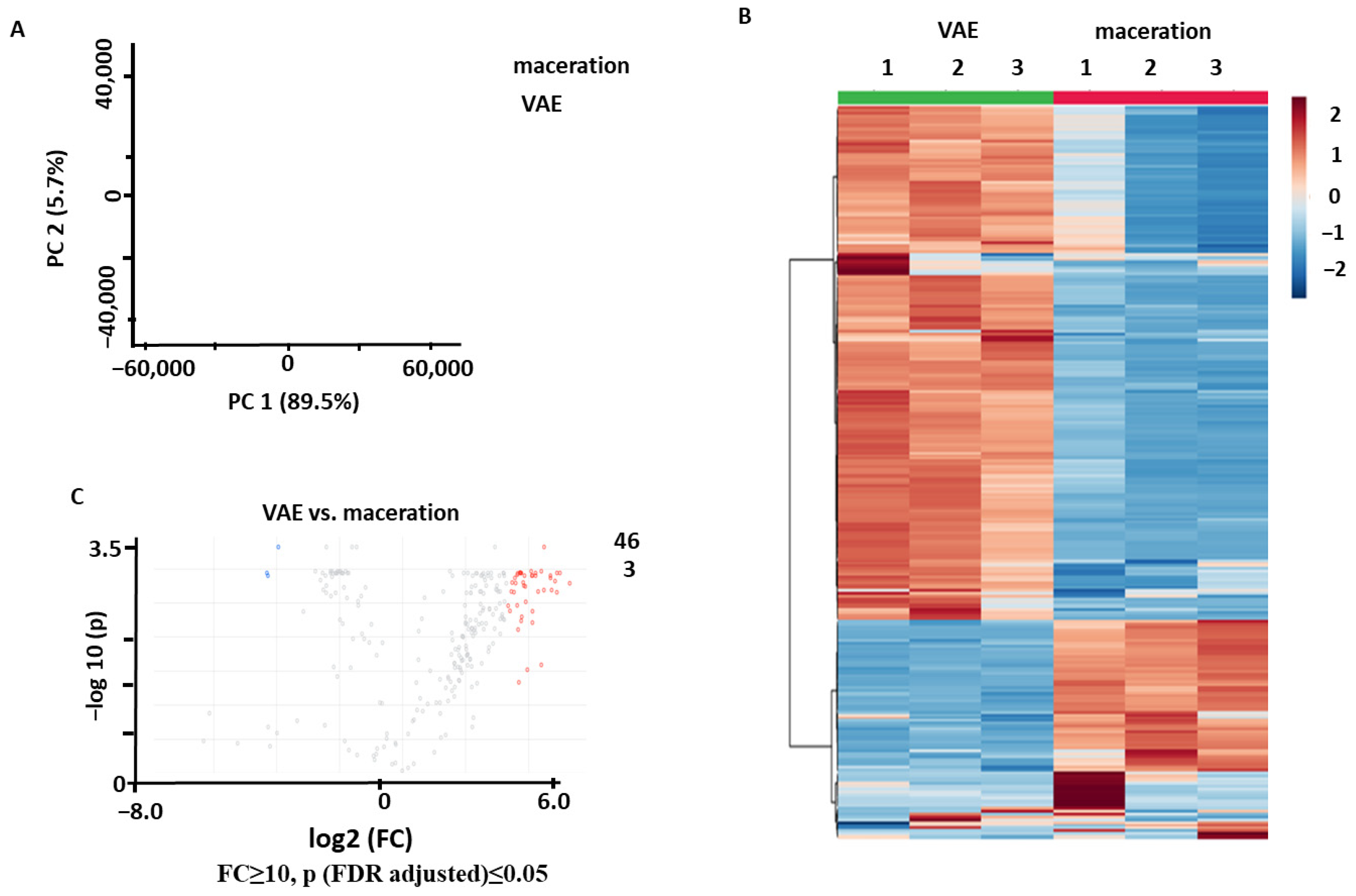
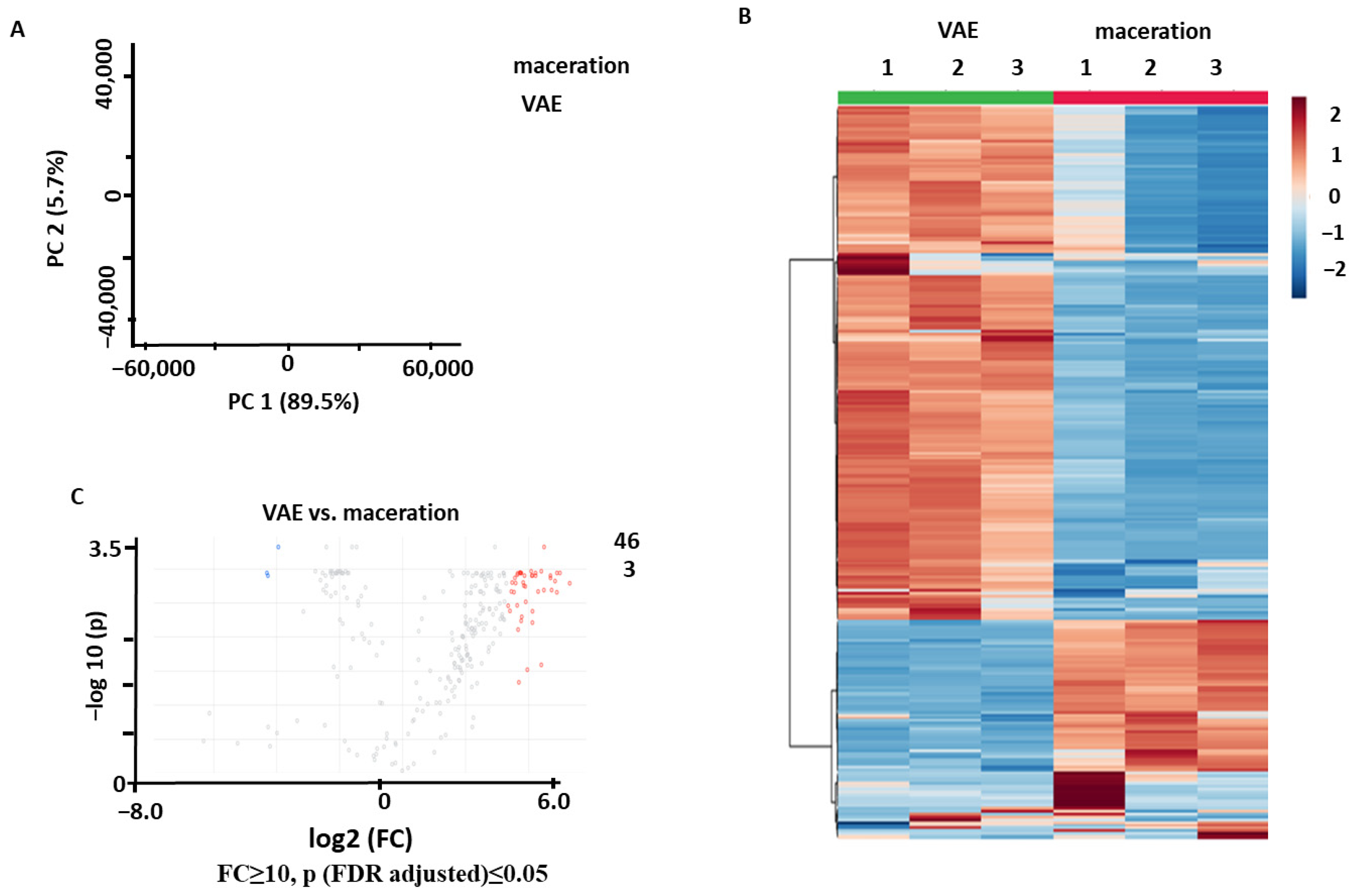
2.2.1. Comparison of VAE and Maceration
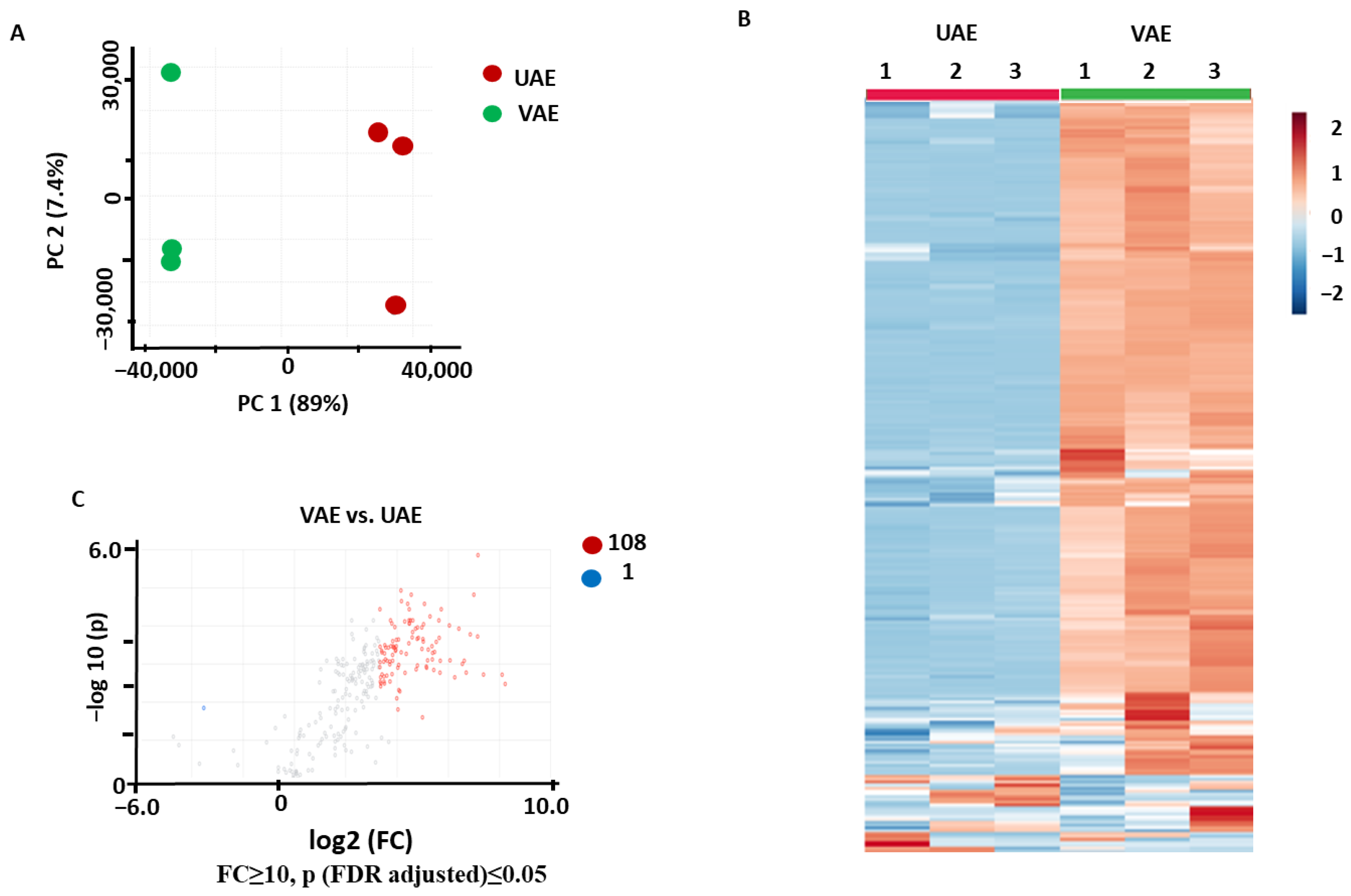
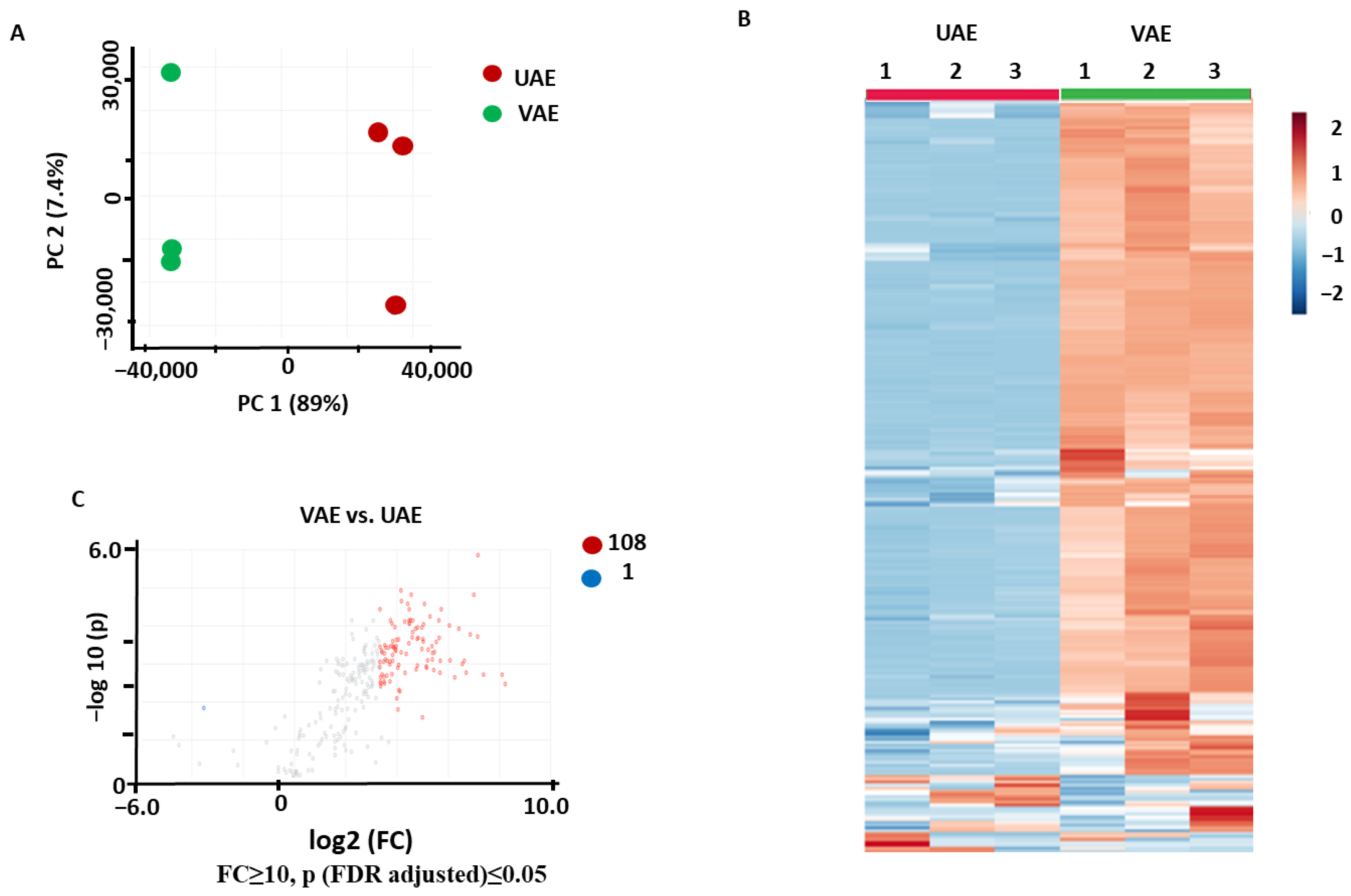
2.2.2. Comparison of VAE and UAE
2.3. Identification of Secondary Metabolites in the NADES Extracts from Aralia elata var. Mandshurica (Rupr. & Maxim.) J. Wen
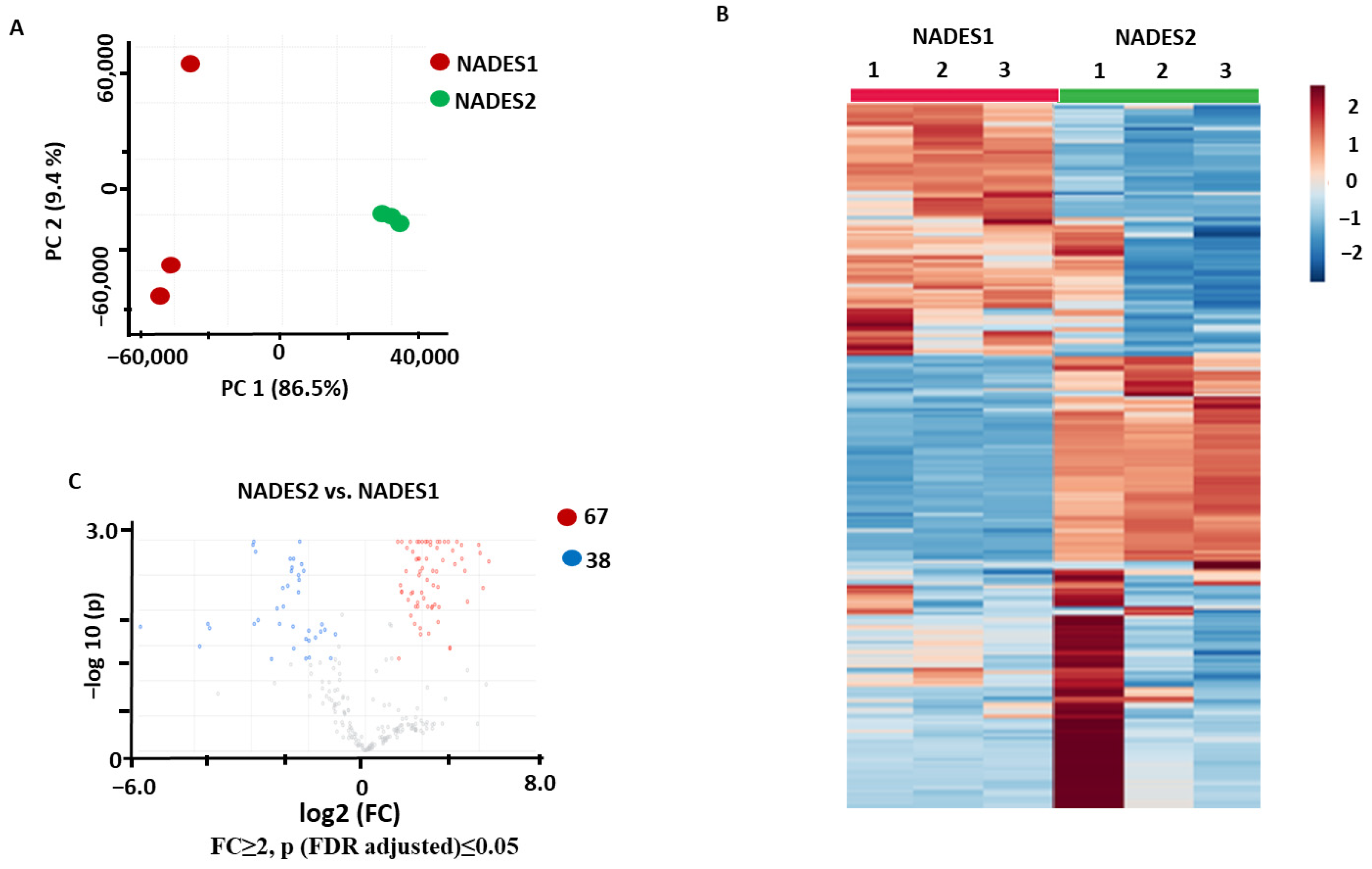
2.3.1. Identification of the Metabolites Annotated as Differentially Abundant in the NADES1 and NADES2 Extracts Obtained by Maceration
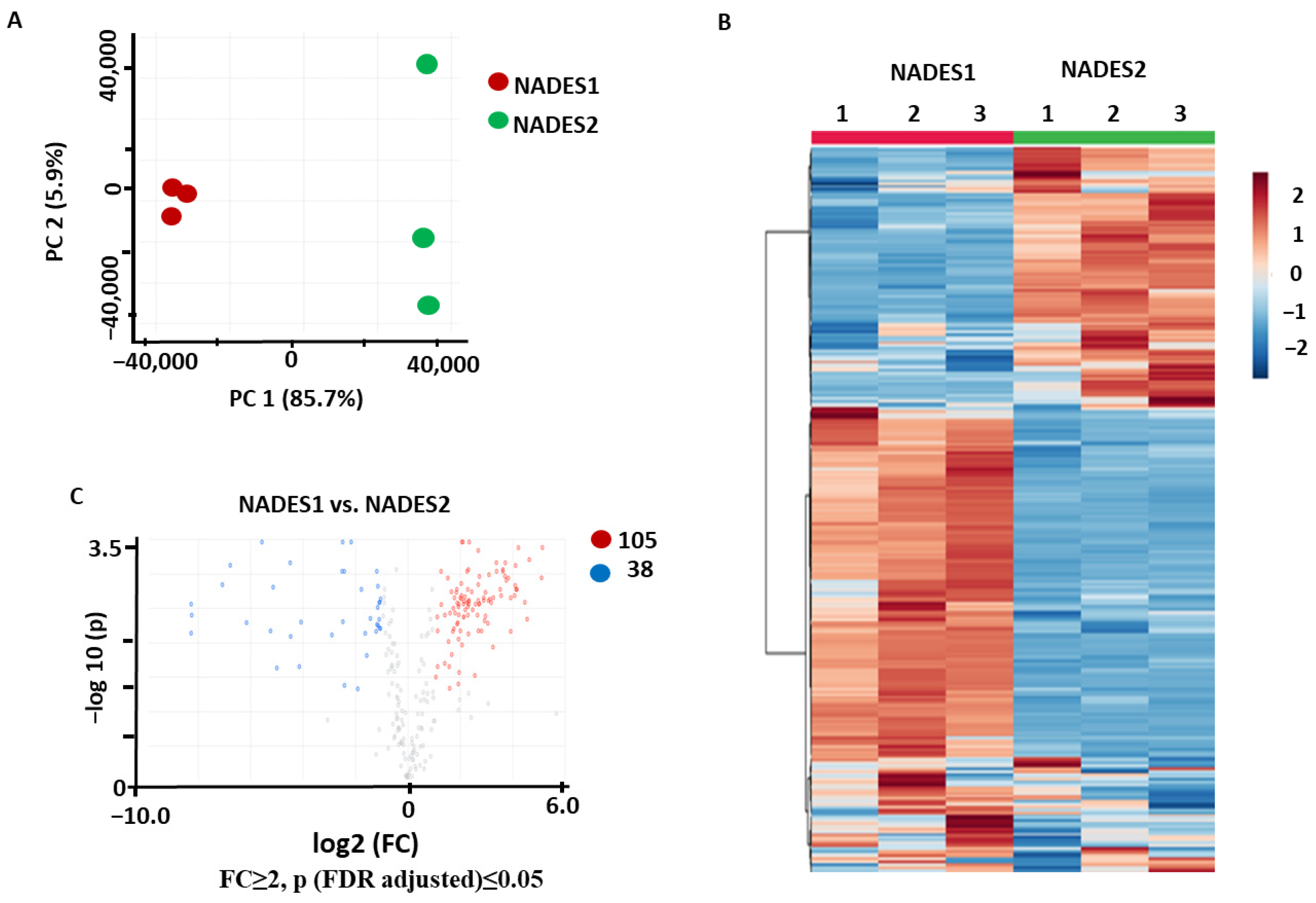
2.3.2. Identification of the Metabolites Annotated as Differentially Abundant in the NADES1 Extracts Obtained by VAE and Maceration
2.4. The Cross-Validation of the Quantitative Results Using the Targeted Relative Quantification Strategy
3. Materials and Methods
3.1. Chemicals
3.2. Raw Material of Plants and the Composition and Preparation of NADESs
3.3. Extraction Procedures
3.4. RP-UHPLC-ESI-LIT-Orbitrap-MS/MS
3.5. Analysis of Secondary Metabolites in the Aralia elata Root Extracts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, Y.H.; Spronsen, J.V.; Dai, Y.; Verberne, M.; Hollmann, F.; Arends, I.W.C.E.; Witkamp, G.J.; Verpoorte, R. Are Natural Deep Eutectic Solvents the Missing Link in Understanding Cellular Metabolism and Physiology? Plant Physiol. 2011, 156, 1701–1705. [Google Scholar] [CrossRef]
- Dai, Y.; Van Spronsen, J.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Natural Deep Eutectic Solvents as New Potential Media for Green Technology. Anal. Chim. Acta 2013, 766, 61–68. [Google Scholar] [CrossRef]
- Fuad, F.M.; Nadzir, M.M.; Harun, A. Hydrophilic natural deep eutectic solvent: A review on physicochemical properties and extractability of bioactive compounds. J. Mol. Liq. 2021, 339, 116923. [Google Scholar] [CrossRef]
- Da Silva, R.F.; Carneiro, C.N.; Cheila, C.B.; de Sousa, C.B.D.C.; Gomez, F.J.V.; Espino, M.; Boiteux, J.; Fernández, M.D.L.Á.; Silva, M.F.; Dias, F.D.S. Sustainable extraction bioactive compounds procedures in medicinal plants based on the principles of green analytical chemistry: A review. Microchem. J. 2022, 175, 107184. [Google Scholar] [CrossRef]
- Wawoczny, A.; Gillner, D. The Most Potent Natural Pharmaceuticals, Cosmetics, and Food Ingredients Isolated from Plants with Deep Eutectic Solvents. J. Agric. Food Chem. 2023, 71, 10877–10900. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Lee, S.; Kang, S.-S.; Shin, H.-S. Selected Commercial Plants: A Review of Extraction and Isolation of Bioactive Compounds and Their Pharmacological Market Value. Trends Food Sci. Technol. 2018, 82, 89–109. [Google Scholar] [CrossRef]
- Flisyuk, E.V.; Belokurov, S.S.; Narkevich, I.A.; Shikov, A.N.; Flisyuk, O.M.; Ivkin, D.Y. Kinetics of Extraction of Dioscine from Vegetable Raw Materials in Vibrocavitation Homogenizer. Drug Dev. Regist. 2020, 9, 77–81. [Google Scholar] [CrossRef]
- Quitério, E.; Grosso, C.; Ferraz, R.; Delerue-Matos, C.; Soares, C. A Critical Comparison of the Advanced Extraction Techniques Applied to Obtain Health-Promoting Compounds from Seaweeds. Mar. Drugs 2022, 20, 677. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; P, N.; Kumar, M.; Jose, A.; Tomer, V.; Oz, E.; Proestos, C.; Zeng, M.; Elobeid, T.; K, S.; et al. Major Phytochemicals: Recent Advances in Health Benefits and Extraction Method. Molecules 2023, 28, 887. [Google Scholar] [CrossRef] [PubMed]
- Shikov, A.N.; Mikhailovskaya, I.Y.; Narkevich, I.A.; Flisyuk, E.V.; Pozharitskaya, O.N. Methods of Extraction of Medicinal Plants. In Evidence-Based Validation of Herbal Medicine; Elsevier: Amsterdam, The Netherlands, 2022; pp. 771–796. [Google Scholar] [CrossRef]
- Mohd Fuad, F.; Mohd Nadzir, M. Ultrasound-Assisted Extraction of Asiaticoside from Centella asiatica Using Betaine-Based Natural Deep Eutectic Solvent. Ind. Crops Prod. 2023, 192, 116069. [Google Scholar] [CrossRef]
- Suresh, P.S.; Singh, P.P.; Anmol; Kapoor, S.; Padwad, Y.S.; Sharma, U. Lactic Acid-Based Deep Eutectic Solvent: An Efficient Green Media for the Selective Extraction of Steroidal Saponins from Trillium govanianum. Sep. Purif. Technol. 2022, 294, 121105. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.; Kang, M.; Li, Z.; Wang, X.; Jing, X.; Han, J. Sustainable ultrasound-assisted extraction of Polygonatum sibiricum saponins using ionic strength-responsive natural deep eutectic solvents. Ultrason. Sonochem. 2023, 100, 106640. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.P.; Pham, T.V.; Tran, T.N.H.; Vo, L.T.V.; Vu, T.T.; Pham, N.D.; Nguyen, D.Q. Ultrasonic-Assisted and Microwave-Assisted Extraction of Phenolics and Terpenoids from Abelmoschus Sagittifolius (Kurz) Merr Roots Using Natural Deep Eutectic Solvents. ACS Omega 2023, 8, 29704–29716. [Google Scholar] [CrossRef] [PubMed]
- Flisyuk, E.V.; Belokurov, S.S.; Narkevich, I.A.; Flisyuk, O.M.; Ivkin, D.Y. The Analysis of Process of Crushing in the Vibrocavitational Homogenizer. Drug Dev. Regist. 2020, 9, 53–58. [Google Scholar] [CrossRef]
- Belokurov, S.S.; Flisyuk, E.V.; Narkevich, I.A.; Luzhanin, V.G.; Shilov, S.V.; Novikova, K.O. Comparative Analysis of Perspective Extragation Methods for Receiving Extractions from Fenugreek Seeds. Drug Dev. Regist. 2019, 8, 49–55. [Google Scholar] [CrossRef]
- Gubina, G. Clinical applications of Aralia mandshurica tincture. In Medicinal Preparations from Plants; Turova, A., Ed.; Science Press: Moscow, Russia, 1988; pp. 263–269. [Google Scholar]
- Garkaev, L.H.; Kvakina, E.B.; Kuzmenko, T.S. Antistress Reactions and Activation Therapy. The Reaction of Activation as a Way to Health through Self-Organization; Processes Imedis: Moscow, Russia, 1998; p. 656. [Google Scholar]
- Markin, V.V.; Markina, L.D. Use of natural adaptogens for students. Sib. Med. Rev. 2007, 45, 54–58. [Google Scholar]
- Markina, L.D.; Markin, V.V. Forecasting of the development of dysadaptable conditions and algorithm of their effective correction. Pac. Med. J. 2008, 3, 30–36. [Google Scholar]
- Shrefova, L.R.; Vasilenko, A.Z.; Gagarinova, V.M. Increasing of nonspecific resistance of the body by means of pharmacy and nutraceutical products –the basis of respiratory diseases prevention. Prev. Clin. Med. 2011, 41, 98–102. [Google Scholar]
- Panossian, A.; Efferth, T. Network Pharmacology of Adaptogens in the Assessment of Their Pleiotropic Therapeutic Activity. Pharmaceuticals 2022, 15, 1051. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, J.; Zeng, Y.; Jin, S.; Liu, W.; Li, Z.; Qin, X.; Bai, Y. Traditional Uses, Phytochemistry, Pharmacology, Toxicity and Quality Control of Medicinal Genus Aralia: A Review. J. Ethnopharmacol. 2022, 284, 114671. [Google Scholar] [CrossRef]
- Petrochenko, A.A.; Orlova, A.; Frolova, N.; Serebryakov, E.B.; Soboleva, A.; Flisyuk, E.V.; Frolov, A.; Shikov, A.N. Natural Deep Eutectic Solvents for the Extraction of Triterpene Saponins from Aralia elata var. mandshurica (Rupr. & Maxim.) J. Wen. Molecules 2023, 28, 3614. [Google Scholar] [CrossRef]
- Gao, D.; Cho, C.-W.; Yang, Z.; Li, X.; Kang, J.-S. Identification and Quantitation of the Bioactive Components in Wasted Aralia elata Leaves Extract with Endothelial Protective Activity. Molecules 2023, 28, 5907. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Kim, O.J.; Kang, S.S.; Jeong, C. Araloside A, an antiulcer constituent from the root bark of Aralia elata. Biol. Pharm. Bull. 2005, 28, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, R.; Xie, X.; Sun, G.; Sun, X. Araloside C protects H9c2 cardiomyoblasts against oxidative stress via the modulation of mitochondrial function. Biomed. Pharm. 2019, 117, 109143. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wang, M.; Liu, X.; Zhang, J.; Xu, X.; Xu, H.; Sun, G.; Sun, X. Araloside C Prevents Hypoxia/Reoxygenation-Induced Endoplasmic Reticulum Stress via Increasing Heat Shock Protein 90 in H9c2 Cardiomyocytes. Front. Pharmacol. 2018, 9, 180. [Google Scholar] [CrossRef]
- Luo, Y.; Dong, X.; Yu, Y.; Sun, G.; Sun, X. Total aralosides of Aralia elata (Miq) seem (TASAES) ameliorate nonalcoholic steatohepatitis by modulating IRE1alpha-mediated JNK and NF-kappaB pathways in ApoE −/− mice. J. Ethnopharmacol. 2015, 163, 241–250. [Google Scholar] [CrossRef]
- Liu, G.; Feng, S.; Sui, M.; Chen, B.; Sun, P. Extraction and Identification of Steroidal Saponins from Polygonatum cyrtonema Hua Using Natural Deep Eutectic Solvent-synergistic Quartz Sand-assisted Extraction Method. J. Sep. Sci. 2023, 46, 2200823. [Google Scholar] [CrossRef]
- Lanjekar, K.J.; Rathod, V.K. Recovery and Separation of Glycyrrhizic Acid from Natural Deep Eutectic Solvent (NADES) Extract by Macroporous Resin: Adsorption Kinetics and Isotherm Studies. Prep. Biochem. Biotechnol. 2024, 54, 39–48. [Google Scholar] [CrossRef]
- Koh, Q.Q.; Kua, Y.L.; Gan, S.; Tan, K.W.; Lee, T.Z.E.; Cheng, W.K.; Lau, H.L.N. Sugar-Based Natural Deep Eutectic Solvent (NADES): Physicochemical Properties, Antimicrobial Activity, Toxicity, Biodegradability and Potential Use as Green Extraction Media for Phytonutrients. Sustain. Chem. Pharm. 2023, 35, 101218. [Google Scholar] [CrossRef]
- Ferreira, C.; Sarraguça, M. A Comprehensive Review on Deep Eutectic Solvents and Its Use to Extract Bioactive Compounds of Pharmaceutical Interest. Pharmaceuticals 2024, 17, 124. [Google Scholar] [CrossRef]
- Shen, L.; Pang, S.; Zhong, M.; Sun, Y.; Qayum, A.; Liu, Y.; Rashid, A.; Xu, B.; Liang, Q.; Ma, H.; et al. A Comprehensive Review of Ultrasonic Assisted Extraction (UAE) for Bioactive Components: Principles, Advantages, Equipment, and Combined Technologies. Ultrason Sonochem. 2023, 101, 106646. [Google Scholar] [CrossRef]
- Koraqi, H.; Aydar, A.Y.; Khalid, W.; Ercisli, S.; Rustagi, S.; Ramniwas, S.; Pandiselvam, R. Ultrasound-assisted extraction with natural deep eutectic solvent for phenolic compounds recovery from Rosa damascene Mill.: Experimental design optimization using central composite design. Microchem. J. 2024, 196, 109585. [Google Scholar] [CrossRef]
- Pavlić, B.; Mrkonjić, Ž.; Teslić, N.; Kljakić, A.C.; Pojić, M.; Mandić, A.; Stupar, A.; Santos, F.; Duarte, A.R.C.; Mišan, A. Natural Deep Eutectic Solvent (NADES) Extraction Improves Polyphenol Yield and Antioxidant Activity of Wild Thyme (Thymus serpyllum L.) Extracts. Molecules 2022, 27, 1508. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martín, M.; Cubero-Cardoso, J.; González-Domínguez, R.; Cortés-Triviño, E.; Sayago, A.; Urbano, J.; Fernández-Recamales, Á. Ultrasound-assisted extraction of phenolic compounds from blueberry leaves using natural deep eutectic solvents (NADES) for the valorization of agrifood wastes. Biomass Bioenergy 2023, 175, 106882. [Google Scholar] [CrossRef]
- Shikov, A.N.; Pozharitskaya, O.N.; Makarov, V.G.; Makarova, M.N. Anti-inflammatory effect of Pinus sibirica oil extract in animal models. J. Nat. Med. 2008, 62, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Vasylishin, M.S.; Budaeva, V.V.; Kukhlenko, A.A.; Karpov, A.G.; Ivanov, O.S.; Orlov, S.E.; Babkin, V.A.; Medvedeva, E.N. Extraction of arabinogalactan from sawdust of Larix sibirica in a rotary-pulsation apparatus. Polzunovsky Bull. 2010, 4, 168–173. [Google Scholar]
- Whaley, A.O.; Whaley, A.K.; Kovaleva, E.L.; Frolova, L.N.; Orlova, A.A.; Luzhanin, V.G.; Flisyuk, E.V.; Shigarova, L.V.; Shikov, A.N. The standardization of officinal medicinal plants used in the Eurasian Economic Union: Comparison with other pharmacopoeias. Phytochem. Rev. 2023, 2023, 1–71. [Google Scholar] [CrossRef]
- Liu, W.; Li, J.; Zhang, X.; Zu, Y.; Yang, Y.; Liu, W.; Xu, Z.; Gao, H.; Sun, X.; Jiang, X.; et al. Current Advances in Naturally Occurring Caffeoylquinic Acids: Structure, Bioactivity, and Synthesis. J. Agric. Food Chem. 2020, 68, 10489–10516. [Google Scholar] [CrossRef] [PubMed]
- Chhetri, D.R. Myo-Inositol and Its Derivatives: Their Emerging Role in the Treatment of Human Diseases. Front. Pharmacol. 2019, 10, 1172. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhang, Z.; Jiang, P.; Zhang, R.; Ni, D.; Yuan, Y.; Zhang, S. Characterization of Chikusetsusaponin IV and V Induced Apoptosis in HepG2 Cancer Cells. Mol. Biol. Rep. 2022, 49, 4247–4255. [Google Scholar] [CrossRef]
- Mckerrecher, D.; Pike, K.G.; Waring, M.J. Heteroaryl Benzamide Derivatives for Use as GLK Activators in the Treatment of Diabetes. EP Patent Application 2,027,113, 25 February 2009. [Google Scholar]
- Xia, P.; Mao, Y.; Liang, Z. Two Important Ginseng Plants in Southeast Asia: A Systematic Review of Their Traditional Uses, Botany, Phytochemistry, and Pharmacology. Acta Physiol. Plant 2022, 44, 105. [Google Scholar] [CrossRef]
- Tang, Y.; Xin, H.; Guo, M. Review on Research of the Phytochemistry and Pharmacological Activities of Celosia Argentea. Rev. Bras. De Farmacogn. 2016, 26, 787–796. [Google Scholar] [CrossRef]
- Muley, B.; Khadabadi, S.; Banarase, N. Phytochemical Constituents and Pharmacological Activities of Calendula Officinalis Linn (Asteraceae): A Review. Trop. J. Pharm Res. 2009, 8, 455–465. [Google Scholar] [CrossRef]
- Parvizpour, S.; Masoudi-Sobhanzadeh, Y.; Pourseif, M.M.; Barzegari, A.; Razmara, J.; Omidi, Y. Pharmacoinformatics-Based Phytochemical Screening for Anticancer Impacts of Yellow Sweet Clover, Melilotus Officinalis (Linn.) Pall. Comput. Biol. Med. 2021, 138, 104921. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Miyase, T.; Noguchi, H. Triterpenoid Saponins from Ilex Kudincha. J. Nat. Prod. 1999, 62, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, S.; Huang, W.; Ma, M.; Chen, X.; Zeng, W.; Liang, K.; Wang, H.; Bi, Y.; Li, X. Design, Synthesis, and Biological Evaluation of Hederagenin Derivatives with Improved Aqueous Solubility and Tumor Resistance Reversal Activity. Eur. J. Med. Chem. 2021, 211, 113107. [Google Scholar] [CrossRef]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-Based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | tR (min) | m/z [M-H]− Observed | m/z [M-H]− Calculated | Elemental Composition [M-H]− | MS2 Fragmentation Patterns—Product Ions, m/z (rel. Intensity) | Δm (ppm) | Assignment | Fold Change, log2(FC) * |
|---|---|---|---|---|---|---|---|---|
| 1 | 2.8 | 487.1089 | 487.1093 | C20H23O14− | 191.0546 (5), 295.0452 (5), 323.0779 (2), 353.0869 (100), 371.0973 (10), 469.0923 (7) | 0.8 | Malonylhydroxy-dihydrocaffeoyl-quinic acid | 2.9 ↓ |
| 2 | 2.9 | 469.0987 | 469.0988 | C20H21O13− | 191.0559 (10), 353.0878 (100) | 0.3 | Malonylcaffeoyl quinic acid | 4.3 ↓ |
| 3 | 3.8 | 517.1586 | 517.1563 | C22H29O14− | 191.0559 (40), 309.0946 (5), 353.0871 (100) | −4.5 | Pentofuranosyl-dihydrocaffeoyl quinic acid | 2.9 ↑ |
| 4 | 4.6 | 577.1634 | 577.1622 | C20H33O19− | 181.0716 (12), 261.0612 (9), 279.0716 (100), 297.0822 (33), 377.1450 (2), 443.1400 (10), 461.1507 (27), 559.1511 (5) | 2.1 | Myo-inositol derivatives | 2.9 ↑ |
| 5 | 5.5 | 225.0761 | 225.0768 | C11H13O5− | 107.0503 (5), 137.0973 (4), 163.0764 (15), 181.0869 (23), 207.0660 (100), 225.0761 (3) | 1.0 | 3-hydroxy-5-(2-methoxy-1-methylethoxy)benzoate | 3.6 ↓ |
| 6 | 6.1 | 249.0550 | 249.0557 | C16H9O3− | 115.0038 (20), 133.0143 (100), 205.0504 (7), 231.0664 (3), 249.0550 (3) | 2.8 | Malic acid derivative | 3.4 ↓ |
| 7 | 6.1 | 705.1675 | 705.1672 | C32H33O18− | 339.0503 (3), 487.1212 (3), 513.1025 (100) | −0.4 | Caffeoylquinic acid dimer | 3.9 ↑ |
| 8 | 8.9 | 243.1237 | 243.1238 | C12H19O5− | 99.0089 (7), 181.1233 (10), 199.1338 (6), 207.1024 (5), 225.1129 (100), 243.1237 (6) | −0.4 | Trihydroxy-dodecadienoic acid | 3.2 ↓ |
| 9 | 12.4 | 961.4589 | 961.4569 | C47H74ClO18− | 565.3505 (3), 631.3851 (3), 733.5586 (3), 763.4248 (10), 793.4357 (8), 925.4802 (100) | −2.1 | Chikusetsusaponin IV (syn. Araloside A) | 5.5 ↑ |
| 10 | 12.8 | 793.4367 | 793.4380 | C42H65O14− | 437.3418 (6), 455.3522 (65), 483.3470 (98), 551.3730 (63), 569.3835 (45), 631.3836 (20), 731.4355 (22), 793.4367 (100) | 1.6 | Oleanolic acid hexuronide–hexoside | 7.9 ↑ |
| 11 | 12.8 | 763.4286 | 763.4274 | C41H63O13− | 437.3411 (5), 455.3524 (30), 523.3785 (27), 569.3837 (65), 613.3735 (100), 632.3838 (15), 763.4286 (45) | −1.6 | Pseudoginsenoside Rp1 | 5.5 ↑ |
| No | tR (min) | m/z [M-H]− Observed | m/z [M-H]− Calculated | Elemental Composition [M-H]− | MS2 Fragmentation Patterns—Product Ions, m/z (rel. Intensity) | Δm (ppm) | Assignment | Fold Change, log2(FC) * |
|---|---|---|---|---|---|---|---|---|
| 12 | 2.9 | 469.0987 | 469.0988 | C20H21O13− | 191.0559 (10), 353.0878 (100) | 0.3 | 5-O-malonil-caffeoylquinic acid | 4.3 ↓ |
| 13 | 9.3 | 1167.4970 | 1167.4996 | C54H84ClO25− | 697.3640 (3), 969.4656 (5), 1131.5182 (100) | 2.2 | 3,16-dihydroxyolean-12-en-23,28-dioic acid 28-O-6-O-3-hydroxy-3-methylglutaryl-hexopyranosyl-hexopyranosyl-hexopyranosyl ester | 3.5 ↑ |
| 14 | 9.4 | 1137.4930 | 1137.4890 | C53H82ClO24− | 823.8554 (3), 939.4536 (5), 975.4535 (5), 1101.5084 (100) | −3.5 | Celosin I | 3.8 ↑ |
| 15 | 9.6 | 1005.4430 | 1005.4467 | C48H74ClO20− | 688.4667 (3), 730.2089 (3), 842.4215 (5), 969.4651 (100) | 3.7 | 3-O-hexopyranosyl-hexopyranosyl-hexuronopyranosyl melilotigenin isomer 1 | 3.7 ↑ |
| 16 | 9.7 | 1007.4580 | 1007.4624 | C48H76ClO20− | 777.4055 (5), 799.3860 (20), 911.4629 (5), 939.4566 (25), 971.4799 (100) | 4.4 | Sophoraflavoside II isomer 1 | 3.6 ↑ |
| 17 | 9.8 | 941.4707 | 941.4752 | C47H73O19− | 465.3363 (3), 537.3576 (15), 583.3629 (100), 627.3525 (35), 669.3629 (15), 733.4151 (5), 777.4045 (40), 819.4150 (10), 941.4707 (5) | 4.8 | Dihydrogypsogenin 3-O-pentopyranosyl-hexpyranoxyl-hexuronopyranoside isomer 1 | 3.7 ↑ |
| 18 | 10.5 | 955.4914 | 955.4908 | C48H75O19− | 455.3535 (5), 551.3747 (5), 569.3853 (12), 613.3745 (6), 748.4362 (7), 793.4387 (100), 834.4436 (8), 955.4914 (20) | −0.6 | Calendulaglycoside C | 5.0 ↑ |
| 19 | 11.3 | 1005.4440 | 1005.4467 | C48H74ClO20− | 404.2202 (3), 572.4813 (3), 842.4193 (3), 942.4771 (3), 969.4661 (100) | 2.7 | 3-O-hexopyranosyl-hexopyranosyl-hexuronopyranosyl melilotigenin isomer 2 | 4.7 ↑ |
| 20 | 11.5 | 1007.4573 | 1007.4624 | C48H76ClO20− | 939.4597 (3), 971.4816 (100) | 5.0 | Sophoraflavoside II isomer 2 | 3.9 ↑ |
| 21 | 11.6 | 971.4841 | 971.4857 | C48H75O20− | 407.3312 (15), 471.3464 (10), 567.3672 (15), 747.4294 (10), 790.4078 (30), 809.4285 (45), 925.4777 (7), 971.4841 (100) | 1.6 | Sophoraflavoside II isomer 3 | 3.7 ↑ |
| 22 | 12.1 | 941.4711 | 941.4752 | C54H87O24 | 565.3526 (10), 609.3419 (7), 745.4152 (87), 777.4048 (3), 807.4152 (30), 941. 4695 (100) | 4.4 | Dihydrogypsogenin 3-O-pentopyranosyl-hexpyranoxyl-hexuronopyranoside isomer 2 | 3.8 ↑ |
| 23 | 12.3 | 961.4589 | 961.4569 | C47H74ClO18− | 455.3523 (35), 551.3732 (40), 565.3505 (3), 731.4357 (100), 763.4248 (10), 793.4357 (8), 925.4808 (65) | 3.8 | Chikusetsusaponin IV (syn. Araloside A) | 4.7 ↑ |
| 24 | 12.6 | 845.4110 | 845.4096 | C42H66ClO15− | 407.3310 (50), 477.3728 (30), 539.3671 (20), 567.3671 (33), 587.3570 (50), 647.3779 (100), 747.4291 (8), 809.4301 (30) | −1.7 | Ilexoside XLVIII | 3.9 ↑ |
| 25 | 12.7 | 777.4057 | 777.4067 | C41H61O14− | 469.3318 (3), 565.3526 (3), 583.3625 (15), 627.3519 (100), 645.3624 (20), 777.4057 (3) | 1.3 | 3-O-hexopyranosyl-pentopyranosylurs-12,18-diene-24,28-dioic acid | 5.3 ↑ |
| 26 | 12.8 | 793.4367 | 793.4380 | C42H65O14− | 455.3522 (70), 483.3470 (100), 537.3574 (30), 551.3730 (65), 569.3834 (45), 613.3732 (20), 631.3835 (25), 731.4354 (25), 793.4367 (95) | 1.6 | Oleanolic acid hexoside–hexuronide | 3.9 ↑ |
| 27 | 12.9 | 779.4216 | 779.4223 | C41H63O14− | 455.3525 (3), 523.3778 (10), 585.3778 (35), 629.3673 (100), 647.3779 (20), 779.4216 (3) | 0.9 | 3-O-pentopyranosyl-hexuronopyranosyl hederagenin | 3.8 ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaleta, A.; Frolova, N.; Orlova, A.; Soboleva, A.; Osmolovskaya, N.; Flisyuk, E.; Pozharitskaya, O.; Frolov, A.; Shikov, A. The Effects of Selected Extraction Methods and Natural Deep Eutectic Solvents on the Recovery of Active Principles from Aralia elata var. mandshurica (Rupr. & Maxim.) J. Wen: A Non-Targeted Metabolomics Approach. Pharmaceuticals 2024, 17, 355. https://doi.org/10.3390/ph17030355
Kaleta A, Frolova N, Orlova A, Soboleva A, Osmolovskaya N, Flisyuk E, Pozharitskaya O, Frolov A, Shikov A. The Effects of Selected Extraction Methods and Natural Deep Eutectic Solvents on the Recovery of Active Principles from Aralia elata var. mandshurica (Rupr. & Maxim.) J. Wen: A Non-Targeted Metabolomics Approach. Pharmaceuticals. 2024; 17(3):355. https://doi.org/10.3390/ph17030355
Chicago/Turabian StyleKaleta, Alyona, Nadezhda Frolova, Anastasia Orlova, Alena Soboleva, Natalia Osmolovskaya, Elena Flisyuk, Olga Pozharitskaya, Andrej Frolov, and Alexander Shikov. 2024. "The Effects of Selected Extraction Methods and Natural Deep Eutectic Solvents on the Recovery of Active Principles from Aralia elata var. mandshurica (Rupr. & Maxim.) J. Wen: A Non-Targeted Metabolomics Approach" Pharmaceuticals 17, no. 3: 355. https://doi.org/10.3390/ph17030355
APA StyleKaleta, A., Frolova, N., Orlova, A., Soboleva, A., Osmolovskaya, N., Flisyuk, E., Pozharitskaya, O., Frolov, A., & Shikov, A. (2024). The Effects of Selected Extraction Methods and Natural Deep Eutectic Solvents on the Recovery of Active Principles from Aralia elata var. mandshurica (Rupr. & Maxim.) J. Wen: A Non-Targeted Metabolomics Approach. Pharmaceuticals, 17(3), 355. https://doi.org/10.3390/ph17030355